
Abstract
Cigarette smoke (CS) exposure is an important risk factor for chronic obstructive pulmonary disease (COPD). MicroRNA-150 (miR-150) is involved in several inflammatory diseases. However, little is known about the role of miR-150 in the pathogenesis of COPD. In this study, we established a CS-related mouse model of COPD and evaluated the impact of miR-150 on CS-induced lung inflammation. We further investigated the effects of miR-150 overexpression on pro-inflammatory cytokine production and apoptosis in airway epithelial cells exposed to CS extract (CSE). It was found that miR-150 was significantly (p < 0.05) downregulated in the lungs of CS-exposed mice, compared to control mice under normal air. The CSE-exposed BEAS-2B airway epithelial cells displayed a four- to six-fold reduction in miR-150 levels, compared to control cells (p < 0.05). Delivery of miR-150 mimic attenuated CS-induced lung inflammation and accumulation of neutrophils, lymphocytes, and macrophages in bronchoalveolar lavage fluid. Moreover, miR-150 overexpression prevented the induction of interleukin-6, tumor necrosis factor alpha, and interleukin-8 expression and nuclear factor kappa B (NF-κB) transcriptional activity in BEAS-2B cells by CSE. Additionally, miR-150 protected BEAS-2B cells from CSE-induced apoptosis, which was associated with reduced p53 expression. Co-expression of p53 restored apoptotic response to CSE in miR-150-overexpressing BEAS-2B cells. Collectively, miR-150 suppresses CS-induced lung inflammation and airway epithelial cell apoptosis, which is causally linked to repression of p53 expression and NF-κB activity. Restoration of miR-150 expression may represent a potential therapeutic strategy for CS-related COPD.
Introduction
Chronic obstructive pulmonary disease (COPD), which is characterized by airway inflammation and destruction of lung parenchyma, is a leading cause of death worldwide. 1 Cigarette smoke (CS) exposure is a major risk factor for COPD. 2 It has been reported that CS exposure causes accumulation of inflammatory cells in the airway, production of numerous pro-inflammatory cytokines, and epithelial and vascular remodeling in a mouse model of COPD. 3,4 Induction of apoptotic death in airway epithelial cells accounts for CS-induced lung injury. 5 Several lines of evidence have linked increased expression of p53 to CS-induced airway epithelial cell apoptosis. 6,7 However, the exact mechanism for CS-induced lung injury has not yet been fully elucidated.
MicroRNAs (miRs) are an important class of small regulatory RNAs, which can bind to the 3′-untranslated region (3′-UTR) of target mRNAs, leading to translational repression or mRNA degradation. 8 Several miRs have been found to be involved in the pathogenesis of COPD. 9,10 For example, a previous study has shown that miR-218-5p is significantly downregulated in patients with COPD compared with never smokers and displays the ability to attenuate CS-induced lung inflammation in animal models. 10 MicroRNA-150 (miR-150) has gained increasing attention due to its critical role in cancer progression. 11,12 Overexpression of miR-150 serves as a poor prognostic factor for non-small cell lung cancer. 11 Ectopic expression of miR-150 facilitates the proliferation and tumorigenesis of gastric cancer cells. 13 Similarly, miR-150 shows the ability to augment the proliferation of lung cancer cells, which is ascribed to downregulation of p53. 14 However, relatively little is known about the role of miR-150 in COPD.
In this study, we established a CS-related mouse model of COPD and investigated the effect of administration of miR-150 mimic on CS-induced lung inflammation. We further studied the effects of miR-150 overexpression on cigarette smoke extract (CSE)-elicited production of pro-inflammatory cytokines and apoptosis in airway epithelial cells. Additionally, the molecular mechanism underlying the action of miR-150 was checked.
Materials and methods
Animals and CS exposure
Adult C57BL/6 mice (6 weeks of age) were purchased from the Experimental Animal Center of Zhengzhou University (Zhengzhou, China). The animals had ad libitum access to food and water. CS exposure was done as described previously. 15 In brief, animals were placed in an inhalation chamber connected to a pump and exposed to 3 days of CS. The smoke was generated by burning 3R4F research cigarettes (University of Kentucky, Lexington, Kentucky, USA), which contain 9.4 mg of tar and 0.726 mg of nicotine per cigarette. Mice were exposed to CS for three consecutive days (five times per day with a 30-min smoke-free interval, 30 min each time). For evaluation of the role of miR-150 in CS-induced lung injury, miR-150 mimic or negative control miR (Genepharm, Shanghai, China) was administered only one time to mice (1 nM per mouse 16,17 ; six mice for each treatment), followed by CS exposure for three consecutive days. Control animals were exposed to normal air. Twenty-four hours after the last CS treatment, animals were killed. One part of lung tissues was processed for hematoxylin and eosin staining, and the other was subjected to gene expression analysis. The animal experimental protocol was approved by the Animal Care and Use Committee of Xi’an Jiaotong University (Xi’an, China).
Preparation of BALF
The lungs of mice were lavaged three times with physical saline after introduction of a cannula to the trachea. Bronchoalveolar lavage fluid (BALF) was centrifuged at 650g for 10 min, and the supernatant was collected for analysis of interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α), and interleukin-8 (IL-8) levels with commercially available enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, Minnesota, USA). The pellet was resuspended with physical saline, and differential cell numbers were determined on cytospins.
Preparation of CSE
CSE was prepared as previously described. 18 In brief, the smoke of 10 cigarettes was bubbled through serum-free Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, California, USA; 50 mL) and sterilized via filtration through a 0.22-μm filter (Millipore, Bedford, Massachusetts, USA). The CSE stock solution was considered as 100% CSE and diluted to indicated concentrations before use.
Cell culture
BEAS-2B cells derived from normal human bronchial epithelium were purchased from Sigma-Aldrich (St. Louis, Missouri, USA) and cultured in DMEM containing 10% fetal bovine serum (Sigma-Aldrich) at 37°C in a 5% carbon dioxide incubator. For CSE exposure, BEAS-2B cells at 80% confluence were incubated with 5 and 15% CSE for 24 h before further experiments.
Quantitative RT-PCR analysis
Total RNA from cells and lung homogenates was extracted using TRIzol reagent (Invitrogen) following the manufacturer’s instructions. Quantification of miR-150 levels by real-time polymerase chain reaction (RT-PCR) was performed using the TaqMan miRNA assay system (Applied Biosystems, Foster City, California, USA). Small nuclear RNA U6 was used as a loading control. For detection of IL-6, TNF-α, and IL-8 mRNA by RT- PCR, cDNA synthesis was performed with random hexamer primers using the Superscript III Reverse Transcriptase Kit (Invitrogen). PCR primers are summarized as follows 19 : IL-6: forward, 5′-AAATGCCAGCCTGCTGACGAAC-3′ and reverse, 5′-AACAACAATCTGAGGTGCCCATGCTAC-3′; TNF-α: forward, 5′-CGAGTGACAAGCCTGTAGC-3′ and reverse, 5′-GGTGTGGGTGAGGAGCACAT-3′; IL-8: forward, 5′-AACTTCTCCACAACCCTCTG-3′ and reverse, 5′-TTGGCAGCCTTCCTGATTTC-3′; β-actin: forward, 5′-ACGGGGTCACCCACACTGTGC-3′ and reverse, 5′-CTAGAAGCATTTG-CGGTGGACGATG-3′. The mRNA levels of IL-6, TNF-α, and IL-8 were normalized to that of β-actin.
Cell transfection
Cell transfection was achieved using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen); miR-150 mimic and negative controls were used at a final concentration of 50 nM. BEAS-2B cells transfected with miR-150 mimic or control miR were cultured for 24 h before exposure to 15% CSE. In some experiments, cells were co-transfected with miR-150 mimic and p53-expressing plasmid or empty vector for 24 h before CSE treatment. The p53-expressing plasmid was purchased from Sino Biological Company (Beijing, China), which encodes the full-length open reading frame of p53 lacking its 3′-UTR.
Luciferase reporter assay
Nuclear factor kappa B (NF-κB)-dependent transcriptional activity was assessed using the reporter plasmid pNF-κB-Luc (Agilent Technologies, Santa Clara, California, USA). BEAS-2B cells were co-transfected with miR-150 mimic or control miR, together with the pNF-κB-Luc plasmid and Renilla luciferase reporter pRL-TK (Promega, Madison, Wisconsin, USA) for 24 h before CSE treatment. After incubation for 24 h, luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega).
Western blot analysis
Cell lysates were prepared using the RIPA Lysis Buffer (Beyotime, Shanghai, China) supplemented with a cocktail of protease inhibitors and phosphatase inhibitors (Roche Applied Science, Indianapolis, Indiana, USA). Protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The membranes were incubated overnight at 4°C with anti-IL-6, anti-TNF-α, anti-IL-8, anti-p53, and anti-β-actin antibodies (all from Santa Cruz Biotechnology Inc., Santa Cruz, California, USA; 1:500 dilution), followed by incubation with secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology Inc.) for 1 h at room temperature. β-Actin was used as a loading control. Protein bands were detected with the enhanced chemiluminescence reagent (Sigma-Aldrich) and quantified with Quantity One software (Bio-Rad Laboratories Inc., Hercules, California, USA).
Apoptosis analysis by flow cytometry
For measurement of apoptosis, the Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (Sigma-Aldrich) was used. In brief, cells were fixed and incubated with FITC-conjugated Annexin V and propidium iodide (PI) in the dark for 30 min. Stained cells were analyzed by a FACSCalibur flow cytometer (Becton Dickinson Biosciences, San Jose, California, USA).
Measurement of caspase 3 activity
The activity of caspase 3 was measured with the Caspase 3 Colorimetric Assay kit (Sigma-Aldrich) as per the manufacturer’s protocol. The assay was based on the release of chromophore molecules by the enzymatic cleavage of Asp-Glu-Val-Asp (DEVD) (Asp-Glu-Val-Asp, a caspase-specific peptide substrate conjugated to reporter ρ-nitroanaline molecules). Briefly, cells were lysed in ice-cold lysis buffer and centrifuged at 14,000g for 10 min at 4°C. Cell lysates were incubated with the substrate solution containing Ac-DEVD-pNA for 2 h at 37°C, and caspase 3 activity was determined by measuring absorbance at 405 nm.
Statistical analysis
The SPSS 19.0 software (SPSS Inc., Chicago, Illinois, USA) was used for statistical analysis. Data are expressed as mean ± standard deviation. Differences in group means were determined using one-way analysis of variance or Student’s t test; p values <0.05 were considered statistically significant.
Results
MiR-150 is downregulated in CS-exposed mouse lungs and human bronchial epithelial cells (HBECs)
The levels of miR-150 in the lung tissues from CS-exposed and control mice were measured by RT- PCR analysis. The results demonstrated that the expression of miR-150 was reduced by 72% in the lungs of mice after exposure to CS (p < 0.05 vs. control mice; Figure 1(a)). We further confirmed the findings in human airway epithelial cells after exposure to CSE. There was a 60 and 75% reduction in miR-150 levels in BEAS-2B cells exposed to 5 and 15% CSE, respectively, compared to control cells (p < 0.05; Figure 1(b)).

MiR-150 is downregulated in CS-exposed mouse lungs and HBECs. (a) RT-PCR analysis of miR-150 levels in lungs from CS-exposed and control mice (n = 6). (b) BEAS-2B cells were exposed to normal air (as a control) or CSE and measured for miR-150 levels. *p < 0.05 versus the control group. miR-150: microRNA-150; CS: cigarette smoke; RT-PCR: real-time polymerase chain reaction; CSE: cigarette smoke extract; HBEC: human bronchial epithelial cell.
Delivery of miR-150 mimic mitigates CS-induced lung inflammation
To check the role of miR-150 in the susceptibility of mice to CS-induced lung injury, we used an acute CS model of COPD; miR-150 mimic was intratracheally administered to mice before exposure to CS. Histologically, CS-exposed mice displayed increased infiltration of inflammatory cells to the lung, compared to control mice exposed to normal air (Figure 2(a)). Of note, delivery of miR-150 mimic attenuated CS-induced lung inflammation. RT-PCR analysis confirmed the increased expression of miR-150 in the lung tissue from CS-exposed mice after administration of miR-150 mimic (Figure 2(b)). In BALF, a significant (p < 0.05) increase in the number of neutrophils (5.4 × 105 vs. 1.1 × 105), lymphocytes (9.4 × 104 vs. 0.22 × 104), and macrophages (13.2 × 104 vs. 0.45 × 104) was observed after CS exposure relative to control values (Figure 2(c)). Such increase was significantly (p < 0.05) abolished by delivery of miR-150 mimic. Measurement of inflammatory mediators in BALF (Figure 2(d)) revealed that CS exposure resulted in a significant (p < 0.05) release of IL-6 (50.2 vs. 14.6 pg/mL), TNF-α (171.5 vs. 51.2 pg/mL), and IL-8 (56.9 vs. 10.2 pg/mL). Administration of miR-150 mimic significantly blocked the induction of these cytokines.

Delivery of miR-150 mimic mitigates CS-induced lung inflammation. Mice were exposed to normal air or CS with or without prior administration of miR-150 mimic or control miR. (a) Representative sections of lungs stained with H&E. Delivery of miR-150 mimic prevented the infiltration of inflammatory cells to the lung. Scale bar = 100 μm. (b) RT-PCR analysis confirmed the upregulation of miR-150 in the lung after administration of miR-150 mimic. (c) Determination of the number of neutrophils, lymphocytes, and macrophages in BALF. (d) Measurement of cytokine levels in BALF by ELISA. *p < 0.05. miR-150: microRNA-150; CS: cigarette smoke; RT-PCR: real-time polymerase chain reaction; BALF: bronchoalveolar lavage fluid; ELISA: enzyme-linked immunosorbent assay; H&E: hematoxylin and eosin.
MiR-150 suppresses the expression of inflammatory cytokines in HBECs induced by CSE
Next, we examined the effect of overexpression of miR-150 on CSE-induced production of inflammatory cytokines in HBECs. As determined by RT-PCR analysis (Figure 3(a)), HBECs cultured with 15% CSE showed a significant (p < 0.05) expression of IL-6 (2.3-fold), TNF-α (3.5-fold), and IL-8 (1.9-fold) transcripts, relative to control cells under normal culture conditions. The induction of these cytokines was less profound in HBECs treated with 5% CSE (data not shown). Therefore, in the following experiments, 15% CSE was used. Transfection with miR-150 mimic significantly prevented the induction of the inflammatory cytokines by CSE. Western blot analysis validated the reduction of IL-6, TNF-α, and IL-8 expression after overexpression of miR-150 (Figure 3(b)). Since NF-κB is a pivotal transcriptional regulator of inflammatory genes, 20 we checked the effect of miR-150 on NF-κB activity. As depicted in Figure 3(c), addition of 15% CSE increased NF-κB transcriptional activity by 4.4-fold in HBECs. Ectopic expression of miR-150 suppressed the CSE-induced NF-κB transcriptional activity by 65% (Figure 3(c)). Collectively, these data suggest that miR-150 acts as a negative regulator of NF-κB activities in airway epithelial cells in response to CS.

miR-150 suppresses the expression of inflammatory cytokines in HBECs induced by CSE. (a) Quantification of the mRNA levels of IL-6, TNF-α, and IL-8 in BEAS-2B cells exposed to 15% CSE with or without pretransfection with miR-150 or control miR. (b) Western blot analysis of the protein levels of IL-6, TNF-α, and IL-8 in BEAS-2B cells treated as in (a). Numbers below the blots represent fold change in protein levels. (c) NF-κB-dependent luciferase reporter assay. BEAS-2B cells were co-transfected with miR-150 mimic or control miR, together with the pNF-κB-Luc plasmid and pRL-TK for 24 h before 15% CSE treatment. The firefly luciferase activity was normalized to that of Renilla luciferase. *p < 0.05. CSE: cigarette smoke extract; HBEC: human bronchial epithelial cell; miR-150: microRNA-150; IL-6: interleukin-6; TNF-α: tumor necrosis factor alpha; IL-8: interleukin-8; NF-κB: nuclear factor kappa B.
MiR-150 targets p53 to suppress CSE-induced apoptosis in HBECs
We also tested the effect of miR-150 overexpression on CSE-induced airway epithelial cell apoptosis. As determined by Annexin V/PI staining, ectopic expression of miR-150 rendered BEAS-2B cells more resistant to CSE-induced apoptotic death, which decreases the percentage of apoptosis from 32.6% to 11.5% (Figure 4(a)). Measurement of caspase 3 activity further validated the antiapoptotic activity of miR-150 in HBECs. As shown in Figure 4(b), exposure to 15% CSE led to 2.7-fold increase in caspase 3 activity in BEAS-2B cells (p < 0.05 vs. control cells under normal air). The elevation of caspase 3 activity was significantly (p < 0.05) abolished by miR-150 overexpression.
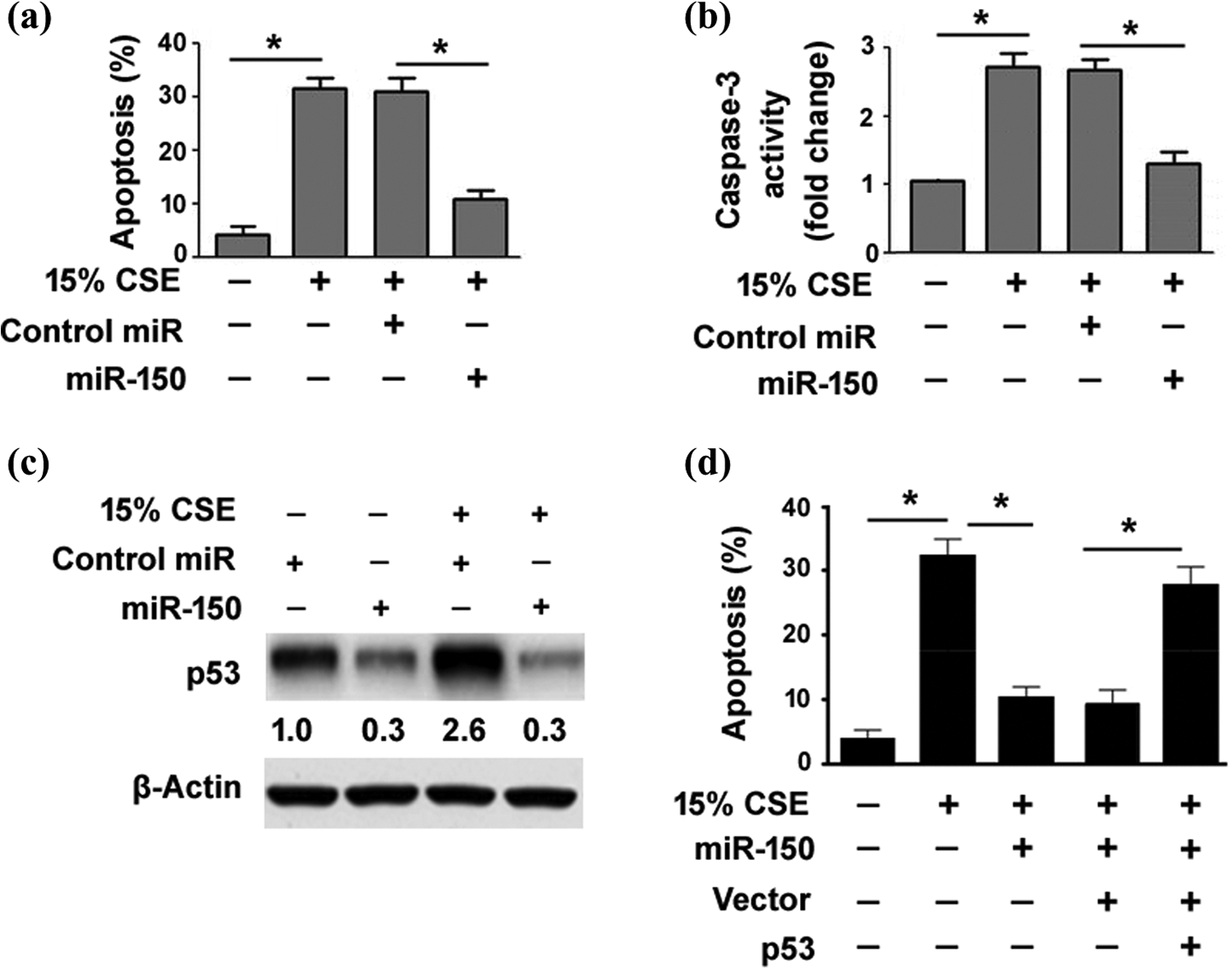
MiR-150 targets p53 to suppress CSE-induced apoptosis in HBECs. (a) Flow cytometric analysis of apoptosis in BEAS-2B cells pretransfected with control miR or miR-150 before CSE exposure. Bar graphs represent the results from three independent experiments. (b) Measurement of caspase 3 activity with the Caspase 3 Colorimetric Assay Kit. CSE-induced elevation of caspase 3 activity was significantly reduced by miR-150 overexpression. (c) Western blot analysis of p53 protein levels in BEAS-2B cells pretransfected with control miR or miR-150 mimic with or without CSE treatment. (d) Detection of apoptosis after Annexin V/PI staining. Co-expression of p53 restored apoptotic response to CSE treatment in miR-150-overexpressing BEAS-2B cells. *p < 0.05. miR-150: microRNA-150; CSE: cigarette smoke extract; HBEC: human bronchial epithelial cell.
It has been reported that miR-150 can repress the expression of p53, a key regulator of cell apoptosis. 14 We asked whether the antiapoptotic activity of miR-150 in HBECs is ascribed to downregulation of p53. To address this issue, we examined the effect of miR-150 on endogenous p53 expression in HBECs. The results showed that miR-150 overexpression significantly (p < 0.05) suppressed the expression of p53 in BEAS-2B cells, even after treatment with 15% CSE (Figure 4(c)). Most importantly, co-expression of a miR-resistant form of p53 restored apoptotic response to CSE treatment in BEAS-2B cells with overexpression of miR-150 (Figure 4(d)). Taken together, these results indicate that miR-150 protects HBECs from CSE-induced apoptosis by targeting p53.
Discussion
Accumulating evidence has suggested the involvement of miR-150 in inflammatory diseases. 21 –23 It was reported that miR-150 is upregulated in human colon with active ulcerative colitis compared to the normal colon. 21 Compared to wild-type controls, miR-150-deficient mice displayed enhanced obesity-associated tissue inflammation. 22 Another study demonstrated that miR-150 has the capacity to reduce the production of inflammatory cytokines through inhibition of the NF-κB pathway. 23 These studies suggest the anti-inflammatory activity of miR-150. In this work, we sought to check role of miR-150 in the pathogenesis of COPD. We found that there was a significant reduction of miR-150 in CS-exposed mouse lungs and CSE-treated HBECs. Moreover, restoration of miR-150 significantly blocked the inflammatory infiltration of the lung after exposure to CS. Consistently, CS-induced accumulation of neutrophils, lymphocytes, and macrophages in BALF was significantly suppressed by delivery of miR-150 mimic. Additionally, the concentrations of pro-inflammatory cytokines including IL-6, TNF-α, and IL-8 in BALF were lowered by overexpression of miR-150. Collectively, our results point toward the anti-inflammatory role for miR-150 in smoke-related COPD.
MiR-150 plays distinct roles in different types of cells. 24 –26 For example, miR-150 impairs the transformation of macrophages to foam cells through facilitating cholesterol efflux. 26 Exogenous miR-150 has been found to promote endothelial cell migration. 24 Our data showed that miR-150 can also coordinate the biology of airway epithelial cells. It was found that overexpression of miR-150 significantly impaired the expression of IL-6, TNF-α, and IL-8 in HBECs in response to CSE. Moreover, miR-150 overexpression rendered HBECs more resistant to CSE-induced apoptosis and reduced the activity of caspase 3. The in vitro observations provide an explanation for the protective activity of miR-150 in the CS-induced COPD mouse model.
Mechanistically, miR-150 overexpression inhibited NF-κB transcriptional activity in CSE-treated HBECs. NF-κB is responsible for the transcriptional activation of many pro-inflammatory cytokines. 20 Therefore, inhibition of NF-κB-dependent transcriptional activity may account for the anti-inflammatory activity of miR-150; p53 has been identified as a direct target gene of miR-150. 14 We found that miR-150 overexpression repressed the expression of p53 in HBECs. Given the well-established role for p53 in cell apoptosis, we hypothesized that miR-150 might exert its antiapoptotic activity in HBECs through downregulation of p53. In support of this hypothesis, co-expression of p53 reversed the antiapoptotic activity of miR-150, as evidenced by increased apoptosis after CSE treatment. Several lines of evidence suggest a cross talk between NF-κB and p53 signaling. 27,28 It has been reported that p53 can stimulate NF-κB activation via phosphorylation of p65 by ribosomal S6 kinase 1. 29 Therefore, it is possible that miR-150-mediated inhibition of NF-κB activity involves downregulation of p53. However, the exact mechanism for the suppression of NF-κB activation by miR-150 needs to be further determined; p53 is a well-defined tumor suppressor, and its inactivation has been linked to tumor development and progression. 30 MiR-150-mediated downregulation of p53 in HBECs suggests a possibility that miR-150 may contribute to lung tumorigenesis.
Although we provide evidence that the protective activity of miR-150 in COPD is associated with downregulation of p53, we cannot exclude the possibility that other target genes also mediate the action of miR-150. Indeed, multiple miR-150 target genes other than p53 have been identified. For example, miR-150 targets CDK3 to inhibit cell proliferation. 31 c-Myb serves as a target in miR-150-mediated B-cell differentiation. 32 Profiling gene expression in HBECs after miR-150 overexpression would aid in deciphering the mechanism by which miR-150 prevents the pathogenesis of COPD.
In conclusion, miR-150 confers protection against CS-induced lung inflammation and attenuates CSE-induced airway epithelial cell apoptosis, which is, at least partially, ascribed to repression of p53. Delivery of miR-150 may represent a potential therapeutic strategy for the treatment of CS-related COPD.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflict of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.