
Abstract
Isoflurane has been demonstrated to induce mitochondrial damage and cell apoptosis. The isoflurane-induced inflammation may be an important reason for this phenomenon. Studies have shown that ulinastatin (UTI) has an anti-inflammatory effect. Our aim was to investigate whether UTI could attenuate isoflurane-induced mitochondrial damage and cell apoptosis by inhibiting inflammation. Human neuroglioma H4 cells were exposed to isoflurane with or without UTI. The ratio of cell apoptosis was evaluated by flow cytometry. β-Amyloid (Aβ) peptide and cleaved caspase 3 expression were evaluated by Western blot analysis. The concentrations of tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β) were detected by sandwich enzyme-linked immunosorbent assays. Mitochondrial structural changes were detected by transmission electron microscopy. Mitochondrial membrane potential (Δψm) was determined by 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1). The activity of the mitochondrial electron transport chain (ETC) complexes I, II, III, and IV was determined by assay kits. UTI attenuated the TNF-α and IL-1β release induced by isoflurane. UTI could also reduce mitochondrial structure damage, mitigate the decrease in Δψm, and improve ETC complexes dysfunction. Furthermore, it decreased cell apoptosis induced by isoflurane in H4 cells. UTI had no effect on isoflurane-induced Aβ expression. UTI may mitigate isoflurane-induced mitochondrial damage and cytotoxicity by inhibiting inflammation.
Introduction
Isoflurane is a common inhaled anesthetic in the clinic, and many studies have shown that isoflurane can cause neuroinflammation and neuron apoptosis in vivo and in vitro, 1 –4 which is related to postoperative neurocognitive disease (PNCD) or Alzheimer’s disease (AD). 5 –7
Mitochondrial damage or dysfunction can cause neuronal apoptosis, 8 and mitochondrial dysfunction may be one of the early triggering events in isoflurane-induced neuronal damage. In our previous study, we found that isoflurane can induce mitochondrial damage and PNCD in old rats. 9 Thus, mitochondrial damage may play an important role in isoflurane-induced cognitive impairment. Many studies have shown the interrelationship between inflammation and mitochondrial damage in neurodegenerative diseases. Inflammatory factors can generate oxidative stress that causes mitochondrial damage 10,11 or affects mitochondrial activity. 12 Therefore, inhibiting inflammation may be an approach to attenuate isoflurane-induced mitochondrial damage and neurotoxicity, thereby preventing or treating PNCD.
Ulinastatin (UTI) is a urinary trypsin inhibitor widely used for treating acute inflammatory response-related diseases in Asia because of its anti-inflammatory effects. 13,14 UTI inhibits the NF-κB pathway and mitogen-activated protein kinases (MAPKs) to mitigate the release of inflammatory cytokines such as tumor necrosis factor α (TNF-α), interleukin 1 (IL-1), and IL-6. 15,16 Studies have indicated that UTI may have a neuroprotective effect. 17 Yano et al. reported that UTI exerted the neuroprotective effect in a focal cerebral ischemia–reperfusion injury model. 18 In this study, we investigated the effects of isoflurane and UTI on β-amyloid (Aβ) peptide expression, cell apoptosis, inflammatory response, and mitochondrial damage in H4 cells to assess whether UTI has a protective effect against isoflurane-induced cytotoxicity.
Materials and methods
Cell lines and culture
The human neuroglioma naive H4 cells from human glioma tissue were provided by the China Center for Type Culture Collection in WuHan. The cells were cultured in high glucose Dulbecco’s modified Eagle’s medium containing 9% heat-inactivated fetal calf serum, penicillin at 100 U mL−1, streptomycin at 100 U mL−1, and 2 mM
Cell groups and treatment
The cell treatment and method were performed in accordance with our previous study. 9 H4 cells were treated at a density of 0.5×106 mL−1 in six-well plates, and the experiments were repeated three times with three wells for each group. The cells were divided into five groups: the control (Con) group, the isoflurane (Iso) group, the UTI 500 group (Isoflurane plus 500 U mL−1 UTI), the UTI 1000 group (Isoflurane plus 1000 U mL−1 UTI), and the UTI 2000 group (Isoflurane plus 2000 U mL−1 UTI); according to grouping, 500, 1000, and 2000 U mL−1 UTI were separately added to the culture medium. UTI was obtained from Tianpu Biochemistry and Medicine Corporation (Guangzhou, People’s Republic of China). Cells were exposed in a sealed plastic box in a 37°C incubator, and the gas was delivered from an anesthesia machine. The control group was exposed to 21% O2 plus 5% CO2 for 6 h. The isoflurane group, UTI 500 group, UTI 1000 group, and UTI 2000 group were exposed to 3% isoflurane plus 21% O2 plus 5% CO2 for 6 h, and a gas analyzer (Capnomac, Helsinki, Finland) was used to continuously monitor the concentrations of inhaled and exhaled gases (oxygen and isoflurane) in the chamber.
Apoptosis analysis by flow cytometry
The percentage of cell apoptosis was detected by an Annexin V/propidium iodide double staining kit (AP101; Multisciences Biotech Co., Ltd, Hangzhou, China). The analysis was performed according to the manufacturer’s protocol.
Measurement of Aβ peptide and cleaved caspase 3 expression in H4 cells by Western blot analysis
The cells were harvested and analyzed for Aβ peptide and cleaved caspase 3 expression and extracts from harvested cells were subjected to Western blot analyses. An anti-Aβ antibody (1:1500 dilution; ab68896; Abcam, Cambridge, UK) was used to detect the monomer of Aβ1-42 (4.3 kDa), and GAPDH (1:1500 dilution) levels were used to normalize protein levels. An anti-cleaved caspase 3 antibody (1:1000 dilution; AC030, Beyotime Biotechnology, Shanghai, China) was used to detect cleaved caspase 3 (17 kDa and 12 kDa), and β-actin (1:1000 dilution) levels were used to normalize protein levels.
Mitochondrial morphological damage observation by TEM
Transmission electron microscopy (TEM) for mitochondrial morphological analysis was performed according to standard instructions. For morphological TEM, H4 neuroglioma cells were fixed in 2.5% glutaraldehyde in phosphate buffer overnight at 4°C.
Assay of the activity of mitochondrial ETC complexes I, II, III, and IV
Mitochondria preparation
Mitochondria were isolated from H4 cells by standard differential centrifugation and resuspended in isolation buffer following the manufacturer’s instructions for the mitochondria isolation kit (C3606; Beyotime Biotechnology, Shanghai, China) and stored on ice. The mitochondria should be used instantly after preparation. Mitochondrial protein content was quantified using a bicinchoninic acid (BCA) protein assay kit.
Determination of activity for complex enzyme I
A total of 5–10 µg of mitochondrial protein was subjected to freezing and thawing at −20°C/20°C three times to reach maximum reaction rate, and 1 mL of buffer containing 2 µg of antimycin A and 60 µmol L−1 coenzyme Q10 (CoQ10) was added to the mix, followed by the addition of 0.13 mmol L−1 of the substrate nicotinamide adenine dinucleotide (NADH) to initiate the reaction. Notably, 5 µmol L−1 rotenone was added to the blank tube to inhibit complex enzyme I activity. The change in NADH absorption at 340 nm was measured within 1 min to determine the activity of compound enzyme I.
Determination of the activity of complex II
A total of 5–10 µg of mitochondrial protein was mixed with 1 mL of buffer containing 88 µmol L−1 2,6-dichlorophenolindophenol (DCPIP), 2 µg of antimycin A and 60 µmol L−1 CoQ10, and then 25 mmol L−1 succinic acid was added to initiate the reaction. The change in DCPIP absorption at 600 nm was measured to determine the activity of compound enzyme II.
Determination of the activity of complex enzyme III
A total of 5–10 µg of mitochondrial protein was mixed with 1 mL of buffer containing 0.5 mmol L−1 ethylenediaminetetraacetic acid and 35.7 µmol L−1 oxidized cytochrome c, and then 100 µmol L−1 ubiquinol 10 was added to initiate the reaction. The change in cytochrome c absorption at 550 nm was measured to determine the activity of compound enzyme III.
Determination of the activity of complex enzyme IV
A total of 20–30 µg of mitochondrial protein and 33 µL of cytochrome c (20 mg L−1) were added to 0.5 mL of KH2PO4 (200 mmol L−1, pH 7.4), 0.375 mL of distilled water, and 25 µL of 2% Triton X-100 and mixed. The change in cytochrome c absorption at 550 nm was measured to determine the activity of compound enzyme IV.
The change in Δψm measurement by JC-1
The change in mitochondrial membrane potential (Δψm) was detected by flow cytometry using a JC-1 dye kit (C2006; Beyotime Biotechnology, Shanghai, China). The JC-1 staining solution and buffer were prepared according to the manufacturer’s instructions. JC-1 (5 μmol L−1) was mixed with the cells for 15 min at 37°C. The stained cells were washed with phosphate-buffered saline, and then a minimum of 1.0 × 104 cells mL−1 per sample was analyzed by flow cytometry to investigate and analyze the intensity of the red and green fluorescence. When cells’ mitochondrial membrane was damaged and depolarized, the color of this dual-emission probe changed from red to green. The percentage of cells with abnormally low Δψm was analyzed.
Measurement of the extracellular concentration of TNF-α and IL-1β by sandwich ELISA
The concentrations of TNF-α and IL-1β in the culture medium were determined with enzyme-linked immunosorbent assays (ELISAs) of human TNF-α (589201; Cayman Chemical, Ann Arbor, Michigan, USA) and IL-1β (583311; Cayman Chemical) according to the manufacturer’s instructions. The absorbance in each well was measured at 450 nm. The concentrations of TNF-α or IL-1β in the test samples were determined to compare results with signals from unconditioned medium spiked with known quantities of TNF-α or IL-1β.
Statistical analysis
The data were statistically analyzed by SPSS20.0. The measured data, such as flow cytometry, Western blot, JC-1, assay of the activity of electron transport chain (ETC) complexes and ELISA, are expressed as the mean ± SD. The Tukey test after one-way analysis of variance was used for intergroup comparisons. The value of p < 0.05 was considered statistically significant.
Results
Effect of UTI on reducing the H4 cell apoptosis induced by isoflurane
After exposure to 3% isoflurane for 6 h, the ratio of cell apoptosis was examined. Isoflurane significantly increased neuroglioma H4 cell apoptosis (p < 0.001). However, 500, 1000, and 2000 U mL−1 UTI treatment all reduced H4 cell apoptosis induced by isoflurane (p < 0.001). Moreover, this antiapoptotic effect was dose dependent, and the greater the dose, the more significant was the effect. Therefore, in the following experiment, we chose 2000 U mL−1 UTI as the research dosage (Figure 1).

Apoptosis of H4 cells treated with isoflurane plus UTI. H4 cells were treated with 3% isoflurane or 3% isoflurane plus UTI at concentrations of 500, 1000, or 2000 U mL−1 for 6 h. The percentages of early and late apoptotic cells were analyzed by flow cytometry (a, b). The H4 cell apoptosis induced by isoflurane can be significantly reduced by UTI, and this effect was dose dependent. The results are expressed as the mean ± SD (n = 3), one-way ANOVA, ***p < 0.001. UTI: ulinastatin; SD: standard deviation; ANOVA: analysis of variance.
Effect of UTI on decreasing the cleaved caspase 3 expression induced by isoflurane
Cleaved caspase 3 was detected to further determine cell apoptosis. After exposure to 3% isoflurane for 6 h, cleaved caspase 3 expression in H4 cells significantly increased (p < 0.01), and treatment with 2000 U mL−1 UTI significantly decreased the level of cleaved caspase 3 expression (p < 0.05) (Figure 2).

Cleaved caspase 3 expression in H4 cells. The expression of cleaved caspase 3 was determined by Western blot (a). The level of cleaved caspase 3 in the isoflurane group was significantly elevated compared with that of the control group and the UTI 2000 group (b). The results are expressed as the mean ± SD (n = 3), one-way ANOVA, *p < 0.05, **p < 0.01, and n.s. means nonsignificant between two groups. UTI: ulinastatin; SD: standard deviation; ANOVA: analysis of variance.
Effect of UTI on suppressing the inflammatory response of H4 cells induced by isoflurane
The concentrations of the inflammatory factors TNF-α and IL-1β in the culture medium were examined to determine the inflammatory response. After isoflurane exposure, the concentrations of TNF-α and IL-1β in the culture medium significantly increased, and after treatment with 2000 U mL−1 UTI, the release of TNF-α (p < 0.05) and IL-1β (p < 0.01) was significantly reduced (Figure 3).

The concentrations of IL-1β and TNF-α in the culture medium. After isoflurane exposure, the concentrations of IL-1β (a) and TNF-α (b) in the culture medium significantly increased in the isoflurane group, and 2000 U mL−1 UTI treatment reduced this effect. The results are expressed as the mean ± SD (n = 3), one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, and n.s. means nonsignificant between two groups. IL-1β: interleukin 1β; TNF-α: tumor necrosis factor α; UTI: ulinastatin; SD: standard deviation; ANOVA: analysis of variance.
Effect of UTI on reversing ETC complex dysfunction induced by isoflurane
In this study, the activities of mitochondrial ETC complexes I (Figure 4(a)), II (Figure 4(b)), III (Figure 4(c)), and IV (Figure 4(d)) were examined after treatment. After exposure to 3% isoflurane for 6 h, the activities of complexes I, II, and III increased (p < 0.001), and the activity of complex IV significantly declined (p < 0.001). With treatment with 2000 U mL−1 UTI, those effects can be recovered (p < 0.001) (Figure 4).
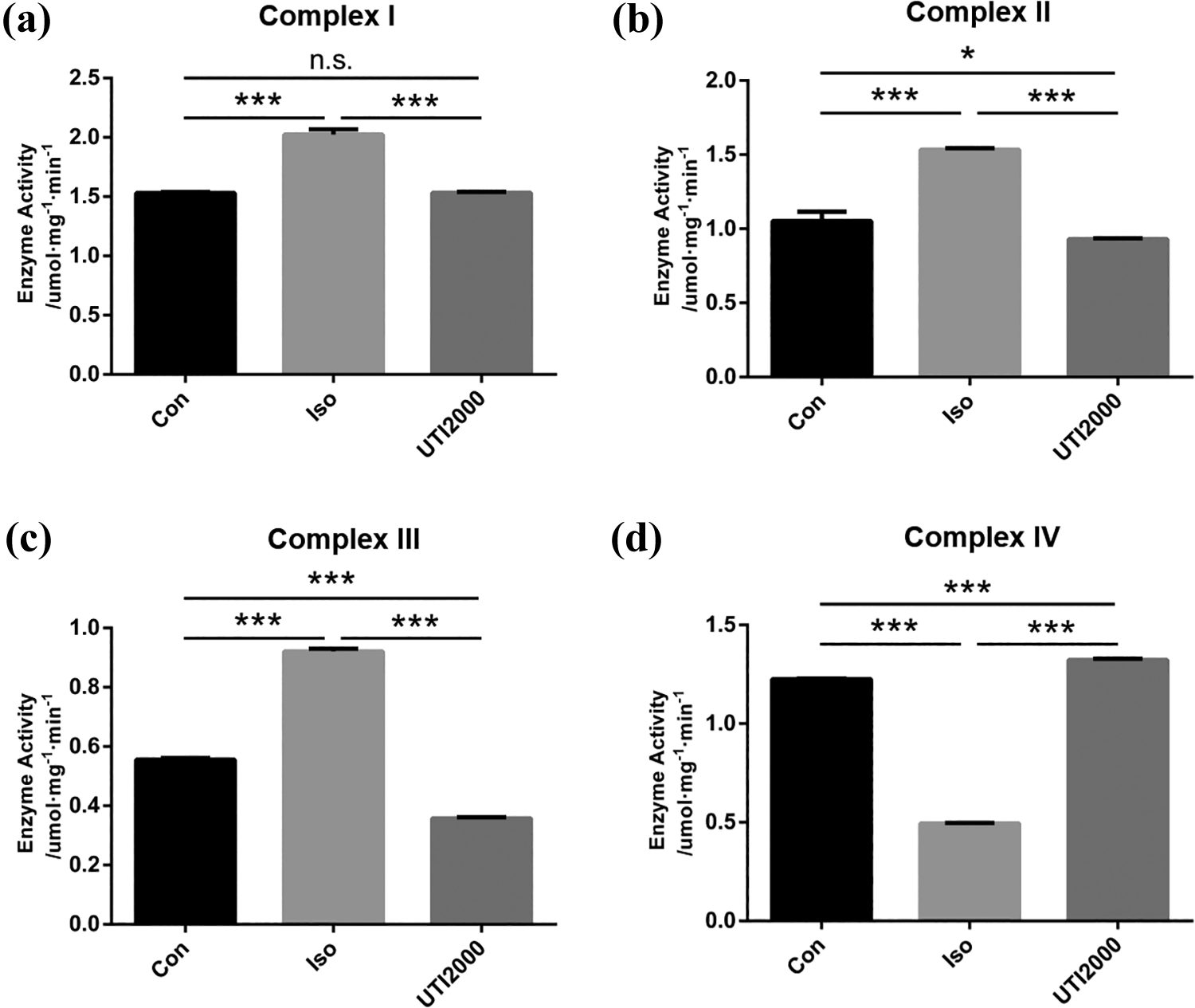
(a to d) The activity of ETC complexes I, II, III, and IV changed in different groups. After exposure to 3% isoflurane for 6 h, the activity of ETC complexes was dysfunctional, while 2000 U mL−1 UTI partially reversed this effect. The results are expressed as the mean ± SD (n = 3), one-way ANOVA, *p < 0.05, ***p < 0.001, and n.s. means nonsignificant between two groups. ETC: mitochondrial electron transport chain; UTI: ulinastatin; SD: standard deviation; ANOVA: analysis of variance.
Effect of UTI on the change in mitochondrial structure and Δψm induced by isoflurane
After exposure to 3% isoflurane for 6 h, the structure of mitochondria and Δψm were examined. The JC-1 result showed that the Δψm in H4 cells significantly declined (p < 0.001) (Figure 5(a) and (b)). Under TEM, the mitochondria in the isoflurane group were swollen, some mitochondria were vacuolated, the density of the matrix was not uniform, the cristae were fractured, and the arrangement was disordered (Figure 5(c)). When treated with 2000 U mL−1 UTI, the Δψm of H4 cells was partially recovered (p < 0.05), and the mitochondrial morphology damage was partially restored. The mitochondria were slightly swollen, and the membrane structure became clear and complete. The matrix was uniform, and the cristae were arranged neatly (Figure 5(a) to (c)).

Mitochondrial damage in H4 cells. The Δψm determined by JC-1 (a). The ratio of H4 cells with declined Δψm induced by isoflurane was attenuated when treated with 2000 U mL−1 UTI (b). UTI also reduced the isoflurane-induced mitochondrial structure damage detected by transmission electron microscopy (c). The objects indicated by red arrows are mitochondria (magnification ×40,000, scale bar in each panel = 1 μm). The results are expressed as the mean ± SD (n = 3), one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, and n.s. means nonsignificant between two groups. Δψm: mitochondrial membrane potential; UTI: ulinastatin; SD: standard deviation; ANOVA: analysis of variance.
Effect of UTI on the Aβ peptide expression induced by isoflurane
The expression of the Aβ peptide was examined to determine the effect of UTI on Aβ peptide metabolism. After 3% isoflurane exposure for 6 h, Aβ expression in the H4 cells significantly increased (p < 0.05), while the level of Aβ peptide expression did not change when treated with 2000 U mL−1 UTI (p > 0.05) (Figure 6).
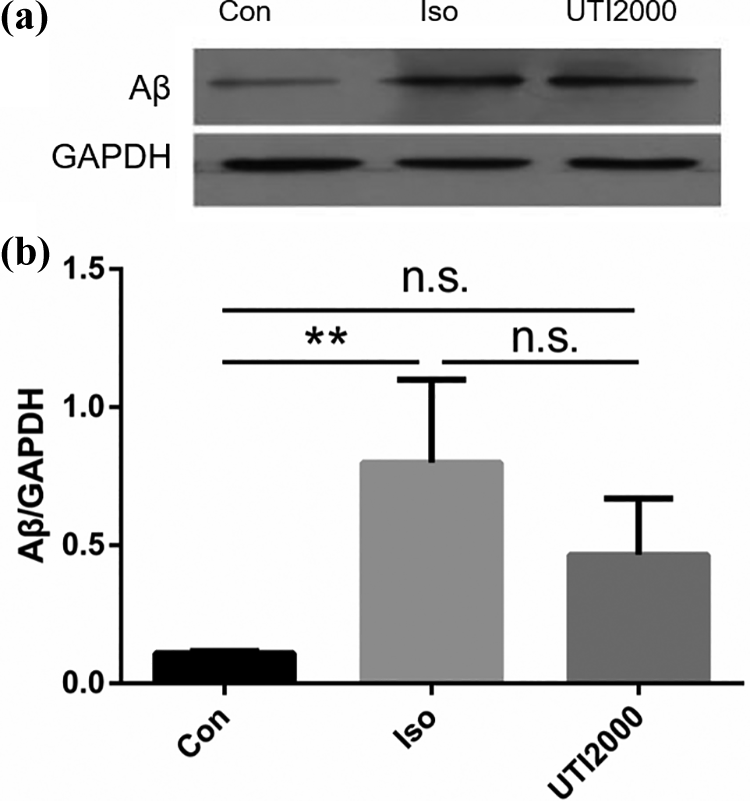
(a and b) Aβ peptide expression in H4 cells. The Aβ peptide expression significantly increased in H4 cells in the isoflurane group, while 2000 U mL−1 UTI treatment did not change the level of Aβ peptide expression. The results are expressed as the mean ± SD (n = 3), one-way ANOVA, **p < 0.01, and n.s. means nonsignificant between two groups. Aβ: β-amyloid peptide; UTI: ulinastatin; SD: standard deviation; ANOVA: analysis of variance.
Discussion
According to the results of our study, UTI attenuates isoflurane-induced mitochondrial damage and cell apoptosis. The data showed that isoflurane can trigger inflammation, induce mitochondrial damage including the activities of ETC complexes, alter Δψm, damage the structure, and cause cell apoptosis in H4 cells. Treatment with 2000 U mL−1 UTI can attenuate these effects, and the mechanism could be through suppressing the inflammatory response. These results suggested that UTI may have a protective effect against isoflurane-induced cytotoxicity by inhibiting inflammation.
Neuroinflammation is thought to play an important role in PNCD or neurodegenerative diseases. The clinical concentration of isoflurane has been shown to increase proinflammatory cytokines in the brain or neurons and to cause learning and memory impairment in animals related to PNCD. 5 –7 Wu et al. demonstrated that isoflurane can specifically affect neurons, leading to an increase in TNF-α generation to cause neuroinflammation in the brains of mice. 6 In our previous study, we found that 1.2% isoflurane could cause an increase in IL-1β in the hippocampus of adult rats, induce a decrease in neuronal density and further impair long-term spatial reference memory and hippocampus-dependent learning and memory. 2 In the present study, we observed that isoflurane can trigger H4 cells to generate TNF-α and IL-1β and induce H4 cell apoptosis. This result is consistent with previous studies.
Mitochondria are the main energy resource for neuron survival, and mitochondrial damage, or dysfunction can cause neuron energy metabolism disorders and death. Mitochondrial dysfunction is thought to be one of the earliest triggering events in isoflurane-induced neuronal damage. 11 In the present study, we observed that after isoflurane exposure, the ETC complexes showed dysfunction, the Δψm was decreased, and the mitochondrial structure was damaged. The Δψm decrease is one of the characteristic events in the early stage of cell apoptosis. The decrease in Δψm can induce mitochondrial permeability transition pore (mPTP) opening, and proteins such as cytochrome c, BAX, and apoptosis-inducing proteins will be released from the mPTP to cause cell apoptosis. While the maintenance of Δψm depends on the electron transfer process, when the ETC complexes are dysfunctional, the proton pump will be destroyed, and the Δψm will decline. 19,20 In addition, mitochondria are considered to participate in neurodegenerative processes. 21,22 The dysfunction of the ETC complexes was found to be an important characteristic in some neurodegenerative diseases; the decreased activity of ETC complex IV is the most common finding in AD, and deficiency of the activity of ETC complexes was observed in Huntington’s disease. 21,23,24 In our previous study, we found that lidocaine can improve isoflurane-induced cognitive impairment by reducing mitochondrial damage. 9 In the present study, we also found that UTI can alleviate isoflurane-induced mitochondrial damage and H4 cell apoptosis. These results indicated that UTI may have the effect of preventing or treating PNCD.
After isoflurane exposure, the activity of complexes I, II, and III increased, and the activity of complex IV decreased. This result suggested that complex IV could be a target of isoflurane-induced mitochondrial damage. Complex IV is the terminal complex of the two mitochondrial ETCs; when it shows abnormal ability, the energy production in cells will greatly decrease, and other ETC complexes could compensate for the increase in energy production under this condition. This could be the reason for the increase in the activity of complexes I, II, and III.
Mitochondria are vulnerable to endogenous and exogenous factor effects. Inflammation can influence mitochondrial function by some pathways. Proinflammatory cytokines, such as TNF, can cause mitochondrial damage by promoting oxidative stress. 10 Toll-like receptors (TLRs) can regulate mitochondrial activity, and the activation of TLR3 can reduce mitochondrial oxygen consumption by opening the permeability transition pore. 25 IL-1β can decrease mitochondrial activity through the production of NO. 26 In the present study, we observed that when treated with UTI, the release of the proinflammatory cytokines TNF-α and IL-1β induced by isoflurane significantly decreased, while mitochondrial damage and cell apoptosis were also mitigated. According to these results, we speculated that the inflammatory response-induced mitochondrial damage may be a reason for isoflurane-induced nerve cell apoptosis, and UTI effectively alleviated these conditions by suppressing the inflammatory response.
Regarding Isoflurane-induced PNCD, the Aβ peptide is considered to be an important reason. In addition to excess soluble Aβ peptide generating and exacerbating neuroinflammation, 27 the Aβ peptide can regulate the synthesis and release of acetylcholine to influence memory, consciousness, and learning dysfunction, 28 and overexpression of the Aβ peptide can extremely damage learning memory and hippocampal volume in mice. 29 In our study, even when the cells were treated with 2000 U mL−1 UTI, the release of TNF-α, and IL-1β were reduced, but the expression of the Aβ peptide did not obviously change. This result indicated that the main effect of UTI in attenuating isoflurane-induced cytotoxicity may suppress the inflammatory response without influencing the Aβ peptide.
There are some limitations of this study. First, we found that UTI can reduce isoflurane-induced mitochondrial damage and apoptosis by inhibiting inflammation, but we did not explore the specific pathway by which UTI regulates inflammation. Second, our study observed that isoflurane mainly affected ETC complex IV, but we did not further reveal the reason and the significance of this phenomenon. Third, all results of the present study were obtained in vitro, and further study should be developed to test the in vivo effects.
In conclusion, our study provided evidence that UTI can alleviate isoflurane-induced mitochondrial damage and cell apoptosis, and the potential mechanism could be through suppressing the inflammatory response in vitro.
Footnotes
Author contributions
DL and XZ contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China [Grant Number 81201022]; the Techpool Research Foundation [Grant Number UF201306]; and the Natural Science Foundation of Guangdong Province [Grant Number 2018A0303130195].