
Abstract
Background:
Ursolic acid (UA) is a natural pentacyclic triterpenoid that is known for its benefits under several pathological conditions. Cisplatin (CP) is among the most preferred chemotherapeutic agents; however, its nephrotoxicity limits its clinical utility.
Purpose:
This study was aimed to determine the role of UA in the reduction of CP-induced nephrotoxicity and mitigation of pro-inflammatory cytokines and apoptosis in a rat model.
Methodology:
Male Wistar rats were randomized into vehicle control, CP (7.5 mg/kg), UA 10 mg/kg, and CP with UA 5 and 10 mg/kg groups. Kidney and blood samples were collected for assessment of renal function, measurement of pro-inflammatory cytokines, apoptosis markers, antioxidant activity, and tissue histology.
Results:
CP significantly increased the levels of serum Cr, BUN, and uric acid; it also induced histological damage reflecting the pathophysiology observed during nephrotoxicity. CP has also shown its pro-oxidant activity in kidney tissue because CP decreased the levels of GSH, SOD, and CAT; it increased the lipid peroxidation as measured by MDA content. In addition, CP significantly upregulated the activity of pro-inflammatory cytokines and expression of apoptotic markers, that is, there were increased levels of IL-1β, IL-6, TNF-α, caspase-3, and caspase-9. Two weeks of continuous treatment of UA showed significant recovery against CP-induced nephrotoxicity; UA decreased the levels of Cr, BUN, and uric acid and ameliorated histological damage. UA also downregulated the activities of IL-1β, IL-6, and TNF-α as well as expression of caspase-3 and caspase-9. Furthermore, CP-induced oxidative stress that was antagonized by UA—the levels of GSH, SOD, and CAT were significantly increased while MDA content was decreased.
Conclusions:
UA has a protective effect against CP-induced nephrotoxicity, which may be due to its antioxidant activity and mitigation of IL-1β, IL-6, TNF-α, and markers of apoptosis.
Introduction
CP is one of the most frequently used and competent chemotherapy medications for various types of solid malignant tumors located in the head, neck, lungs, cervix, ovarian, testicular, and stomach. 1 CP-treated chemotherapy accounts for approximately 90% of the maximum cure rate in testicular cancer. 2 Despite its cancer chemotherapeutic action, CP exerts many undesirable side effects including nephrotoxicity; 3 this limits its use in chemotherapy. CP induces nephrotoxicity due to the accumulation of CP and its metabolites in the kidney tubules leading to tubular injury and cell death. 4 CP-induced nephrotoxicity evolves from the activation of multiple pathways such as reactive oxygen species (ROS)–induced oxidative stress, inflammatory mediators, and apoptosis; these are crucial cellular events and lead to CP-induced nephrotoxicity. 5
CP occurs in an uncharged molecular state in the blood due to high chloride ion concentrations. 6 CP enters into tubular cells by passive diffusion and loses its two chloride groups to become a positive electrophilic compound leading to oxidative stress. Numerous studies have confirmed the role of CP in ROS production, inflammation, and tubular injury. 4 ROS and inflammation pathophysiology are also well-documented in many disease conditions including nephrotoxicity in animal models. 7 ROS-induced free radicals like superoxide, hydroxyl, and peroxide radicals are important mediators of tissue injury. ROS production activates several damaging signaling pathways and can modify patient prognosis. 8
CP-induced pro-inflammatory damage upregulates cytokine production within kidney tissue, which is a well-accepted immunological event in nephrotoxicity. Important inflammatory mediators include IL-1β, IL-6, and TNF-α that are expressed in a nephrotoxicity-induced animal model development. 9 These pro-inflammatory mediators are recognized to play a crucial role in CP-induced nephrotoxicity. 10 Besides enhanced ROS and pro-inflammatory mediators, CP could encourage apoptotic cell death in kidney tubules via activating caspase-dependent pathways. 11 Of these, caspase-3 and -9 are more prominent in CP-induced apoptotic cell death. 12 Hence, attenuation of ROS, inflammatory changes, and caspase-dependent events could be a promising therapeutic target that may help to avert the side effects of CP and prevent nephrotoxicity during cancer chemotherapy.
Recently, antioxidant compounds were administrated to those undergoing CP therapy in the hope of reducing CP-induced nephrotoxicity.13,14 The natural antioxidant UA is a pentacyclic triterpene compound derived from holy basil, apple, cranberries, rosemary, and oregano. UA has exhibited various pharmacological activities such as antioxidant, anti-inflammatory, and anti-apoptotic.15,16 However, the nephron-protective mechanisms of UA against CP-induced kidney injury are not yet entirely understood. UA can potentially prevent the nephrotoxic effects of CP. 17 We determined here how UA can control CP-induced nephrotoxicity by reducing oxidative stress, inflammation, and controlling caspase-3 and -9 activities in a model for nephrotoxicity.
Materials and methods
Chemicals
UA (CAS No. 77.52-1) and CP (CAS No. 15,663-27-1) were purchased from Sigma Aldrich (USA). ELISA kits such as IL-1β (CAT No. ab100768), IL-6 (CAT No. ab234570), and TNF-α (CAT No. ab46070); colorimetric kits for caspase-3 (CAT No. ab39401) and caspase-9 (CAT No. ab65608) were purchased from Abcam, UK. Biochemical kits such as Cr, BUN, and uric acid were obtained from Randox, UK. All other chemicals and solvents used during the experiment were laboratory grade and purchased from commercial sources.
Animals
All animal work was approved by the Standing Committee for Scientific Research Ethics and Jazan University (reference no. SARC/EC/106). Adult male Wistar rats (200–240 g) were acquired from the Jazan University Medical Research Centre (Kingdom of Saudi Arabia). Prior to experimental work, the animals were acclimatized in the institutional animal house under a 12-h light–dark cycle and controlled conditions (humidity 45–55%, room temperature 25 ± 2°C) with standard autoclaved laboratory diet and water provided ad libitum.
Experimental design
Rats were randomized into five groups of six animals in each:
Group 1: Vehicle (0.1% Tween 80)-treated normal control.
Group 2: Single i.p. dose of CP 7.5 mg/kg-treated positive control.
Group 3: UA 10 mg/kg (intragastric-i.g.) alone treated.
Group 4: UA 5 mg/kg (i.g.) followed by single dose of CP.
Group 5: UA 10 mg/kg (i.g.) followed by single dose of CP.
UA was reconstituted in 0.1% Tween-80 and administered once per day for 14 days in groups 3, 4, and 5. 17 Nephrotoxicity was induced in group 2, 4, and 5 animals via single i.p. dose of CP (7.5 mg/kg) on the 10th day of treatment. 12 Doses of CP were prepared by dissolving 7.5 mg of CP in 1 ml of 5% DMSO/saline and administered as 0.1 ml/100 gm; 10 mg UA was dissolved in 2 ml 0.1% of Tween 80 and administered as 0.1 and 0.2 ml/100 gm of animal in their respective groups.
Sample collection and preparation of tissue homogenate
On day 15th, blood samples were collected by an ocular puncture under light ether anesthesia. 12 Blood pathology biomarkers were analyzed by separating serum from whole blood through centrifugation at 3000 × g for 10 min and stored at −20°C. After blood collection, all animals were sacrificed according to ethical guidelines, and both kidneys were isolated to measure oxidative stress, pro-inflammatory cytokines, and apoptosis markers. Kidney homogenate (10% w/v) was prepared in a chilled sodium phosphate buffer (10 mM, pH 7.4) along with a cocktail of 10 μl/ml protease inhibitor. Homogenate was prepared using Ultra Turrax T-25 (IKA, Germany) homogenizer and centrifuged at the speed of 800 × g for 5 min at 4°C to eliminate cell debris from the homogenate. Post-mitochondrial supernatant (PMS) were separated by centrifuging homogenate at 10,500 × g for 15 min at 4°C. 12
Histopathological examination
Kidney tissue samples were fixed in 10% formalin solution (neutral buffered). Before sectioning, formalin-fixed tissues were processed through gradient alcohol dehydration (by submerging tissues in alcohol) and xylol permeabilization. Subsequently, the tissues were embedded in paraffin and sectioned to a 5-mm thickness using a microtome followed by hematoxylin and eosin staining for histological evaluation under a light microscope at 40X magnification.
Estimation of renal antioxidant activity
The sample protein (homogenate and PMS) content was normalized to total cellular protein using the bicinchoninic acid solution (BCA) methodology as described by Lowry et al. (1951). 18 Lipid peroxidase (LPO) was determined by following the methods described by Utley et al. (1974) 19 with minor variations. The absorbance of LPO was determined spectrophotometrically (UV-1601, Shimadzu, Japan) at 535 nm and stated as nmol MDA formed/h/mg protein using a molar extinction coefficient (MEC) of 1.56 × 105 M−1 cm−1. Levels of GSH were determined as described by Jollow et al. (1974) 20 with minor alterations. The optical density was measured immediately at 412 nm. A MEC value of 13.6 × 103 M−1 cm−1 was used to calculate the content of GSH as DTNB conjugate formed per mg protein. SOD was estimated by determining the auto-oxidation of (−)-epinephrine at 480 nm as described by Stevens et al. (2000). 21 The SOD activity was calculated as nmol (−)-epinephrine protected using a MEC of 4.02 × 103 M−1 cm−1. CAT activity was determined according to the method of Claiborne (1985) 22 using an MEC of 43.6 × 103 M−1 cm−1.
Assessment of renal function
Renal functions were determined using biochemical estimation kits for Cr, BUN, and uric acid (Randox, UK) according to the manufacturer’s instructions. The sample absorbance was observed at 490 nm for Cr, 580 nm for BUN, and 520 nm for uric acid, respectively.
Assay of pro-inflammatory cytokines
Separate ELISA assays for IL-1β, IL-6, and TNF-α were performed according to the manufacturer’s guidelines (Abcam, UK). A duplicate set of each sample was also assayed. Absorbance was recorded at 450 nm using a microplate reader (ELx 800TM BioTek, USA).
Assay for caspase-3 and -9
Analysis for apoptosis markers caspase-3 and -9 were performed using colorimetric analysis. The end reactions for caspase-3 and -9 were quantified by PNA light emission as measured at 405 nm using an ELISA reader (ELx 800TM BioTek, USA). The concentrations of caspase-3 and -9 were determined according to the manufacturer’s instructions.
Statistical analysis
Data analysis was performed by using GraphPad Prism software version 8.3.1 (GraphPad Software); statistics were calculated using a one-way ANOVA with a Tukey’s multiple comparison test as a post hoc test. p-values were considered significant if p < 0.05.
Results
Histopathological findings
Microscopic examination of normal kidney histology of control animals showed intact glomeruli and regularly shaped renal tubules reflecting a typical kidney structure (Figure 1(a)). CP treatment of positive control animals disrupted the kidney architecture versus normal controls. The histological observations clearly showed that CP treatment induced renal tubular lesions with severe degenerative changes to the glomerular basement membrane including coagulative necrosis, hemorrhage, congestion, vacuolation, and edema. We characterized coagulative necrosis if there were an eosinophilic cytoplasm and nuclear pyknosis (Figure 1(b)). Histopathological examination clearly showed that UA (5 and 10 mg/kg) significantly improved CP-induced kidney damage (Figures 1(c) and (d)). UA 10 mg/kg alone did not affect the morphology of the kidney versus the normal control group indicating the safety of UA on kidney tissue (Figure 1(e)). 1–51Hematoxylin and Eosin (H&E) staining represents (a) normal control (NC) group; normal kidney with clear glomerular basement membrane-G and normal renal tubules (proximal convoluted tubules-P; Distal Convoluted tubules-D. (b) CP group; toxicity with necrosis in G and P and D with debris in lumen. (c) CP and UA group (UA-5+CP); no significant but slight improve in G, P, and D. (d) CP and UA group (UA-10+CP); improvement in G, P, and D. (e) UA-10 group (UA-10); no significant changes in G, P, and D.
Effect of UA on renal function
Effect of UA treatment on serum Cr, BUN, and uric acid against nephrotoxicity induced by CP.
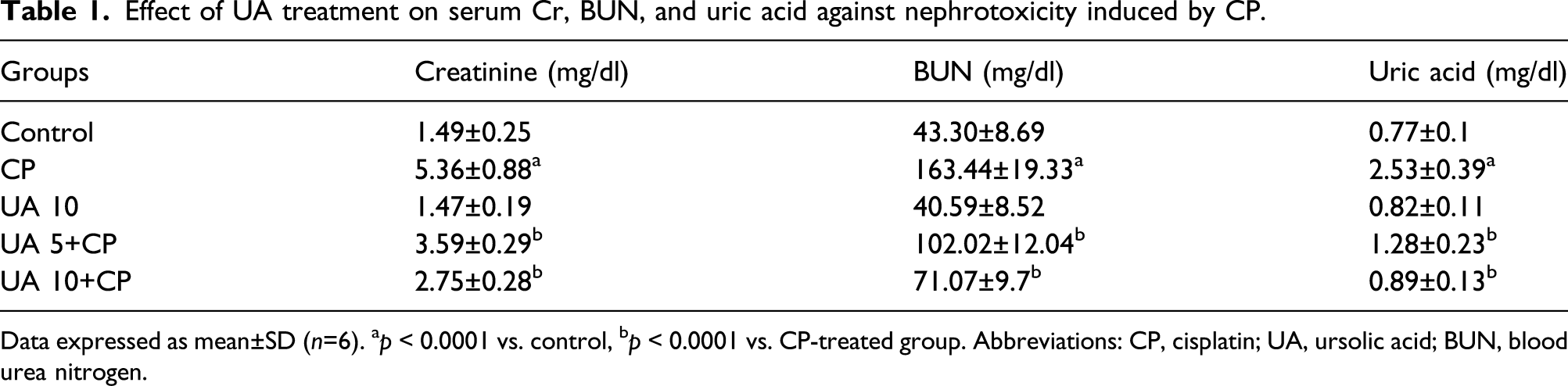
Data expressed as mean±SD (n=6). ap < 0.0001 vs. control, bp < 0.0001 vs. CP-treated group. Abbreviations: CP, cisplatin; UA, ursolic acid; BUN, blood urea nitrogen.
Effect of UA on renal antioxidant activity
Effect of UA treatment on CP-induced oxidative stress in renal tissue.

Data expressed as mean±SD (n=6). ap < 0.0001 vs. control group. bp < 0.0001; cp < 0.001; dp <0.01; ep <0.05 vs. CP-treated group. Abbreviations: CP, cisplatin; UA, ursolic acid; MDA, malondialdehyde; GSH, glutathione; SOD, superoxide dismutase; CAT, catalase.
Effect of UA on pro-inflammatory cytokines
During nephrotoxic induction by CP, the levels of IL-1β (1569.43 ± 41.61) (Figure 2(a)) and IL-6 (2417.52 ± 65.07) (Figure 2(b) were significantly increased (p < 0.0001) in positive control animals. On the contrary, 14 days of continuous treatment with either dose of UA led to a significant decrease in the levels of IL-1β and IL-6 (p < 0.0001). The level of TNF-α (361.67 ± 10.5) was significantly higher (p < 0.0001) in CP-treated positive control versus normal control animals (273.33 ± 4.76 [Figure 2(c)]. As a positive control, the levels of TNF-α were significantly decreased (p < 0.0001) with either UA 5 mg/kg (305.83 ± 7.33) or 10 mg/kg (285.84 ± 5.42) against CP. However, no significant cytokine production difference was seen between the control group and animals treated with UA 10 mg/kg alone. Effects of UA treatment on renal IL1-β (a), IL-6 (b), and TNF-α (c) levels induced by CP. Data expressed as mean±SD (n=6). ap < 0.0001 vs. control, bp < 0.0001 vs. CP-treated group. Abbreviations: CP, cisplatin; UA, ursolic acid; IL-1β, interleukin-1β; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α.
Effect of UA on caspase-3 and caspase-9
The levels of caspase-3 and -9 were significantly increased (p < 0.0001) in the CP-treated group (Figures 3(a) and (b)). Treatment with either UA 5 or 10 mg/kg in group 4 and 5 significantly decreased (p < 0.0001) the levels of caspase-3 and -9 against CP-induced nephrotoxicity. Again, no significant changes were observed in the UA 10-alone treated group compared to the control animals. Effect of UA treatment on renal apoptotic markers caspase-3 (a) and caspase-9 (b) upregulated by CP. Data expressed as mean±SD (n=6). ap < 0.0001 vs. control group. bp < 0.0001 vs. CP-treated group. Abbreviations: CP, cisplatin; UA, ursolic acid.
Discussion
This study evaluated the potential protective effect of UA administration against CP-induced nephrotoxicity using a rat model. Simultaneously, we attempted to decipher the possible mechanism underlying UA’s protective ability by analyzing oxidative stress, pro-inflammatory mediators, and the degree of cell apoptosis. We found that UA administration exerted an impressive protective effect against CP-induced nephrotoxicity. The reduction in CP-induced nephrotoxicity may be due to reduced oxidative status; suppression of pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α; and reduced caspase cascade activity.
CP has a low molecular weight that makes it capable of accumulating at a higher concentration in the kidney, which results in nephrotoxicity. 23 As per literature, the recommended doses of CP to induce nephrotoxicity in rats vary between 5 and 16 mg per kg where toxicity peaked between 3 days and 8 days. In preliminary studies, we found that a 7.5 mg/kg dose of CP was sufficient to produce highest toxicity on the 5th day followed by a gradual decline in toxicity effect.12,24,25 In this study, significant increases in serum Cr, BUN, and uric acid levels after exposure to CP were an indicator of nephrotoxicity and in agreement with earlier reports. 26 These variations within the serum are also reflected in the histopathology of kidney tissues because CP treatment induces visible histological structural damage to glomerular basement membranes and renal tubules versus normal control animals. Treatment of UA significantly decreased serum Cr, BUN, and uric acid levels and protected animals from kidney damage induced by CP.
Numerous studies demonstrate the role of oxidative stress in CP-induced nephrotoxicity and the reno-protective effects by different antioxidants and oxidant stress inhibitors for CP-nephrotoxicity. Accordingly, CP accumulates in the renal epithelium and is metabolized by CYP450, which then converts it into highly reactive metabolites.27,28 CP primarily targets mitochondria resulting in the loss of mitochondrial protein import due to modifications of sulfhydryl (SH) groups. This leads to reduced calcium uptake and reduced mitochondrial membrane potential. 29 Thus, mitochondrial dysfunction leads to increased intracellular calcium levels, which activates NADPH oxidase stimulating ROS production. 30 CP generates free radicals such as hydrogen peroxide radicals and superoxide anions in renal tubular cells. Superoxide anions further convert it into a highly reactive hydroxyl radicals leading to DNA damage.12,30,31
Excessive accumulation of free radicals also damages the membrane’s lipid components by peroxidation causing lipid peroxidation, which surpasses the natural ability of antioxidant defenses within kidney cells. 10 We assessed MDA levels, which is a distinguished oxidative stress marker of lipid peroxidation. We identified a significant reduction in MDA levels in rats treated with UA during CP-induced damage. This further explained UA’s capacity to inhibit mitochondrial lipid peroxidation within renal tubular cells. The intracellular redox homeostasis is maintained by the thiol group (–SH) containing molecules. 32 The high affinity of CP to thiol groups forms a CP–SH complex that leads to depletion of reduced glutathione (GSH) and plays a vital role in ROS scavenging. The stable CP–SH complex formation also decrease other antioxidant enzyme activity including SOD, and CAT. 31 UA was assessed for alterations in its redox status and found that it acts as an antioxidant in restoring the thiol status. 33 In this study, a single dose of CP significantly decreased kidney GSH activity but was restored by UA treatment. In addition, UA effectively prevented the reduction in SOD activity against CP as observed in diabetic nephropathy. 34 CAT is a peroxisomal enzyme that catalyzes hydrogen peroxide breakdown and plays a key role in antioxidant defense of GSH and SOD. Here, CAT activity was significantly increased in UA-treated animals under CP treatment. Increased CAT may be related to UA’s ability to increase CAT mRNA expression leading to increased CAT protein synthesis and quenching of hydrogen peroxide radicals. 33
Several pro-inflammatory cytokines such as IL1-β, IL-6, and TNF-α are upregulated in the kidney due to activation of the inflammatory cascade triggered by CP. The upregulation of pro-inflammatory cytokines by CP is due to the elevation of intracellular ROS in the kidney, which in turn activates the transcription factor NF-κB.35,36 Activation of NF-κB leads to induction of inflammatory signaling cascades and an increase in the renal expression of prototypical inflammatory mediator TNF-α. TNF-α promotes the generation of other inflammatory cytokines especially IL-1β and IL-6.28,37 We identified increased levels of IL1-β, IL-6, and TNF-α in rats treated with CP, which reflects similar findings from previous reports.12,38
UA confers strong anti-inflammatory activity by suppressing multiple signaling pathways leading to pro-inflammatory cytokine production such as TNF-α, IFN-γ, IL-1β, IL-2, IL-6, and MCP-1.34,39 In this study, we found that UA-induced a dose-dependent reduction in levels of TNF-α, IL-1β, and IL-6. This suggests that UA’s ability to alleviate oxidative stress and reduce an inflammatory response may be related. As discussed earlier, NF-ĸB signaling triggers inflammation by upregulating the TNF-α expression in renal epithelial cells. 40 The transcription of TNF-α gene occurs once NF-κB (p65) is translocated to the nucleus where it binds to its DNA target sites leading to upregulation of IL-1β and IL-6. A series of investigations revealed that UA is a potent inhibitor of NF-ĸB39,40,41 as activated by intracellular ROS production of the kidneys during CP treatment. Hence, the observed reduction in TNF-α, IL-1β, and IL-6 levels after UA treatment strengthens its anti-inflammatory role.
Accumulating evidence suggests that CP-induced nephrotoxicity is mediated via apoptotic signaling pathways; this is also linked to ROS-induced oxidative stress.4,25 Apoptosis is programmed cell death and is predominantly mediated by the caspase pathway. Caspases are initiators or executioners of apoptosis. Caspase-8 and -9 are the initiator caspases leading to downstream activation of caspase-3. Downstream signaling occurs once caspase-3 is activated leading to apoptosis activation in the renal tubules.30,42 Excessive ROS can also induce apoptosis through TNF receptor or FAS-activated extrinsic pathway and intrinsic mitochondrial (Bax) pathway controlled by Bcl-2 family.37,43 Intrinsic regulation of apoptosis occurs by binding membrane ligands (Fas-L and TNF-α) to plasma membrane death receptors (Fas and TNF); this process in turn induces activation of caspase-8 and eventually caspase 3 leading to apoptosis.12,32
Bcl-2 and Bax are apoptotic proteins that regulate cell apoptosis. Bcl-2 pore-stabilizing proteins stabilize the barrier function of mitochondrial membranes to inhibit apoptosis thereby prolonging cell viability. Bax (Bcl-2-associated X protein) is mainly located in the cytoplasm and functions as a pore-destabilizing protein that antagonizes the Bcl-2 apoptotic factor. The balance between Bax and Bcl-2 expression regulates apoptosis.12,44 CP-induced ROS activates caspase-9 and -3 through the translocation of pro-apoptosis protein Bax from the cytosol to the mitochondria regulating the release of mitochondrial cytochrome C (an apoptosis-inducing factor) and endonuclease G, which activate apoptosis and induce DNA damage, respectively.28,45 In this study, we observed that CP-induced upregulations of caspase-3 and -9 in kidney tissue were reversed by UA. The suppression of caspase-3 and -9 activities in kidney tissue by UA may be due to UA’s antioxidant capabilities and potent anti-inflammatory activity as explained by Peng et al. (2016). 40
A plethora of studies have revealed the anticancer effect of UA on different cancer cell lines derived from the liver (HepG2, Hep3B, Huh7), intestines (BGC823 and SGC7901), B16F-10 melanoma cells, and human hepatoma cell SMMC-7721.46,47 Scientists verified that the anticancer effect of UA is due to its enhanced pro-apoptotic effects, activation of the caspase cascade (by activation of caspase-3, 8, and 9), mitochondrial dysfunction, upregulation of apoptotic proteins such as p53 and Bax, and downregulation of anti-apoptotic proteins such as Bcl-2 in these cancer cell lines. 39
Benefits of UA and CP combinations have been studied in a variety of cancers. Chemosensitization of UA on CP-resistant HepG2/DDP cells have confirmed the therapeutic capability of UA in overcoming CP-resistant hepatocellular carcinoma. 48 Synergism of UA with CP was more effective in inhibition of cervical cancer (SiHa) cell proliferation and cell apoptosis. 49 On CP-resistant oral carcinoma, UA causes selective cell death of human CP-resistant CAR cells rather than normal cells. 50 In addition, enhancement of CP-induced apoptosis by UA has been studied. The data suggest improved efficacy of cancer treatment by significantly reducing the doses of chemotherapeutic drugs without compromising the treatment results. 51 Overall, UA can be designated as a double-edged sword with diverse benefits such as in direct chemoprevention, as an additive to chemotherapeutic agents, and for prevention of chemotherapy-induced adverse effects such as nephrotoxicity; nevertheless, this work needs further investigation.
Conclusion
In conclusion, UA provides protective role against CP-induced nephrotoxicity, which may be due to its antioxidant capacity and mitigation of pro-inflammatory cytokines and apoptosis. However, further studies are required to fully elucidate the mechanism.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.