
Abstract
Inflammatory responses have been demonstrated to contribute to the neuronal death following cerebral ischemia. This study was to investigate the repairing effects and potential mechanisms of (Z)-7,4′-dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside (DHAG), a compound with neuroprotective effects, on cerebral ischemia-reperfusion (I/R) injury in rats. Cerebral I/R model was established with middle cerebral artery occlusion method in Sprague Dawley rats and then rats were treated with DHAG (1 and 2 mg/kg) for 7 days. The volume of cerebral infarction was detected by triphenyltetrazolium chloride staining. The apoptosis in ischemic brain tissues was detected by terminal deoxynucleotidyl transferase dUTP nick end labeling assay. Oxidative stress markers and inflammatory factors were detected by enzyme-linked immunosorbent assay. Protein expression was detected by Western blot. DHAG treatment significantly alleviated the cerebral I/R injury and decreased apoptosis in brain tissues. Moreover, DHAG treatment significantly inhibited oxidative stress and reduced inflammatory responses, associating with decreasing the protein expression of phosphorylated Janus kinase 1/phosphorylated signal transducer and transcriptional activator 1. These results demonstrated neuroprotective properties of DHAG and highlighted it as a potential therapeutic agent against injury of cerebral IR.
Keywords
Introduction
Cerebral infarction, also known as ischemic stroke which causes brain tissue necrosis by transient or permanent occlusion of the cerebral blood vessels and insufficient blood supply to the brain, is a leading cause of death and disability worldwide. As a major type of stroke, cerebral ischemia is accounting for almost 80% of stroke cases. 1 Restoration of blood supply, referred as “reperfusion,” is virtually a desired goal for acute stroke treatment. Spontaneous reperfusion occurs commonly after stroke, in about 50–70% of patients with ischemic stroke. 2 Clinical data demonstrate that the earlier the treatment of patients with ischemic stroke, the greater the chance of being rescued. However, there is no effective and efficient therapeutic strategy in clinic because of its complicated and still unclear pathological mechanism. Therefore, there is a greater need to search for novel neuroprotective agents.
Ischemia-reperfusion (I/R) injury is a common feature of ischemic stroke. When I/R injury occurs, endothelial injury and dysfunction leads to the damage of blood–brain barrier (BBB). Additionally, the free heme released from methemoglobin during cerebral I/R is a toxic component to endothelial cells, cortical neurons, and astrocytes. 3 –5 The main mechanisms of I/R injury include oxidative stress, neuroinflammatory responses, mitochondrial mechanisms, platelet activation and aggregation, and BBB disruption, which ultimately lead to brain edema or hemorrhagic transformation. 6 Overproduction of pro-inflammatory factors can stimulate the generation of oxidants, resultantly causing subsequent peroxidative damage by increasing the mitochondrial membrane permeability, cytochrome-c release, activating caspase-related apoptosis, and eventually causing neuron death and neurological dysfunctions. 7
As an intracellular signal transduction pathway, the Janus kinase–signal transducer and transcriptional activator (JAK-STAT) has been clarified as one of the important pathways for cytokine signal transduction, not only in inflammatory responses but also in oxidative stress, cell damage, and apoptosis. 8 The JAK-STAT pathway has been reported to be involved in the pathological processes of neurological diseases, such as cerebral ischemia injury and central nervous system tumors. 9
Supplementation of exogenous antioxidants from herbal plants has been used to treat disorders by alleviating the oxidative damage. 10,11 (Z)-7,4′-Dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside (DHAG), a new compound isolated from the endophytic fungus Penicillium citrinum of Bruguiera gymnorrhiza, exerted potent neuroprotective activities against 1-methyl-4-phenylpyridinium-induced oxidative damage in PC12 cells. 12 DHAG was also reported to mitigate photoreceptor cell degeneration in Rd10 mouse model and present anxiolytic activity via counteracting the oxidative stress and inflammatory responses. 13,14 However, whether DHAG could alleviate the cerebral I/R injury remains to be further studied. This study was to investigate the neuroprotective effects of DHAG in middle cerebral artery occlusion (MCAO) rat model and its underlying mechanisms.
Materials and methods
Chemicals and reagents
Cytochrome-c assay kit, caspase-3 and caspase-9 activity assay kits were purchased from R&D Systems (Minneapolis, Minnesota, USA). Glutathione (GSH), superoxide dismutase (SOD), and malondialdehyde (MDA) assay kits were purchased from Jiancheng Institute of Biological Engineering (Nanjing, China). Interleukin-1β (IL-1β), IL-6, and tumor necrosis factor α (TNFα) assay kits were purchased from Thermo Fisher (Waltham, Massachusetts, USA). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kit was purchased from Roche (Mannheim, Germany). DHAG (purity >98%) was purchased from WuXi AppTec Company (Shanghai, China).
Experimental procedures and treatment
A total of 120 male Sprague Dawley (SD) rats, weighing 180–220 g, were purchased from Vital River Ltd (Beijing, China). Rats were maintained in humidity (40–70%) and temperature-controlled (23 ± 2°C) facility on 12:12 hour light:dark cycles under specific pathogen-free condition. Rats had ad libitum access to food and water. Animal experiments were conducted in compliance with the Chinese legislation on the Use and Care of Animals and approved by the Institutional Animal Care and Use Committee of Shanghai Pudong Hospital.
The MCAO method was used to establish the cerebral I/R injury model. 15,16 Briefly, rats were fasted for 12 h before surgery with water accessible and then anesthetized with 3% pentobarbital (45 mg/kg) by intraperitoneal injection. After that, a ventral midline neck incision was made, followed by the identification and careful free dissection of the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) from surrounding tissues. A surgical bolt was inserted into the ICA after the proximal ligation of the CCA and ECA until a mild resistance was felt, indicating the occlusion of the middle cerebral artery (MCA), which resulted in a transient cessation of blood flow and subsequent brain infarction in the area supplied by the MCA. The skin was closed with a 4-0 silk suture, followed by 2 h of ischemia. The bolt was then withdrawn to initiate a reperfusion process. In sham group, rats were anaesthetized and the carotid artery was exposed, without bolt insert. The body temperature of rats was maintained at 37 ± 0.5°C during the whole procedure.
Rats were divided randomly into four groups (n = 20): sham group, model group, and DHAG groups (1 and 2 mg/kg). The dosages of DHAG were based on our preliminary study and literature. 13,14 Rats in DHAG groups received a daily oral dose of 1 or 2 mg/kg, respectively, for 7 days. Rats in the control group and model group received vehicle (0.5% carboxymethylated cellulose) during the same period. The rats were treated at 30 min after ischemia. At the end of the experiment, rats were euthanized by inhaling air flow of CO2 first and then the death was confirmed by cervical dislocation.
Neurological function score
Neurological function score was assessed using a five-point scale (0–4) Bederson’s test by a blinded researcher. 17
Triphenyltetrazolium chloride staining
After treatment for 7 days, whole brains were quickly removed and frozen at −20°C for 30 min. Coronal sections (2 mm) were sliced and incubated with 2% triphenyltetrazolium chloride (Sigma-Aldrich, St. Louis, Missouri, USA) for 30 min at 37°C. After washing, sections were then fixed in 4% paraformaldehyde for 24 h, and photographed. The infarct volume was analyzed by ImageJ 1.43 software.
Brain tissue water content measurement
After treatment for 7 days, whole brains were quickly removed and the wet weight (WW) was immediately weighed. Brains were then dried in a gravity oven at 100°C for 24 h to obtain a dry weight (DW). Water content = (WW − DW)/WW × 100%.
TUNEL assay
After treatment for 7 days, whole brains were quickly removed and fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS, pH 7.4) overnight, dehydrated by gradient sucrose, and transferred into 30% sucrose solution for cryoprotection. Then, 10-µm thickness sections were made using the Tissue-Tek® Cryo3 ® Flex Microtome (Sakura, The Netherlands). The apoptosis was detected by TUNEL staining according to the manufacturer’s instructions. Sections were rinsed and visualized with 3,3′-Diaminobenzidine. The number of TUNEL-positive cells was counted under the microscope.
Cytochrome-c release measurement
After treatment for 7 days, whole brains were quickly removed, weighed and homogenated in PBS (pH 7.4) and centrifuged at 3000 r/min at 4°C for 15 min to remove cellular debris. Supernatants were kept at −80°C and used for further tests. Cytochrome-c was measured according to the manufacturer’s instruction. After reaction with reagents, the optical density was measured by a microplate reader at 450 nm.
Caspase activity measurement
Brain tissue lysate was prepared as mentioned above. Caspase-3 (Ac-DEVD-Amc, 390/475 nm) and caspase-9 (Ac-LEDH-Afc, 400/505 nm) activities were measured by the assay kit (R&D Systems) with the microplate reader according to the manufacturer’s instruction.
MDA and GSH level, SOD activity measurement
Brain tissue lysate was prepared as mentioned above. MDA, GSH, and SOD assay was conducted by colorimetric kit according to the manufacturer’s instructions.
Cytokine and nitric oxide measurement
Brain tissue lysate was prepared as mentioned above. Cytokine levels of IL-1β, IL-6, and TNFα were measured by enzyme-linked immunosorbent assay (ELISA) kits (Thermo Fisher) according to the manufacturer’s instruction. Nitric oxide (NO) was measured by Griess assay (Sigma-Aldrich, St. Louis, Missouri, USA). 18
Western blot analysis
Protein from whole brains was quantified using the bicinchoninic acid assay kit (Beyotime, Shanghai, China). Samples with equal amount of protein (50 μg) were loaded on 4–12% sodium dodecyl sulfate polyacrylamide gel (Thermo Scientific, Waltham, Massachusetts, USA). After electrophoresis, protein was transferred to polyvinylidene difluoride membranes, which were then blocked with 1% bovine serum albumin for 1 h and incubated with the primary antibodies (anti-cleaved caspase-3 rabbit pAb ab49822, 1:1000; anti-cleaved caspase-9 (D353) rabbit pAb orb159342, 1:1000; anti-Bax rabbit mAb ab32503, 1:3000; anti-Bcl-2 rabbit pAb ab196495, 1:1000; anti-Nrf2 rabbit pAb ab92946, 1:1000; anti-NADPH oxidase 1 rabbit pAb ABIN2782754, 1:500; anti-NF-κB (phosphor S536) rabbit pAb ab86299, 1:3000; anti-NF-κB rabbit pAb ab16502, 1:500; anti-STAT1 rabbit pAb ab31369, 1:1000; anti-STAT1 (phospho S727) rabbit mAb ab109461, 1:3000; anti-STAT2 rabbit mAb ab32367, 1:3000; anti-STAT2 (phospho Y690) rabbit pAb ab53132, 1:500; anti-STAT3 rabbit mAb ab68153, 1:1000; anti-STAT3 (phospho S727) rabbit pAb ab30647, 1:1000; anti-JAK1 rabbit mAb ab133666, 1:3000; anti-JAK1 (phospho Y1022) rabbit pAb ABIN2857773, 1:500; anti-Claudin 1 rabbit pAb ab15098, 1:500; anti-Occludin rabbit mAb ab216327, 1:1000; anti-β-actin rabbit pAb ab8227, 1:3000) at 4°C overnight. The membranes were washed three times, followed by incubation with the secondary antibody (goat anti-rabbit IgG H&L HRP, ab6721, 1:5000) at room temperature for 2 h. Membranes were washed and exposed to PierceTM enhanced chemiluminescence substrates (Thermo Scientific, Waltham, Massachusetts, USA), followed by the X-ray film development.
Statistical analysis
Data were expressed as mean ± standard deviation and analyzed with SPSS 19.0 software (SPSS Inc., Chicago, Illinois, USA). All comparisons were made by one-way analysis of variance followed by least significant difference post hoc analysis. The value of p < 0.05 was considered as significant difference.
Results
DHAG treatment ameliorated brain damage caused by I/R
Comparing with sham group, MCAO rats had higher neurological function scores, larger cerebral infarct size and more water content (p < 0.01). However, comparing with model group, the neurological function score, cerebral infarct size and water content were significantly lower after treatment with DHAG (p < 0.05), indicating the brain damage was significantly ameliorated (Figure 1).
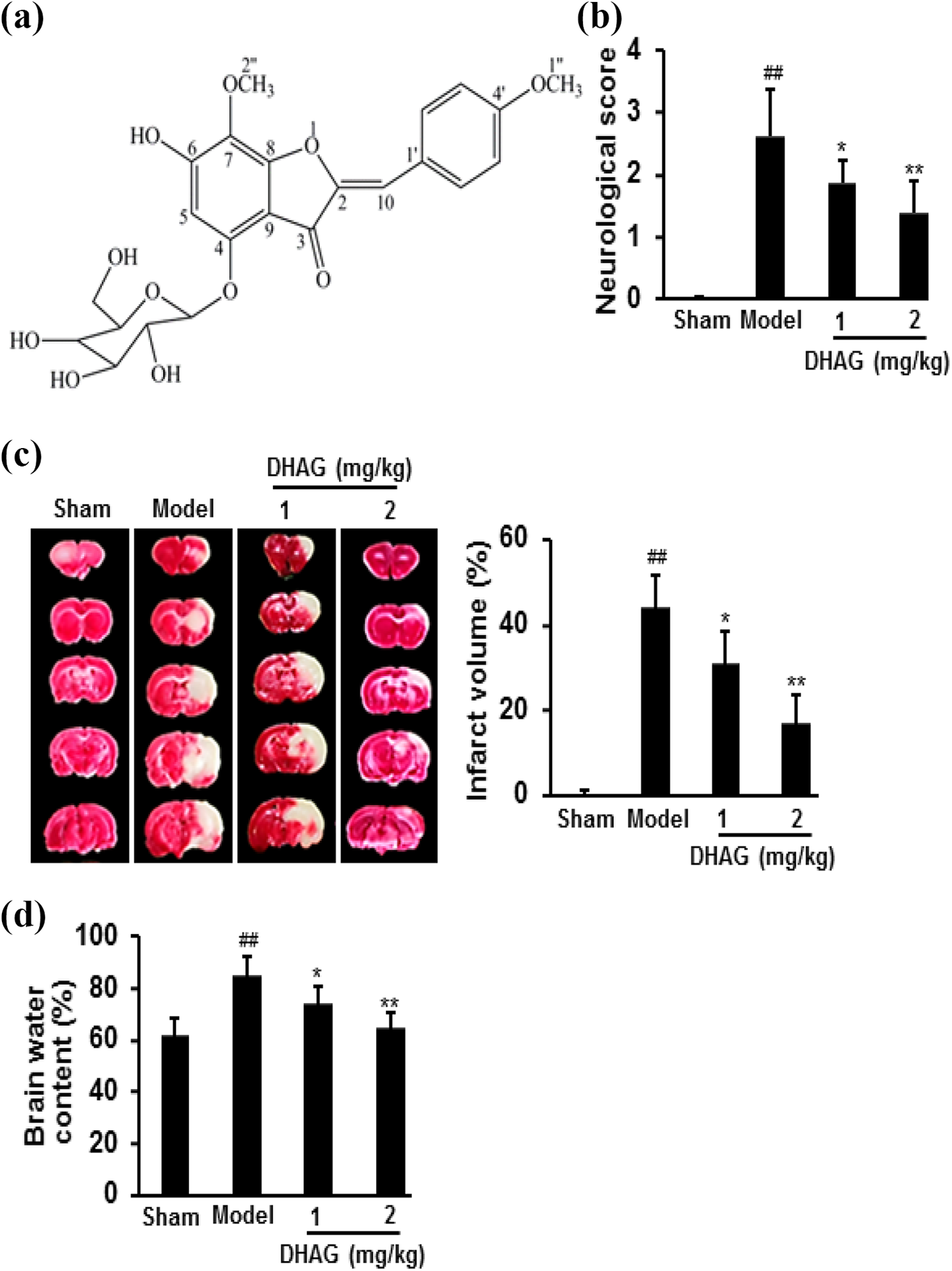
DHAG (a) treatment significantly ameliorated brain damage caused by I/R, shown by neurological function score (b), TTC staining and cerebral infarct size (c), and brain water content (d). Data were expressed as mean ± SD (n = 20 for (b); n = 5 for (c) and (d)). ## p < 0.01 versus sham group; *p < 0.05, **p < 0.01 versus model group. DHAG: (Z)-7,4′-dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside; I/R: ischemia-reperfusion; TTC: triphenyltetrazolium chloride; SD: standard deviation.
DHAG treatment decreased apoptosis and increased tight junction in brain tissues
Comparing with sham group, I/R significantly induced apoptosis in brain tissues (p < 0.01). However, treatment with DHAG significantly decreased cytochrome-c release, caspase activities, as well as DNA fragmentation (p < 0.05). Moreover, proapoptotic protein Bax and caspase expression significantly decreased, but antiapoptotic protein Bcl-2 significantly increased after DHAG treatment (p < 0.05). Furthermore, the tight junction protein Claudin 1 and Occludin expression significantly increased after DHAG treatment, comparing with model group (p < 0.05) (Figure 2). These biochemical markers were in good correlation with the neurological function score, cerebral infarct size, and water content as shown in Figure 1.

DHAG treatment decreased apoptosis and increased tight junction in brain tissues induced by I/R. Apoptosis was measured by DNA damage in whole brain (a), cytochrome-c release (b), caspase activities (c), and apoptosis-related proteins (d). The tight junction was measured by the protein expression (e). Data were expressed as mean ± SD (n = 5 for (b) and (c); n = 3 for (d) and (e)). ## p < 0.01 versus sham group; *p < 0.05, **p < 0.01 versus model group. DHAG: (Z)-7,4′-dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside; I/R: ischemia-reperfusion; SD: standard deviation.
DHAG treatment decreased the oxidative stress in brain tissues
Oxidative stress significantly increased after I/R (p < 0.01). However, treatment with DHAG significantly increased GSH levels and SOD activities, and reduced the production of MDA, together with increasing the protein expression of nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and decreasing the protein expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1, indicating DHAG treatment effectively decreased the oxidative stress (p < 0.05) (Figure 3).

DHAG treatment inhibited oxidative stress in brain tissues. The oxidative stress markers were measured by MDA content (a), SOD activity (b), GSH level (c), and oxidative stress-related protein expression (d). Data were expressed as mean ± SD (n = 5 for (a) to (c); n = 3 for (d)). ## p < 0.01 versus sham group; *p < 0.05, **p < 0.01 versus model group. DHAG: (Z)-7,4′-dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside; MDA: malondialdehyde; SOD: superoxide dismutase; GSH: glutathione; SD: standard deviation.
DHAG treatment decreased inflammatory responses in brain tissues
Comparing with sham group, levels of IL-1β, IL-6, TNFα, and NO in brain tissues of the model group significantly increased (p < 0.01). However, levels of inflammatory factors were significantly lower in the brain tissues after DHAG treatment (p < 0.05). Furthermore, DHAG treatment significantly decreased the protein expression of p-NF-κB (p < 0.05) (Figure 4).

DHAG inhibited inflammatory responses in brain tissues. Inflammatory responses were measured by NO (a), IL-1β (b), IL-6 (c), TNFα (d), and NF-κB protein expression (e). Data were expressed as mean ± SD (n = 5 for (a) to (d); n = 3 for (e)). ## p < 0.01 versus sham group; *p < 0.05, **p < 0.01 versus model group. DHAG: (Z)-7,4′-dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside; NO: nitric oxide; IL-1β: interleukin-1β; IL-6: interleukin-6; TNFα: tumor necrosis factor α; NF-κB: nuclear factor kappa B; SD: standard deviation.
DHAG treatment inhibited the protein expression of p-JAK1 and p-STAT1 in brain tissues
To identify the action mechanisms of DHAG, the protein expression of JAK/STAT was examined. The relative protein expression level of p-JAK1/p-STAT1 significantly decreased after DHAG treatment, compared with model control, while not changing JAK1/STAT1 (p < 0.05). Moreover, DHAG treatment did not change the protein expression of p-STAT2 and p-STAT3 (Figure 5).
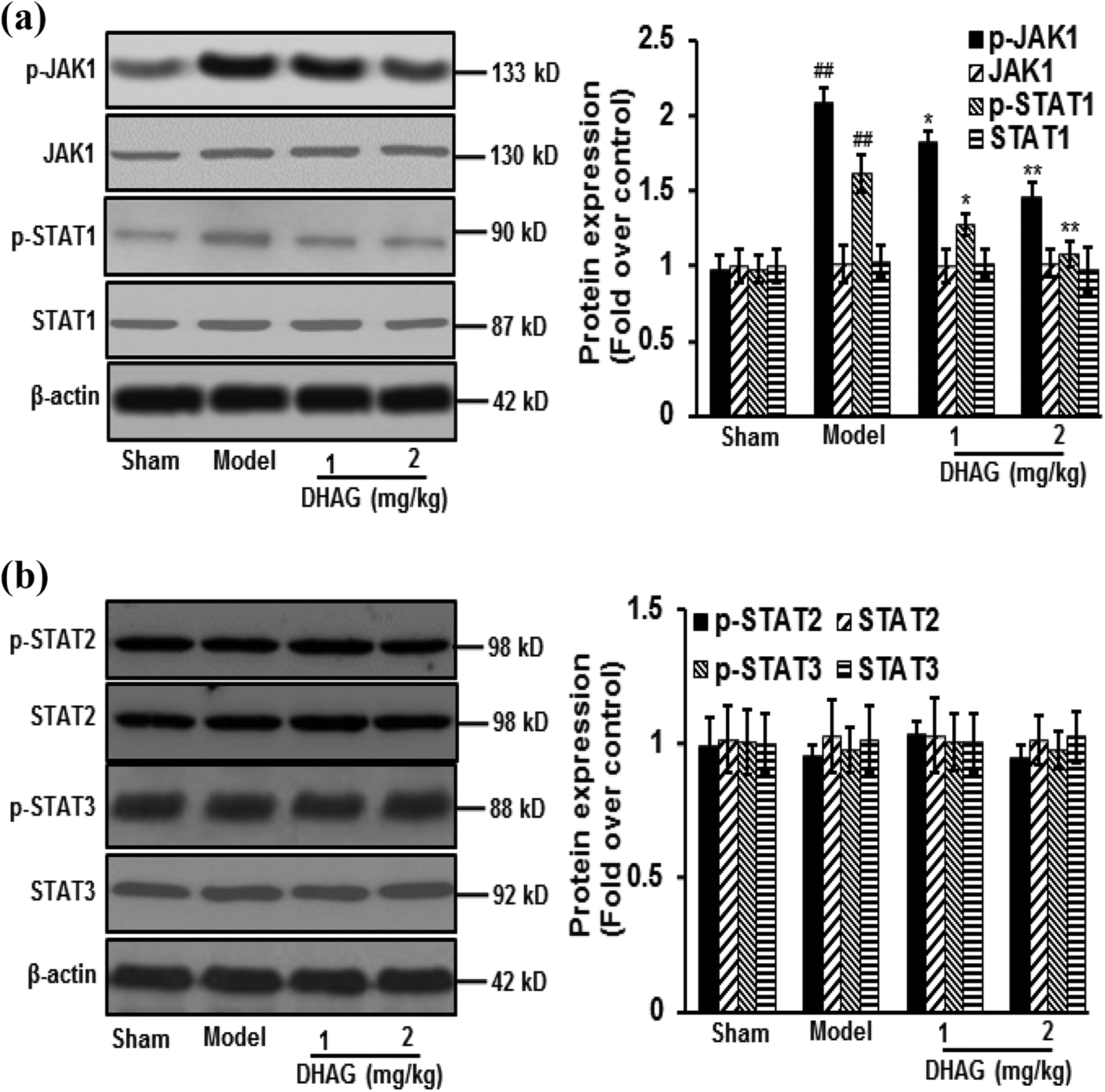
DHAG treatment decreased p-JAK1/p-STAT1 protein expression. DHAG treatment decreased the protein expression of p-JAK1/p-STAT1 (a); DHAG treatment didn’t change the protein expression of STAT2 and STAT3 (b). Data were expressed as mean ± SD (n = 3). ## p < 0.01 versus sham group; *p < 0.05, **p < 0.01 versus model group. DHAG: (Z)-7,4′-dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside; p-JAK1/p-STAT1: phosphorylated-Janus kinase 1/phosphorylated-signal transducer and transcriptional activator 1; SD: standard deviation.
Discussion
Stroke has become the fourth leading cause of death in the world. 19 Cerebral ischemia in patients with stroke can lead to severe cognitive impairment or even “vascular dementia,” which would severely affect the life quality of patients and increase mortality. Excessive perfusion leads to the failure of body’s self-regulatory functions and in turn increases the area of cerebral edema to aggravate cerebral ischemia, which is due to the increased production of free radicals and pro-inflammatory cytokines causing secondary damage to the brain. 20 Therefore, simple thrombolytic therapy has not been the best approach for treating I/R injury. In this study, a similar clinical stroke model, namely MCAO, was used to investigate the protective effects of DHAG. This model has little trauma, strong repeatability, and stability, which is close to clinical pathophysiological changes. 21 Our data demonstrated that DHAG treatment significantly ameliorated brain damage, shown by decreased neurological function score, reduced cerebral infarct size as well as water content in MCAO rats. Moreover, DHAG treatment decreased apoptosis in brain tissues through reducing oxidative stress and inhibiting inflammatory responses, which may be associated with downregulating p-JAK1/p-STAT1.
The Bcl-2 family exerts its proapoptotic (Bax, Bak, and so on) or antiapoptotic (Bcl-2, Bcl-XL) activities mainly through the mitochondrial pathway. When cells are stimulated, intracellular Bax transfers to the mitochondrial outer membrane and forms Bax/Bcl-2 heterodimerization with Bcl-2 to induce the release of cytochrome-c from the mitochondria to the cytoplasm, thereby inducing downstream apoptosis cascade pathway. 22,23 Cytochrome-c release from mitochondria would activate caspases to facilitate the formation of apoptosome complex. 24 As an executor, caspase-3 activates factors of DNA fragmentation, which in turn activate endonucleases to cleave nuclear DNA to cause cell death. 25 DNA double-stranded breaks and caspase-3/9 activities have been considered as biochemical hallmarks for apoptosis. 26,27 In this study, I/R caused brain injury, accompanied by more cytochrome-c release, DNA fragmentation, and increased activities of caspase-3/9. Proapoptotic protein of Bax and caspsase-3/9 expression increased, but antiapoptotic protein of Bcl-2 expression decreased. Moreover, the tight junction protein Claudin 1 and Occludin expression significantly decreased. 28 However, the above effects were largely reversed by DHAG treatment.
Neurons are particularly susceptible to oxidative stress-related damage because of their poor antioxidative defenses. 29 Therefore, the imbalance between the intracellular oxidative and antioxidative defense systems requires the supplement of external antioxidants as potential therapeutics. As a by-product of lipid peroxidation under oxidative stress, MDA is well-known as a widely used marker for oxidative damage. 30 Nrf2 is an important cellular defense against oxidative stress, which can be activated to bind with antioxidant response elements to induce protein expression of cytoprotective targets, such as phase II detoxifying enzymes, antioxidant proteins, and molecular proteasome/chaperones. 31,32 In this study, DHAG treatment activated Nrf2 to trigger the expression of antioxidative genes to restore homeostasis, evidenced by reduced levels of MDA, increased SOD activities as well as GSH levels, and decreased protein expression of NADPH oxidase 1, a major enzyme responsible for production of superoxide by transferring electrons across the membrane from NADPH to molecular oxygen. 33
Moreover, a number of cytokines, such as IL-1β, IL-6, and TNFα, will be produced after cerebral I/R injury. IL-1β, IL-6, TNFα, and NO are all important pro-inflammatory factors with many biological effects. Overproduction of pro-inflammatory factors can lead to cell death, tissue damage, and ultimately organ failure. 34 Excessive pro-inflammatory factors are found after I/R and these factors stimulate the production of oxidants in neurons with subsequent peroxidative damage to macromolecules. Moreover, as a mediator and regulator of inflammatory responses, NO possesses cytotoxic properties and has damaging effects on host tissues. NO is also a potent neurotransmitter at the neuron synapses, which contributes to the apoptosis regulation. 35,36 During inflammation, intracellular responsive cascades such as NF-κB become activated to increase expression of pro-inflammatory products. NF-κB has been considered as a prototypical pro-inflammatory signaling pathway. 37,38 In this study, DHAG treatment significantly reduced inflammatory factor levels and controlled inflammatory responses, which may be through downregulating the protein expression of p-NF-κB.
Studies have demonstrated that pro-inflammatory factors and oxidative stress utilize the JAK/STAT signal pathway to induce cell proliferation or apoptosis. 39 JAK/STAT signaling pathway activated in CNS disorders participates in a variety of neuroinflammatory pathological processes and regulating JAK-STAT signaling pathways can alleviate neuronal damage caused by inflammation. 40 Our study showed that DHAG treatment inhibited the expression of p-JAK1/p-STAT1 proteins in brain tissues.
Some study limitations should be mentioned. Firstly, it is not clear whether DHAG is capable of crossing the BBB. So pharmacokinetic assays are needed to detect the concentration of DHAG in different organs/tissues in the future and effects of DHAG on specific cells or a mix of all need to be further investigated. Secondly, although clear effects were observed with the DHAG treatment, its activities need to be confirmed by other animal models. Finally, it is uncertain whether data from rats accurately reflect molecular changes in human.
In conclusion, I/R resulted in cerebral injury in MCAO rats. DHAG was effective in ameliorating brain damage through inhibiting oxidative stress and inflammatory responses, providing the scientific rationale to develop DHAG as a therapeutic agent against I/R-induced cerebral damage.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Pudong Hospital-Fudan University School of Pharmacy Strategic Cooperation Fusion Fund (no. RHJJ2017-06).