
Abstract
Tectorigenin (TEC) is an effective compound that derived from many plants, such as Iris unguicularis, Belamcanda chinensis and Pueraria thunbergiana Benth. Evidence suggested that TEC has anti-tumor, anti-oxidant activity, anti-bacterial and anti-inflammatory effects. In addition, there has some evidence indicated that TEC is a potential anti-stroke compound; however, its specific roles and associated mechanism have not yet been elucidated. In the present study, we aimed to investigate the anti-inflammatory, anti-oxidant activity and anti-apoptosis effects of TEC on oxygen-glucose deprivation/reperfusion (OGD/R)-induced HT-22 cells, and clarified the relevant mechanisms. Here, we observed that TEC significantly promoted cell survival, impeded cell apoptosis, inhibited ROS and inflammatory cytokines IL-1β, IL-6, TNF-α production in OGD/R-induced HT-22 cells. Moreover, TEC activated PI3K/AKT signal pathway, increased PPARγ expression and inhibited NF-κB pathway activation in OGD/R-induced HT-22 cells. Further studies indicated that PPARγ inhibitor GW9662 activated NF-κB pathway after TEC treatment in OGD/R-induced HT-22 cells. Also, PI3K/AKT inhibitor LY294002, PPARγ inhibitor GW9662 and NF-κB activator LPS both reversed the effects of TEC on OGD/R-induced HT-22 cell biology. Taken together, this research confirmed that TEC benefit to HT-22 cell survival and against OGD/R damage through the PI3K/AKT and PPARγ/NF-κB pathways. These results indicated that TEC might be an effective compound in the treatment for ischemic brain injury.
Keywords
Introduction
Ischemic stroke (IS) account for about 70–80% percent of all strokes, and also characterized by high disability, long-term morbidity at the national level, and mortality. 1 Ischemic strokes were considered to be thromboembolic and atherosclerotic events and occurred when thrombosis which blocked cerebral vessel, and leaded to severe pathological processes, such as subsequent infarction and cellular apoptosis. 2,3 Finding ways to restore blood flow and neuronal protection is a common principle of IS treatment. 4 Recently, using mechanical thrombectomy to therapy ischemic stroke, has achieved highly effective; however, compared to the successful recanalization rate, the functional independence after treatment is barely satisfactory. 5 Moreover, the thrombolytic drug recombinant tissue plasminogen activator has been proven to be effective in only 5% of IS patients because of ischemia-reperfusion (I/R) injury which induced by blood restoration after ischemia. 6 Thus, it is very important to alleviate I/R injury for the treatment of IS.
Traditional Chinese medicines, or natural products, have been proven to be an important source of pharmaceutical formulations and is widely used by almost one-fifth of the world’s population. 7,8 It has fewer complications and low toxicity profile compared with western medicines, thus, the study regarding to traditional Chinese medicines has attracted more and more attention. 9,10 Tectorigenin (TEC), an active component of the traditional medicine, is an O-methoxylated isoflavone which isolated from Iris unguicularis, Belamcanda chinensis, and Pueraria thunbergiana Benth. 11,12 It is now well established that TEC exert pharmacological actions in types of disease, such as anti-proliferation effects, 13 anti-inflammatory effects, 14,15 anti-tumor effects, 16,17 anti-bacterial effects, 18 selective estrogen receptor modulation 19 and free radical neutralization. 20 Regarding neuroprotection, evidence indicated that TEC promoted MPP+-induced SH-SY5Y cell survival and reduced oxidative stress. 21 However, little information is available to reveal the effects and the potential molecular mechanism of TEC on ischemic stroke and cerebral I/R injury.
According to the published estimates, peroxisome proliferator activated receptor gamma (PPARγ) and nuclear factor-kappa B (NF-κB) pathways have proven to be associated with cerebral I/R injury. 22 –24 There has convincing evidence that NF-κB was activated in neurons with cerebral ischemia and play a pivotal role in inflammatory cascade reaction and brain damage. 25 PPARγ exert anti-inflammatory and anti-oxidant effects in neurodegenerative disease, including Alzheimer’s disease and Parkinson’s disease, and ischemic stroke. 26 –28 In addition, there has some evidence indicated that PPARγ can inhibit NF-κB pathway activation. 29
Based on the results from this study, we hypothesized that TEC play an unquestionable role in cerebral I/R injury through inhibiting OGD/R-induced HT-22 cytotoxicity, apoptosis, ROS production and inflammatory cytokines IL-1β, IL-6, TNF-α secretion. We also demonstrated that PI3K/AKT and the PPARγ/NF-κB pathways were associated with the effects of TEC in OGD/R-induced HT-22 cell model.
Methods
Chemicals
TEC purchased from Sigma-Aldrich (St. Louis, MO, USA), The CAS number was 548-77-6 and the purity was determined over 98% by HPLC. The chemical structure showed in Figure 1. The PPAR-γ inhibitor GW9662, and NF-κB activator lipopolysaccharides (LPS, E. coli 0111: B4) were also purchased from Sigma-Aldrich.

The chemical structure of TEC.
Cell culture and in vitro I/R model
Mouse hippocampal neuron line HT22 purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were maintained in DMEM containing 10% (v/v) FBS (Gibco, Waltham, MA, USA), 100 ng/mL streptomycin, and 100 μ/mL penicillin. Cells cultured in a saturated and humidified incubator with 5% (v/v) CO2 and 95% (v/v) air.
For establishing in vitro I/R model, HT22 cells were treated with oxygen-glucose deprivation/reperfusion (OGD/R). HT22 cells were maintained into glucose-deprivation DMEM buffer and cultured in hypoxic chamber with 1% (v/v) O2/ 94% (v/v) N2/5% (v/v) CO2 for 2 h at 37°C. Moreover, subsequently, cells cultured in standard culture medium and normal culture condition (5% CO2 and 95% air) as described above for 24 h as described. 30
Cell treatment
Cells were seeded in a 24-well plate with 1 × 104 cells per well, and pre-treated with 0, 1, 5, 10 or 20 μM TEC for 1 h before the exposure of OGD/R. In addition, HT22 cells were pre-treated with 10 μM TEC and 20 μM PPARγ inhibitor GW9662 for 1 h and then exposure with OGD/R, or pre-treated with 10 μM TEC for 1 h and then exposure with LPS (400 μg/ml) and OGD/R, or pre-treated with 10 μM TEC for 1 h and then exposure with PI3K/AKT inhibitor LY294002 (20 μM) and OGD/R.
CCK-8 assay
Cell viability was determined by using a Cell Counting Kit-8 (CCK8, Dojindo, Kumamoto, Japan) according to the manufacturer’s protocol. HT22 cells plated into 96-well plates and treated with TEC or transfected as above. For measuring cell viability, the CCK-8 reagent was added to each well and incubated for another 4 h. In order to determine cell viability, absorbance at 450 nm was read and recorded by a microplate reader.
Lactate dehydrogenase (LDH) assay
Cytotoxicity of TEC on OGD/R-induced HT22 cells was assayed by LDH Cytotoxicity Detection kit (Thermo Fisher Scientific, Houston, TX, USA) according to the manufacturer’s instructions. Briefly, HT22 cells were treated and cultured as described above, and then the culture medium was collected and transferred to 96-well plate at 50 μl/well; subsequently, the reaction mixture was added and incubated for 30 min at room temperature. Optical density (OD) at 490 nm was counted by a microplate reader to indicate the LDH activity.
Caspase-3 activity
HT22 cells were treated and cultured as described above. Caspase-3 Colorimetric Assay Kit (Abcam, Cambridge, MA, USA) was used to determine caspase-3 activity according to the manufacturers’ protocols. Absorbance at 450 nm was measured using a microplate reader.
ELISA
HT-22 cells were treated and transfected as above. The supernatant of the cell culture was collected and inflammatory cytokines IL-1β, IL-6, TNF-α production were determined by the ELISA kit (R&D Systems, Minneapolis, MN, United States) as recommended by the manufacturer.
Measurement of ROS generation
The production of intracellular ROS in cells was checked by 6-carboxy-2070-di-chlorodihydrofluorescein diacetate (DCFH-DA) (Sigma, St. Louis, MO, USA). Briefly, cells were seeded in 96-well black plates and exposure to specific treatment. Cells were then incubated with 20 μM DCFH-DA in serum-free medium at 37°C for 30 min in the dark. Cells were washed and the fluorescence at 488 nm excitation and 530 nm emission was measured by using a fluorescence microplate reader.
RT-PCR
Total RNA extracted from HT-22 cells by RNAzol reagent (Molecular Research Center, Cincinnati, OH, USA) and the concentration was calculated. Isolated RNAs were reverse transcribed into cDNAs by using a PrimeScript™ RT Master Mix (TaKaRa). Relative mRNA expression levels of PPARγ, IL-1β, IL-6 and TNF-α were assayed by using a SYBR Premix ex Taq (Takara Bio, Inc., Otsu, Japan), 7500 Real-Time PCR system (Applied Biosystems, Carlsbad, USA) according to the manufacturer’s instructions. The primers used in this study as shown in Table 1. β-actin was used as control and 2−ΔΔCt method was used to analysis relative gene expression.
PCR primers sequences used in RT-PCR.

Western blot
Total protein was extracted by using cell lysis buffer (Beyotime, Shanghai, China) and the protein concentration was determined by using the Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific Inc.) Thirty micrograms of protein in each sample (30 µg/lane) were loaded and separated by 10% SDS-PAGE and subsequently transferred to 0.45 mm PVDF membranes (Millipore, Bedford, MA, USA). β-actin (ab8227, 1:2000), PPARγ (ab45036, 1:500), AKT (ab8805, 1:500), pAKT (ab38449, 1:500), p65 (ab16502, 0.5 µg/ml), p-p65 (ab86299, 1:5000), and cytochrome c (ab133504, 1:5000
Statistical analysis
In this study, GraphPad Prism version 5 software (GraphPad Software, Inc., USA) was used to analyze the data. Data were presented as means ± standard deviation (SD). Comparisons between groups were determined by one-way analysis of variance (ANOVA) and followed by Bonferonni’s post hoc test. P < 0.05 was considered statistically significant. All experiment was performed in triplicates, independently.
Results
TEC exposure suppressed OGD/R-induced HT-22 cell injury
To mimic cerebral ischemia-reperfusion injury in vitro, HT-22 cells were treated with OGD/R, and cell survival and LDH release was determined. As shown in Figure 2A and 2B, OGD/R treatment impeded cell survival and promoted cell LDH release, the differences were significant when compared with the control group (p < 0.05). Further studies confirmed that different concentration TEC exposure all contributed to OGD/R-induced HT-22 cell survival, and 1, 5, 10 or 20 μM TEC treatment increased cell survival to 49.6%, 67.2%, 82.6% and 87.4%, respectively. Moreover, we next confirmed that TEC exposure markedly inhibited LDH release in OGD/R-induced HT-22 cell injury, when compared with the OGD/R group (Figure 2A and 2B, p < 0.05).

TEC exposure increased OGD/R-induced HT-22 cell survival and inhibited LDH release. Cells were seeded in a 24-well plates with 1 × 104 cells per well, and pre-treated with 0, 1, 5, 10 or 20 μM TEC for 1 h before the exposure of OGD/R. (A) HT-22 cell survival was determined by CCK-8 assay; (B) LDH Cytotoxicity Detection kit was used to measure LDH release. All experiment was performed in triplicates, independently. * indicated p < 0.05 compared with control, # indicated p < 0.05 compared with OGD/R.
TEC treatment alleviated OGD/R-induced cell apoptosis
We next investigated whether pretreatment with TEC (10 μM) has protective effects on OGD/R-induced HT-22 cell apoptosis. Therefore, cytochrome C protein expression and caspase-3 activity have been determined, the results was presented as Figure 3. The results suggested that OGD/R induced cytochrome C expression compared with the control group (Figure 3A, p < 0.05). Moreover, regarding OGD/R injury, the protein expression of cytochrome C was significantly decreased in TEC treatment group (Figure 3A, p < 0.05). We further investigated the effect of TEC on caspase-3 activity in OGD/R-induced HT-22 cells. As shown in Figure 3B, OGD/R exposure increased caspase-3 activity, whereas TEC pretreatment significantly decreased caspase-3 activity when compared with the OGD/R group (p < 0.05). Further studies suggested that OGD/R exposure also inhibited Bcl-2 and increased cle-caspase-3 protein expression, whereas TEC pretreatment markedly increased BCl-2 expression and decreased caspase-3 expression when compared with the OGD/R group (p < 0.05). Thus, these results showed that TEC treatment inhibited OGD/R induced apoptosis in HT-22 cells.

TEC exposure decreased OGD/R-induced HT-22 cell apoptosis. Cells were pre-treated with 10 μM TEC for 1 h before the exposure of OGD/R. (A) Cytochrome C protein expression was measured by Western blot; (B) Caspase-3 activity was determined by Caspase-3 Colorimetric Assay Kit; (C) Bcl-2 and cle-caspase-3 protein expression were measured by Western blot. All experiment was performed in triplicates, independently. * indicated p < 0.05 compared with control, # indicated p < 0.05 compared with OGD/R.
TEC treatment reduced OGD/R-induced ROS production
To determine the effects of TEC on oxidative stress, OGD/R

TEC exposure suppressed OGD/R-induced ROS production in HT-22 cells. Cells were pre-treated with 10 μM TEC for 1 h before the exposure of OGD/R.
TEC exposure inhibited OGD/R-induced cell inflammation
Next, we investigated the anti-inflammatory effects of TEC in OGD/R-induced HT-22 cells. OGD/R treatment resulted in a significant elevation of interleukin 1β (IL-1β), interleukin 6 (IL-6) and tumor necrosis factor (TNF-α) in HT22 cells. And, IL-1β, IL-6 and TNF-α level were all remarkably reduced in HT22 cells after treatment with TEC compared with OGD/R group as measured by ELISA (Figure 5A, p < 0.05). For confirmation the inhibitory effects of TEC on OGD/R-induced HT-22 cells, we next assessed IL-1β, IL-6 and TNF-α mRNA levels by RT-PCR. As shown in Figure 5B, TEC exposure indeed impeded IL-1β, IL-6 and TNF-α mRNA expression which induced by OGD/R in HT22 cells. Above all, these results suggested that TEC play anti-inflammatory roles in OGD/R-induced HT-22 cells.

TEC exposure suppressed OGD/R-induced inflammation in HT-22 cells. Cells were pre-treated with 10 μM TEC for 1 h before the exposure of OGD/R. (A) IL-1β, IL-6 and TNF-α level were measured by ELISA; (B) IL-1β, IL-6 and TNF-α level were measured by RT-PCR. All experiment was performed in triplicates, independently. * indicated p < 0.05 compared with control, # indicated p < 0.05 compared with OGD/R.
TEC play negative role in OGD/R-induced PI3K/AKT inactivation
To further investigate the depth molecular mechanisms of the neuroprotection effect of TEC in OGD/R-induced HT-22 cells, western blot was used to measure the protein expression of pAKT. As shown in Figure 6, we observed that OGD/R arrangement resulted into a significant reduction in pAKT expression, while TEC exposure partially eliminated the inhibitory role of OGD/R on pAKT expression (Figure 6, p < 0.05). Moreover, the total AKT level was not obviously change in the different groups (Figure 6, p > 0.05). Further studies suggested that PI3K/AKT inhibitor LY294002 treatment reversed the effects of TEC on OGD/R-induced HT22 cell survival, LDH release, cell apoptosis, ROS production, and inflammation (Figure 7A–E, p < 0.05). These results indicated that PI3K/AKT signal pathway involved in the effects of TEC on OGD/R-induced HT22 cell.

TEC exposure promoted OGD/R-induced PI3K/AKT activation. Cells were pre-treated with 10 μM TEC for 1 h before the exposure of OGD/R. Western blot was used to measure the protein expression of AKT and pAKT. All experiment was performed in triplicates, independently. *indicated p < 0.05 compared with control, # indicated p < 0.05 compared with OGD/R.

PI3K/AKT inhibitor LY294002 (LY) reversed the effects of TEC on OGD/R-induced HT22 cells. HT22 cells were pre-treated with 10 μM TEC or TEC + 20 μM LY for 1 h and then exposured with OGD/R. (A) HT-22 cell survival was determined by CCK-8 assay; (B) LDH Cytotoxicity Detection kit was used to measure LDH release; (C) Caspase-3 activity was determined by Caspase-3 Colorimetric Assay Kit; (D) ROS production was measured by DCFH-DA assay; (E) IL-1β, IL-6 and TNF-α level were measured by RT-PCR. All experiment was performed in triplicates, independently. * indicated p < 0.05 compared with control, # indicated p < 0.05 compared with OGD/R, $ indicated p < 0.05 compared with OGD/R + TEC.
TEC contributed to the expression of PPARγ/NF-κB pathways in OGD/R-induced HT-22 cells
We also evaluated the level of PPARγ, p65 and p-p65 protein expression in OGD/R-induced HT-22 cells which treated by TEC with the concentration at 10 μM. As compared to control group, PPARγ expression was markedly decrease after OGD/R treatment (Figure 8, p < 0.05), while its expression was obviously increase in OGD/R + TEC treatment group when compared with the OGD/R treatment alone (Figure 8, p < 0.05). The effect of TEC on NF-κB pathway is opposite to that of PPARγ. The results suggested that OGD/R exposure induced p-p65 expression, whereas TEC treatment inhibited its expression in OGD/R induced HT22 cells (Figure 8, p < 0.05). Further studies confirmed that PPARγ inhibitor GW9662 partially abolished OGD/R-induced p-p65 expression. The above results showed that TEC inhibited NF-κB pathways activation dependent on the expression of PPARγ in OGD/R-induced HT-22 cells.
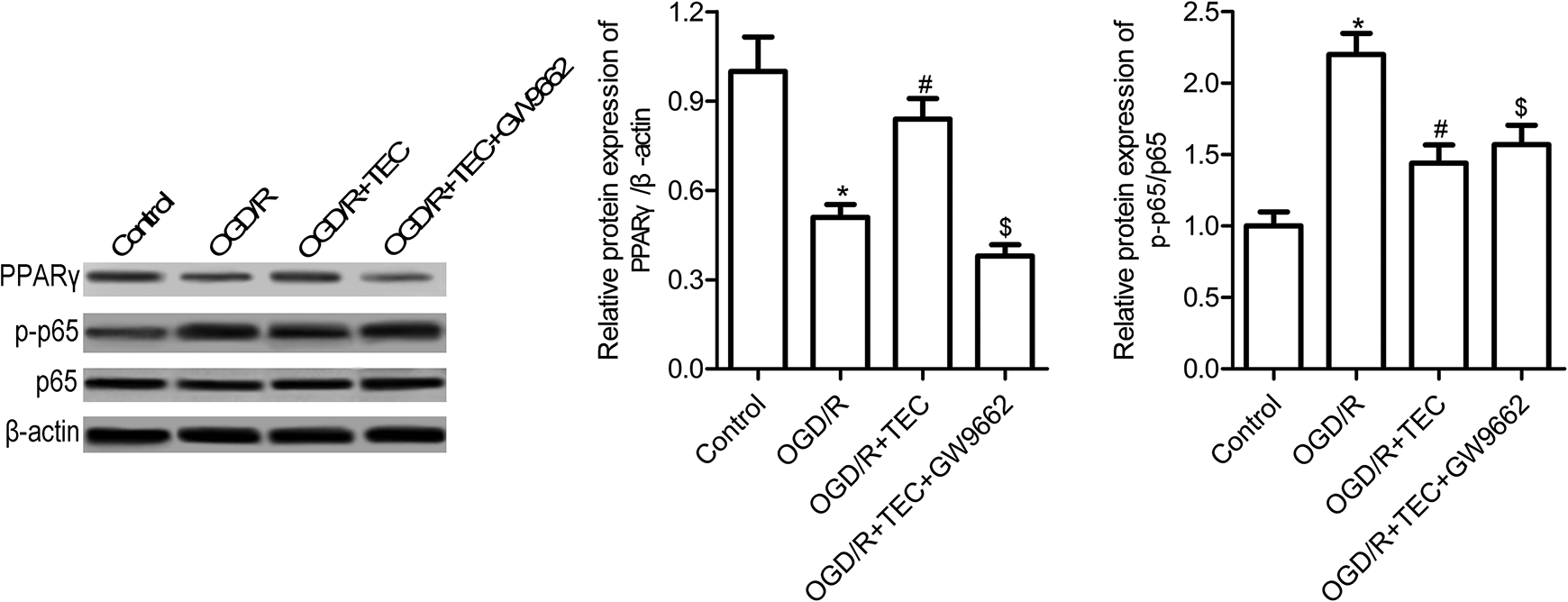
TEC exposure mediated PPARγ/NF-κB pathways. HT22 cells were pre-treated with 10 μM TEC or TEC + 20 μM PPARγ inhibitor GW9662 for 1 h and then exposured with OGD/R. Western blot was used to measure the protein expression of PPARγ, p65 and p-p65. All experiment was performed in triplicates, independently. * indicated p < 0.05 compared with control, # indicated p < 0.05 compared with OGD/R, $ indicated p < 0.05 compared with OGD/R + TEC.
PPARγ inhibitor or NF-κB activator abolished TEC mediated neuroprotection
Finally, to determine whether PPARγ/NF-κB pathways involved in the role of TEC, we detected the effect of PPARγ inhibitor GW9662 and NF-κB activator LPS on TEC function. As shown in Figure 8, compared with the OGD/R + TEC group, GW9662 or LPS treatment inhibits TEC induced HT22 cell survival, increased LDH release, cell apoptosis and ROS production, and promoted IL-1β, IL-6 and TNF-α secretion in HT22 cells treated with OGD/R (Figure 9A–E, p < 0.05). These results indicated that GW9662 or LPS treatment reversed the effects of TEC on OGD/R-induced HT22 cell biology. TEC plays neuroprotective effects in OGD/R-induced HT22 cell dependent on PPARγ/NF-κB pathways.

PPARγ/NF-κB pathways contributed to the effects of TEC on OGD/R-induced HT22 cells. HT22 cells were pre-treated with 10 μM TEC or TEC + 20 μM PPARγ inhibitor GW9662 for 1 h and then exposured with OGD/R, or pre-treated with 10 μM TEC for 1 h and then exposured with LPS (400 μg/ml) and OGD/R. (A) HT-22 cell survival was determined by CCK-8 assay; (B) LDH Cytotoxicity Detection kit was used to measure LDH release; (C) Caspase-3 activity was determined by Caspase-3 Colorimetric Assay Kit; (D) ROS production was measured by DCFH-DA assay; (E) IL-1β, IL-6 and TNF-α level were measured by RT-PCR. All experiment was performed in triplicates, independently. * indicated p < 0.05 compared with control, # indicated p < 0.05 compared with OGD/R, $ indicated p < 0.05 compared with OGD/R + TEC.
Discussion
Cerebral ischemic injury is a serious cerebrovascular disease, which is accompanied by brain tissue damage, neurological deficits, inflammation, and cellular apoptosis. 31,32 Blood reperfusion resulted in more severe secondary tissue damage and additional adjacent brain tissue injury. 33 In addition, there are few therapy options for alleviating cerebral I/R injury, thus, the effective agents that contributed to the injury are urgent to explore. In the present study, we used OGD/R-induced HT-22 cells to mimic cerebral I/R injury in vitro and yielded three main findings: Firstly, we observed that tectorigenin (TEC) exerted neuroprotective effects in I/R injury in vitro through significantly increased OGD/R-induced HT-22 cell viability, suppressed cell apoptosis, ROS production and cell inflammation; Secondly, we confirmed that TEC mediated PI3K/AKT and the PPARγ/NF-κB pathways in OGD/R-induced HT-22 cells; Finally, we demonstrated that PPARγ/NF-κB pathways is crucial in the function of TEC.
Isoflavones have been widely used in the cosmetic, food, and medicinal sectors. As the bioactive compounds, isoflavones exert pharmacological and physiological function, such as anti-free radical, cardiovascular protection, anti-cancer, anti-oxidation, and neuroprotective effects in cerebral ischemia stroke, Parkinson disease and Alzheimer disease. 34 –39 TEC, a compound of flowers of Pueraria lobata, may be a potential anti-stroke isoflavones and play roles in the prevention and treatment of stroke. 37 Also, emerging studies have revealed that TEC contributed to Parkinson’s disease through inhibiting MPP+-induced SH-SY5Y cytotoxicity, ROS production and apoptosis. 21 In agreement with the earlier reports, the study present here confirmed that TEC exerts neuroprotective effects in OGD/R-induced HT-22 cell damage. We discovered that TEC treatment impeded OGD/R-induced HT-22 cell injury, apoptosis, and oxidative stress. Moreover, evidence suggested that inflammatory response has been implicated in cerebral ischemic injury when the cerebral blood flow is blocked. 40,41 TEC has anti-inflammatory effects in types of diseases. 11,42 in the present study, we observed that TEC exposure markedly suppressed OGD/R-induced HT-22 cell inflammatory cytokines secretion.
Further clarification of the associated molecular mechanism of tectorigenin underlying the OGD/R-induced HT-22 cell biology is of great importance. Jeong et al. confirmed that flavonoids could inhibit PI3K/Akt signaling pathway in immunodeficiency virus type 1 (HIV-1)-infected cytoprotective macrophages. 43 However, Wang et al. confirmed that TEC can restore the impaired PI3K signaling in endothelium with insulin resistance. 44 Also, it is shown by other studies that TEC can ameliorate hyperglycemia through regulating the PPARγ and NF-κB signaling pathways. 45 In the present study, we found that TEC could promote PI3K/Akt signaling pathway activation. Our study also examined the PPARγ expression and NF-kB p65 protein phosphorylation, results indicated that TEC can mediated PPARγ/NF-κB signaling pathways.
In conclusion, TEC markedly impeded OGD/R-induced HT-22 cell damage, increased cell survival, inhibited cell apoptosis, oxidative stress and inflammation. We also confirmed that PI3K/Akt and PPARγ/NF-κB signaling pathways involved in the function of TEC.
Footnotes
Authors’ note
Li Yao and Meili Yang contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.