
Abstract
Smoking-associated chronic obstructive pulmonary disease is characterized by inflammation, changes affecting small airways, and development of emphysema. Various short- and long-term models have been introduced to investigate these processes. The aim of the present study was to identify markers of early epithelial injury/adaptation in a short-term animal model of cigarette smoke exposure. Initially, male BALB/c mice were exposed to smoke from one to five cigarettes and lung changes were assessed 4 and 24 hr after smoking cessation. Subsequently, animals were exposed to smoke from five cigarettes for 2 consecutive days and lungs investigated daily until the seventh postexposure day. Lung homogenates cytokines were determined, bronchioloalveolar fluid cells were counted, and lung tissue was analyzed by immunohistochemistry. Exposure to smoke from a single cigarette induced slight pulmonary neutrophilia. Smoke from two cigarettes additionally induced de novo expression of tight junction protein, claudin-3, by alveolar duct (AD) epithelial cells. Further increases in smoke exposure induced epithelial changes in airway progenitor regions. During the recovery period, the severity/frequency of epithelial reactions slowly decreased, coinciding with the switch from acute to a chronic inflammatory reaction. Claudin-3 and Clara cell 10 kDa protein were identified as possible markers of early tobacco smoke–induced epithelial injury along ADs.
Introduction
Chronic obstructive pulmonary diseases (COPDs) or airway diseases are the group of pulmonary diseases characterized by increased airflow resistance due to obstruction at any level of the bronchial tree. The group is comprised of several separate entities: emphysema of various etiologies, chronic bronchitis, asthma, and bronchiectasis. Clinically, chronic bronchitis and emphysema present in the same patient are referred to as COPD. The main etiological factor for COPD is smoking (Husein 2010).
Smoking-associated COPD is one of the leading health problems today and the fifth most common cause of death worldwide (Halbert et al. 2006; Pauwels and Rabe 2004), with one of the five cigarette smokers developing COPD (Taylor 2010). COPD is a multiorgan disease with renal and hormonal abnormalities, muscle wasting, osteoporosis, and anemia (Celli and McNee 2004). Its pulmonary component is characterized by partially irreversible airflow limitation recognized as a progressive decrease in forced expiratory volume in 1 sec (FEV1; Rabe et al. 2007). The main pathological characteristics of smoking-associated COPD are inflammation along the bronchus and bronchioles, small airway and vascular remodeling, and development of centrilobular emphysema (O’Donnell et al. 2004; Wright and Churg 2006).
Tobacco smoke contains many diverse toxic chemicals including a high proportion of reactive oxygen (ROS) and nitrogen species (RNS), lipopolysaccharide (LPS), nicotine, and nitrosamines. Inhalation of tobacco smoke triggers a complex cell signaling cascade involving numerous cytokines. A correlation between exposure to tobacco smoke and the increased number of neutrophils in bronchoalveolar lavage fluid (BALF) has been described in humans (Pesci et al. 1998) and animal models (Vlahos et al. 2010). In mouse models of cigarette smoke–induced inflammation, it has been shown that the initial neutrophil influx is mediated largely by keratinocyte-derived chemokine (KC) and macrophage inflammatory protein (MIP)-2. In addition, elevated levels of tumor necrosis factor (TNF)-α and interleukin (IL)-1β were found in BALF of experimental animals exposed to cigarette smoke for 10 days (Betsuyaku et al. 2008). Apart from neutrophils, macrophages and lymphocytes have been detected in BALF, as well as in sputum of COPD patients, when compared to smokers without COPD (Pesci et al. 1998).
Disease progression has been shown to correlate with the changes affecting small airways (Hogg et al. 2004). These are manifested as an increased proportion of small airways harboring neutrophils, macrophages, CD4- and CD8-T lymphocytes, and B-lymphocytes and the formation of lymphoid aggregates adjacent to the bronchiole wall. Emphysema in COPD develops as a consequence of a disrupted balance between tissue damage and the ability to restore the pulmonary architecture. Proteases, released into pulmonary tissue during inflammation by infiltrating neutrophils, macrophages, and alveolar macrophages, play a pivotal role in tissue destruction (Vlahos et al. 2006).
Decreased lung function, measured as FEV1 and emphysema in COPD patients, has been linked to increased levels of sputum neutrophils (Stanescu et al. 1996) as well as to a Th1-response (Grumelli et al. 2004), suggesting that lung tissue damage may be driven by innate and adaptive immune responses. In addition, a direct toxic effect of cigarette smoke on epithelial cells has been documented (Broekema et al. 2009; Lin et al. 2012).
In vitro cigarette smoke exposure induces DNA damage in bronchial epithelial cells, while in the pulmonary adenocarcinoma cell line (A549) cigarette smoke induces apoptosis and necrosis. In primary alveolar epithelial cells (AEC), cigarette smoke induces irreversible growth arrest (Thorley and Tetley 2007). These in vitro experiments showed that apart from inflammation, parenchymal damage can be induced by the directtoxic effect of cigarette smoke on various types of epithelial cells. Recently, mechanisms involved in the intrinsic maintenance of pulmonary epithelial integrity (Voelkel and Taraseviciene-Stewart 2005) and repair have attracted increasing attention (Bourbon et al. 2009; Thorley and Tetley 2007). Therefore, the scientific focus in studies on the etiology of COPD is slowly shifting toward endogenous pulmonary progenitor cells.
Therapeutic strategies in COPD are currently based mainly on the suppression of inflammation and indirectly target the development of emphysema. Antagonists or inhibitors of inflammatory mediators, however, have not yielded satisfactory results in COPD patients (Barnes 2008). Therapies based on enhanced regeneration of resident pulmonary stem cells and allograft bone marrow–derived mesenchymal stem cells have been suggested as a possible therapeutic approach to COPD (Hackett, Knight, and Sin 2010; Sueblinvong and Weiss 2010).
Various animal models, using a multitude of approaches, have been employed to investigate COPD. Genetically modified animals have been engineered to assess the importance of specific pathways in the pathobiology of COPD. In wild-type animals, inflammation and emphysema have been induced, among others, by noxious chemicals, such as tobacco smoke. Short-term tobacco smoke exposure is mainly used to study the inflammatory response, while long-term models (3- to 6-month tobacco smoke exposure) were designed to study emphysema (Wright, Cosio, and Churg 2008). The aim of the present study was to identify markers of early epithelial injury/adaptation using immunohistochemistry in a short-term tobacco smoke exposure COPD animal model. In order to determine the minimal smoke exposure needed to induce inflammation and/or epithelial alterations BALB/c mice were exposed to smoke from one to five cigarettes, and animals were sacrificed 4 and 24 hr postexposure. Aiming to explore an early postexposure recovery process, we exposed animals to smoke from five cigarettes for 2 consecutive days and sacrificed them daily until the seventh postexposure day. Cell-specific markers for macrophages (F4/80), epithelial cells (pan-cytokeratin [CK]), and marker for Clara cells (Clara cell 10 kDa protein) were used to determine the type of cells with altered morphology. The presence of cell injury caused by oxidative stress was investigated since it has been shown that oxidative stress plays a role in cell injury caused by tobacco smoke in humans and experimental animals (Churg, Cosio, and Wright 2008). Possible cellular adaptation to injury was approached by exploring the cell proliferation using Ki67 marker since increased proliferation rate following naphthalene, a constituent of tobacco smoke, has been documented in animal model (Oliver et al. 2009). Impairment of airway epithelial barrier function induced by cigarette smoke has been shown in in vitro settings (Heijink et al. 2012). We, therefore, explored the expression pattern of several members of claudin family, a tight junction protein family variously distributed in pulmonary cell types (Kaarteenaho-Wiik and Soini 2009). In lungs, claudin-3, claudin-4, and claudin-8 are involved in tightening the epithelial barrier (Krause et al. 2008; Coyne et al. 2003). Collected data could provide insight into changes preceding the development of emphysema, as well as the suitability of the short-term tobacco smoke exposure model for the assessment of novel therapeutics facilitating the regeneration of resident pulmonary stem cells.
Materials and Methods
Animals
Male BALB/c mice, 8 weeks old, were obtained from Charles River, Germany. Mice were maintained under standard laboratory conditions (temperature 23–24°C, relative humidity 60 ± 5%, 15 air changes per hr, artificial lighting with a circadian cycle of 12 hr). Pelleted food and tap water were provided ad libitum. All procedures on animals were approved by the ethics committee of GlaxoSmithKline Research Centre Zagreb Limited and performed in accordance with the European Economic Community Council Directive 86/609.
Cigarette Smoke Exposure
After acclimatization, mice were randomly divided into two main groups, cigarette smoke exposed (smoking groups) or sham exposed (control group). For whole body cigarette-smoke exposure, animals were placed inside a 2.0 l Plexiglas chamber (Braintree Scientific Braintree, MA) and exposed to the smoke produced by an indicated number of 3R4F reference cigarettes without filter (University of Kentucky, Lexington, KY; for details, see http://www.ca.uky.edu/refcig/3R4F%20Preliminary%20Analysis.pdf). Smoke was introduced into the chamber by a peristaltic pump (Masterflex L/S, Cole Parmer, Vernon Hills, IL) at a rate of 40 to 80 ml/min. Simultaneously, fresh air was insufflated at a rate of 0.4 to 0.8 l/min resulting in 10 to 20% saturation with cigarette smoke (Figure 1). Sham control animals were exposed to room air.
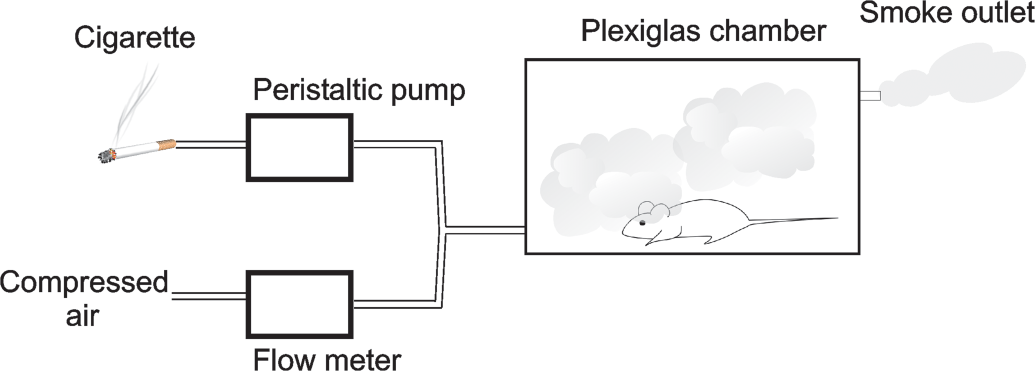
Illustration of smoke exposure system. Cigarette smoke is driven via silicone tube from a lit cigarette to an exposure chamber by a peristaltic pump at a rate 40–80 ml/min. Simultaneously, compressed air, regulated by a flowmeter, is driven via another silicone tube to an exposure chamber resulting in 10–20% saturation with cigarette smoke.
For histological evaluation, mice (five per group) were exposed to cigarette smoke from one to five cigarettes. Cigarettes were lit consecutively except for exposure to five cigarettes, which was divided into two periods (3 + 2 cigarettes) separated by a 2-hr interval. Four and 24 hr after the last exposure to cigarette smoke, mice were killed and bronchoalveolar lavage was performed with cold phosphate-buffered saline (PBS) in a total volume of 1 ml (three lavages with 0.4, 0.3, and 0.3 ml). Following bronchoalveolar lavage, lungs were excised and fixed in 10% buffered neutral formalin fixative.
To study pulmonary changes during a 7-day recovery period, mice (five per group) were exposed to smoke twice daily (3 + 2 cigarettes) for 2 consecutive days. Between each cigarette, a 1-min pause without smoke was introduced. The lungs and BALF were collected 24 hr after the last smoke exposure for a total of 7 days.
For measurement of inflammatory mediators in the lung tissue, mice (n = 70) were divided into fourteen groups. On the first day, animals were exposed to cigarette smoke from three cigarettes followed by a 2-hr break after which they were exposed to an additional two cigarettes. The same procedure was repeated on the second day. Animals were killed and lungs collected 0.5, 1, and 2 hr after exposure to the first three cigarettes and 0.5, 1, 2, and 4 hr after exposure to the additional two cigarettes on both days. Lungs were stored at −80°C until homogenization.
Histology and Immunohistochemistry
Lungs were inflated with 10% formalin, fixed for 48 hr and whole lung with ventral surface down was paraffin-embedded (Kittel et al. 2004). Slides were stained with hematoxylin-eosin and periodic acid-Schiff (PAS). Immunohistochemical staining was performed using the following primary antibodies: F4/80 (Acris, Germany, BM4008G, 1:50), claudin-3 (GeneTex, Irvine, CA, GTX15102, 1:50), claudin-4 (Abcam, UK, ab53156, 1:250), claudin-8 (Santa Cruz, Santa Cruz, CA, sc-33065, 1:100), Clara cell 10 kDa protein (CC-10; Santa Cruz, Santa Cruz, CA, sc-9772, 1:100), CK, (Novus Biologicals, Littleton, CO, NB600-579, 1:25), Ki67 (Santa Cruz, Santa Cruz, CA, sc-7846, 1:50), and nitrotyrosine (Antibodies-online, Germany, ABIN339841, 1:100). Antibody binding was detected by secondary biotinylated antibodies, streptavidin-horseradish peroxidase conjugate, and diaminobenzidine chromogen (HRP-DAB System, R&D Systems, Minneapolis, MN) according to the manufacturer’s recommendations. Staining localization and intensity was examined by an observer blinded to the experimental design and subjectively graded according to the severity (0, absent; 1, minimal; 2 mild; 3, moderate; 4, marked) and distribution (1, focal; 2, locally extensive; 3, multifocal; 4, multifocal and coalescing; 5, diffuse) criteria. Scored parameters included alveolar macrophage hyperplasia, peribronchial mononuclear cells and neutrophils, perivascular mononuclear cells and neutrophils, and bronchioalveolar duct junction (BADJ)/alveolar duct (AD) inflammation, and epithelial changes.
Measurement of Inflammatory Mediators in Lungs
Lungs were homogenized on ice in PBS with protease inhibitors (1 µg/ml leupeptin, 2 µg/ml aprotinin, 1 µg/ml pepstatin, and 17 µg/ml phenylmethylsulfonyl fluoride); 4 ml of PBS with protease inhibitors was added per gram of lung tissue. Homogenates were centrifuged (4°C, 2,500 g, 15 min) and stored at −80°C until analysis.
Samples were analyzed using xMAP coated microsphere technology (Luminex, Austin, TX), which enables simultaneous measurement of multiple biomarkers. Concentrations of granulocyte macrophage colony stimulating factor (GM-CSF), interferon γ (IFNγ), IL-1α, IL-1β, IL-10, IL-12p40, IL-12p70, IL-13, IP-10, keratinocyte-derived chemokine (KC), LPS-induced chemokine (LIX), leukemia inhibitory factor (LIF), monocyte chemoattractant protein (MCP)-1, macrophage colony stimulating factor (M-CSF), monokine-induced by interferon gamma (MIG), MIP-1α, MIP-1β, MIP-2, regulated upon activation, normal T-cell expressed (RANTES), and TNF-α were determined with Milliplex MAP Kit (Millipore, Billerica, MA) according to the manufacturer’s protocol. In brief, 25 µl of samples were incubated with antibody-coated microparticles for 16 hr at 4°C. Afterward, washed beads were incubated with biotinylated detection antibody cocktail for 1 hr at room temperature, washed, and incubated for 30 min with streptavidin-phycoerythrin conjugate. After the final wash, the microparticles were resuspended in buffer and analyzed with Luminex 200 (Luminex, Austin, TX) and STarStation software v2.3 (Applied Cytometry Systems, Sheffield, UK) using five-parameter, logistic-curve fitting.
Concentrations of analytes in lung homogenates were further normalized to protein concentration in the samples and expressed as picograms of analyte per milligram of protein.
Determination of Protein Concentration
Protein concentration in lung homogenates was determined by BCA Protein Assay (Thermo Fisher Scientific) according to the manufacturer’s recommendation.
Statistical Analysis
Cell counts and cytokine concentrations are presented as means ± standard error of the mean (SEM), while histological scores are presented as median ± range. To define statistically significant differences between sham and cigarette smoke–exposed mice, the data were subjected to one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA). The level of significance was set at p < .05 in all cases.
Results
Exposure to Cigarette Smoke of Differing Intensity
Cell count in BALF
Exposure to cigarette smoke from one to five cigarettes did not significantly increase the total cell number in BALF when compared to sham control, determined 24 hr after the last cigarette (Figure 2A). However, the percentage of neutrophils rose significantly after exposure to only one cigarette and only slightly increased following exposure to smoke from one to five cigarettes (Figure 2B). Consequently, the percentage of macrophages significantly decreased in BALF from all cigarette smoke–exposed animals (Figure 2C), while the absolute number of macrophages decreased only slightly (data not shown).
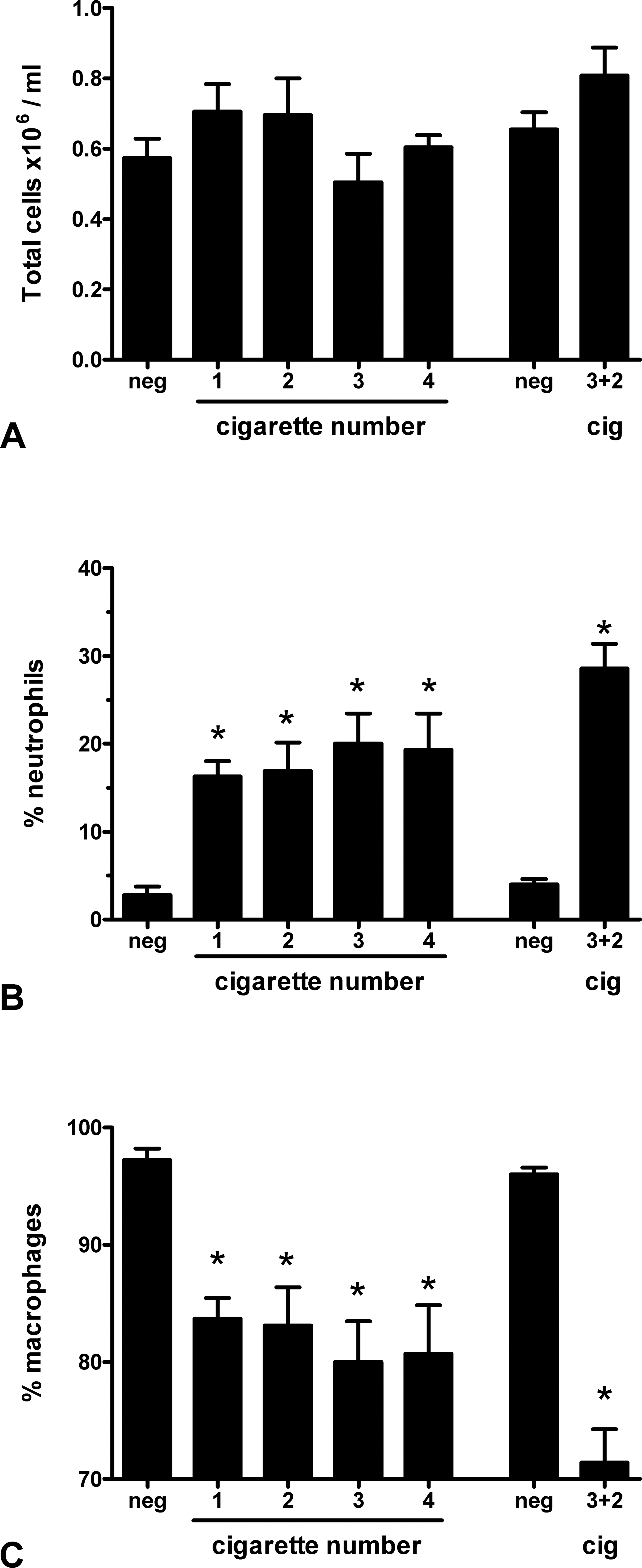
Total cell number (A), and neutrophil (B) and macrophage (C) percentage in BALF 24 hr after exposure to air (neg) or smoke from one to five cigarettes. Data are presented as means ± standard error of the mean (SEM), *p < .05, one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test.
Cytokines in lung homogenates
Of nineteen measured cytokines, eight were present below or around the limit of detection of the kit used (GM-CSF, IFNγ, IL-10, IL-12p70, IL-13, MIP-1α, MIP-1β, and TNF-α). In addition, concentrations of IL-1α, IL-1β, IL-12p40, IP-10, M-CSF, MIG, and RANTES were weakly increased (data not shown). One hour after exposure to cigarette smoke from the first three cigarettes, a slight increase of KC and MCP-1 concentrations in lung homogenates was detected (Figure 3A and D). Concentrations of both cytokines gradually increased from the first exposure to cigarette smoke, reaching their highest levels on the second day of exposure to cigarette smoke, that is, following exposure to six to ten cigarettes. On the other hand, concentrations of MIP-2 and LIX were significantly elevated after exposure to smoke from the first three cigarettes (Figure 3B and C). Exposure of animals to cigarette smoke of additional cigarettes on the second day did not further increase the concentrations of these two cytokines.
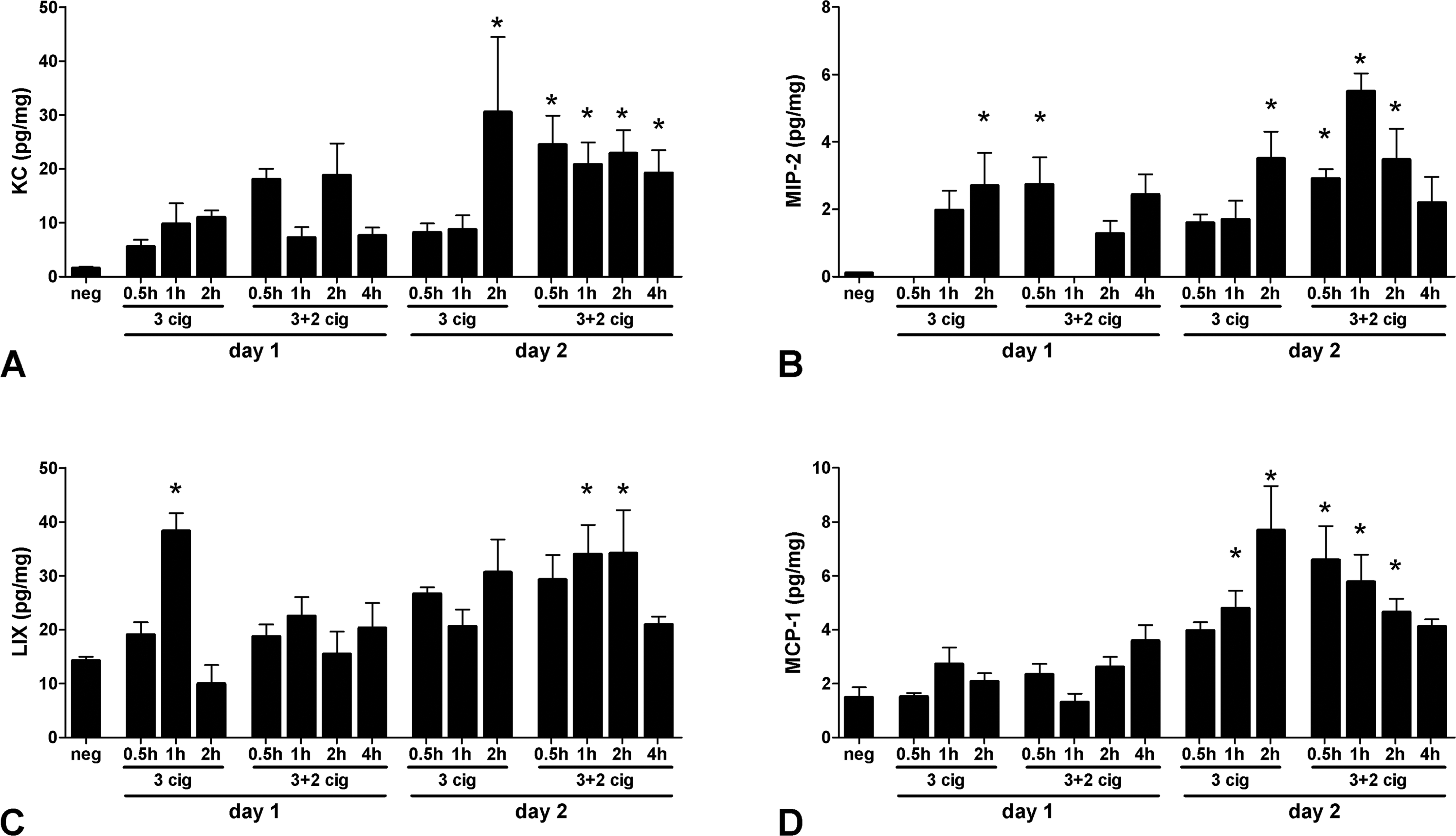
Cytokine concentrations in lung homogenates following exposure to air (neg) or cigarette smoke. Concentrations are given for 0.5, 1, and 2 hr after exposure to three cigarettes and 0.5-, 1-, 2-, and 4-hr postexposure to five (3 + 2) cigarettes (day 1) followed by cytokine concentrations measured at same time points after exposure to three and five (3 + 2) cigarettes for 2 consecutive days (day 2). Data are presented as means ± standard error of the mean (SEM), *p < .05, one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test.
Histology
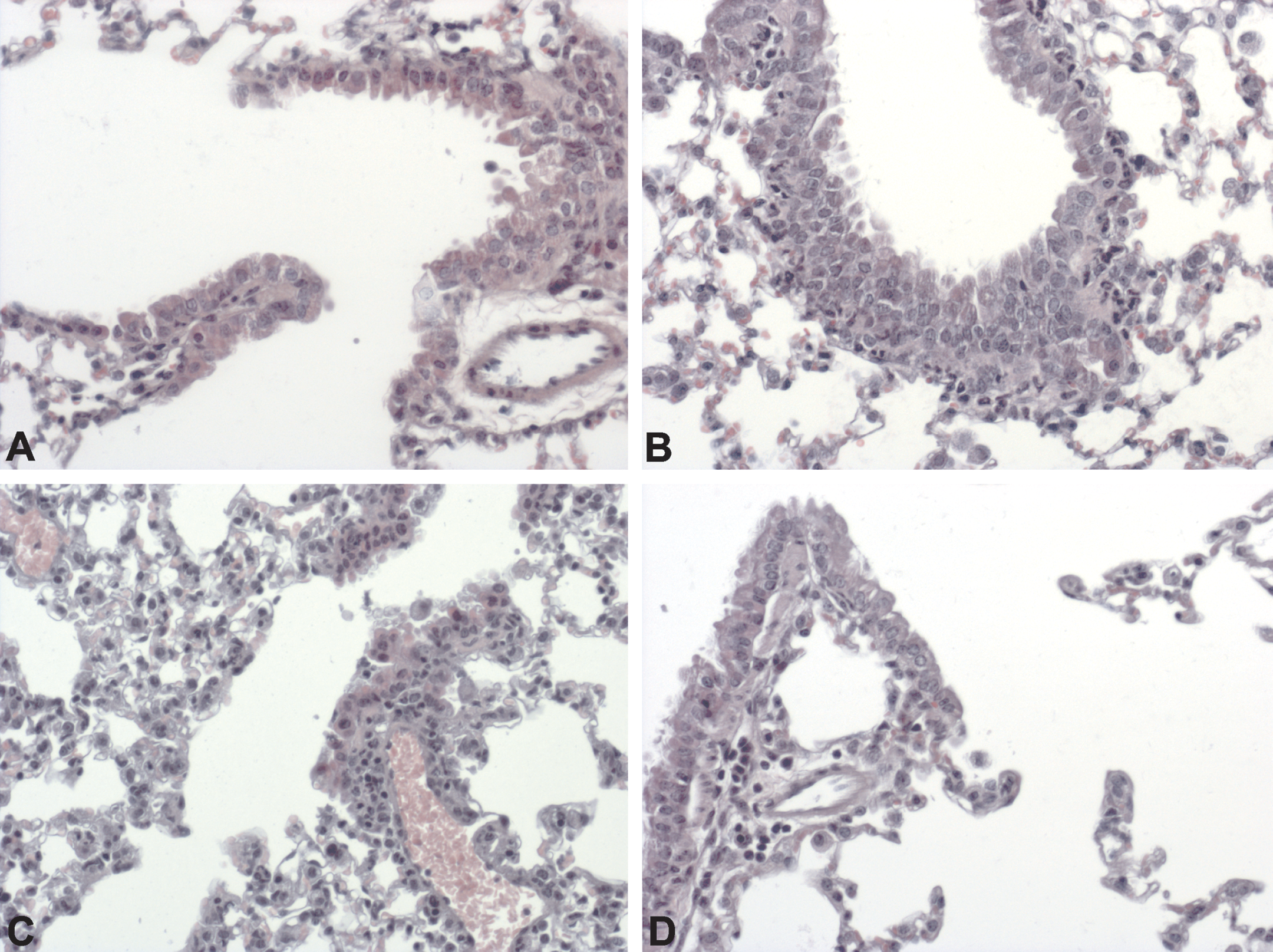
Vasodilatation and blood stasis appeared 4 hr after exposure to a single cigarette. Exposure to smoke from two or more cigarettes caused minimal accumulation of cells in interstitium, scarce edema (Figure 4B, in relation to sham controls, Figure 4A), single-cell loss in the bronchial epithelium, and desquamation of cells in the bronchial lumen. In addition, 4 hr after exposure to smoke from 3 + 2 cigarettes, pulmonary edema developed, while in bronchial epithelium rare mitotic figures were observed.
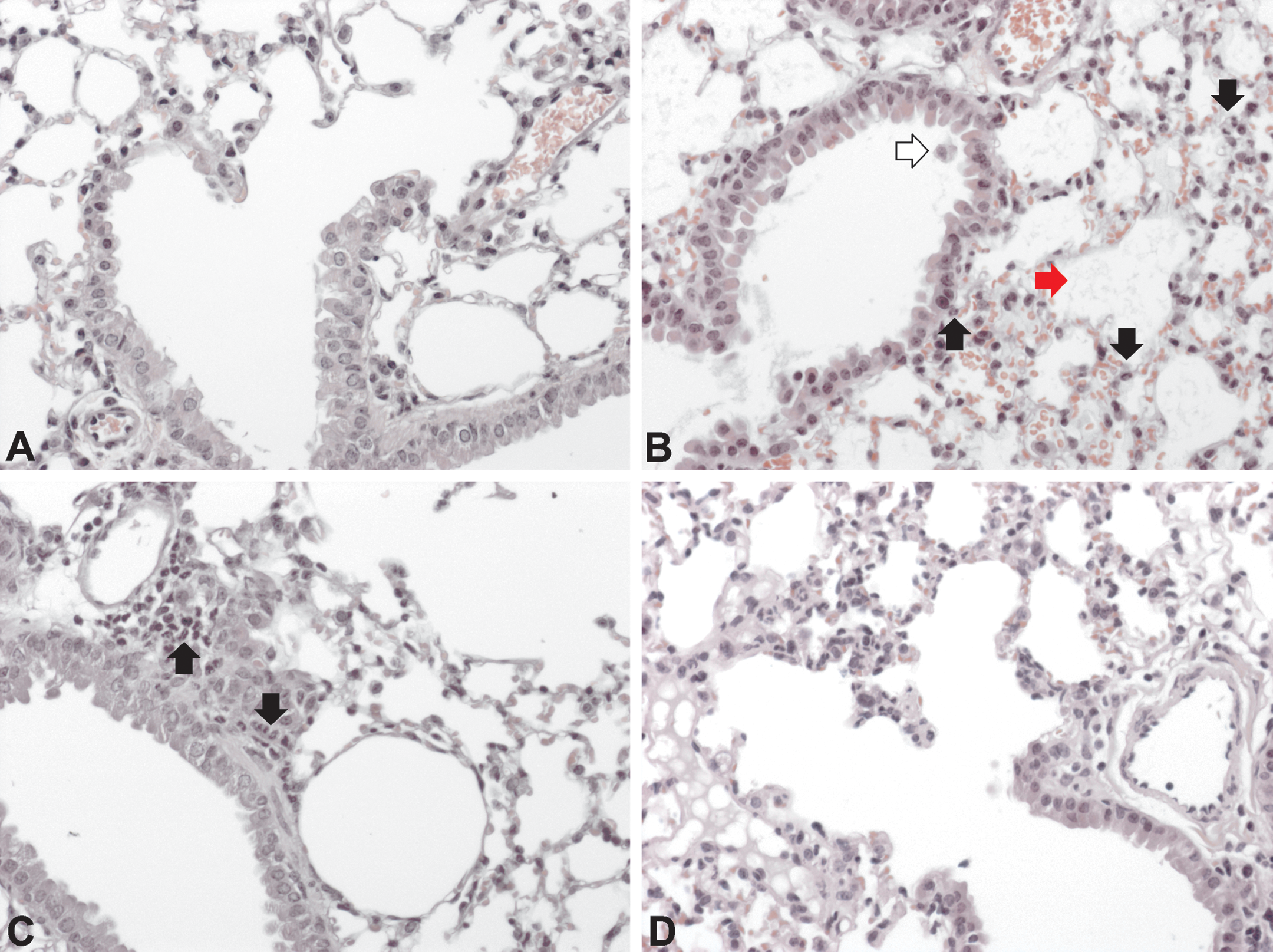
Representative photomicrographs of lung tissue of sham- (A) and cigarette smoke–exposed animals (B, C, and D). Four hours after exposure to smoke from a single cigarette (B), interstitial accumulation of neutrophils (black arrow), edema (red arrow), and single-cell loss in the bronchial epithelium (white arrow) could be observed. A day after exposure to smoke from four cigarettes (C), neutrophils (black arrow) accumulated along bronchioles and arterioles. Twenty-four hours after exposure to five cigarettes (D) prominent interstitial accumulation of neutrophils was observed. A, B, C, and D: HE, magnification 200×.
A day after exposure to smoke from one to five cigarettes, neutrophils accumulated along bronchioles/ADs, adjacent arteries/arterioles, and to a lesser degree in interstitium (Figure 4C and D). Exposure to smoke from two or more cigarettes resulted in de novo claudin-3 positive staining of epithelial cells lining the ADs 24 hr postexposure (Figure 5B and C). Within the same time frame, following exposure to smoke from 3 + 2 cigarettes, single large cells with hyperchromatic nuclei appeared along ADs spread diffusely in the pulmonary tissue.
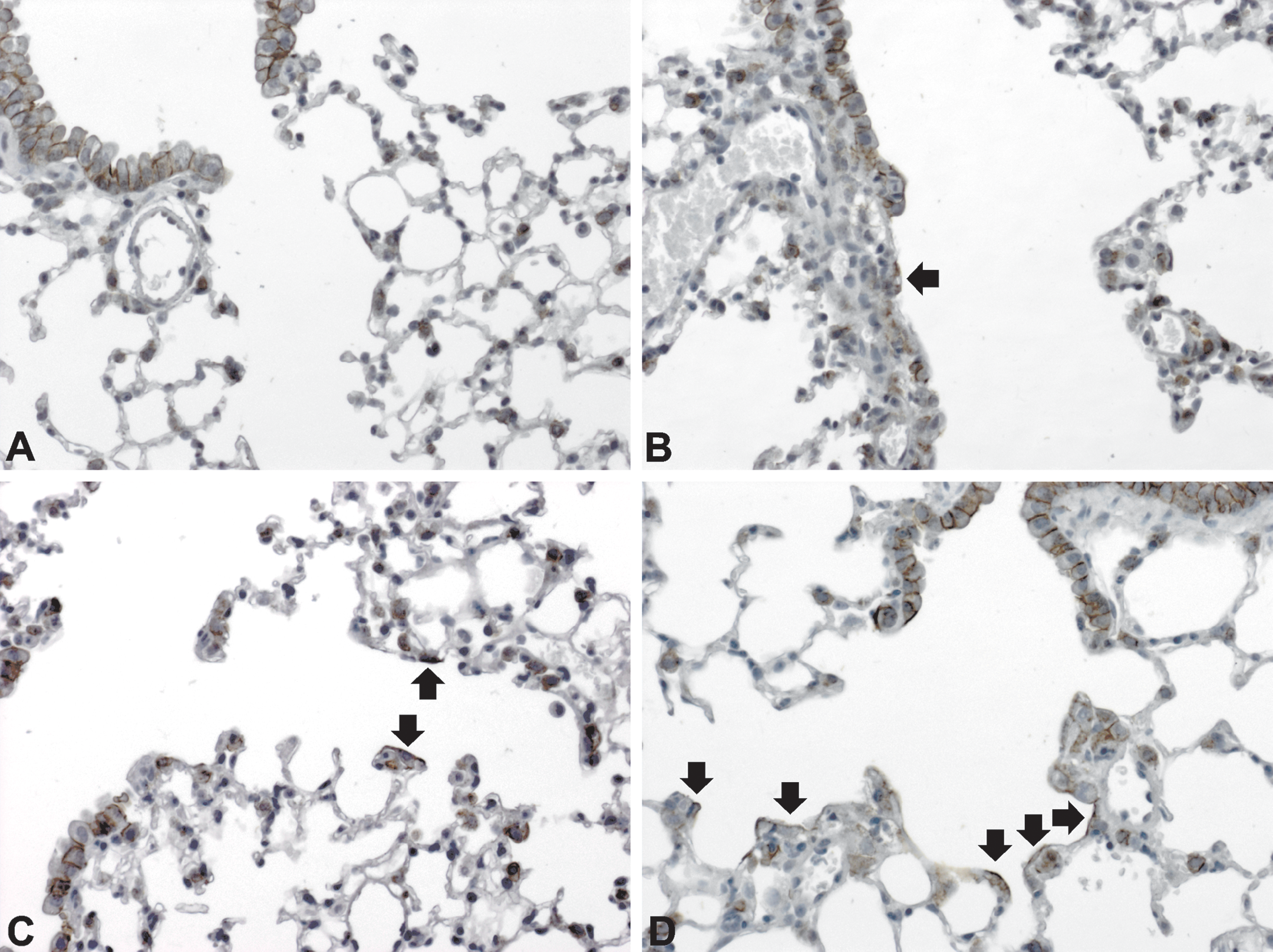
Claudin-3 expression in lungs of sham- (A) and cigarette smoke–exposed animals (B–D). In normal lungs, epithelial cells in conducting airways expressed claudin-3, while alveolar duct lining cells were negative. The transition was abrupt. AEC-II cells were stained along the cellular membrane (A). De novo claudin-3 staining cells along alveolar ducts (arrow) could be observed a day after exposure to smoke from three (B) or four cigarettes (C). Following exposure to smoke from 3 + 2 cigarettes for 2 consecutive days (D), most alveolar duct lining cells expressed claudin-3 on their surface. A, B, C, D:, magnification 200×.
Exposure to Smoke from 3 + 2 Cigarettes for 2 Days Followed by a 7-day Recovery Period
Cell count in BALF
Two-day exposure to cigarette smoke from a total of ten cigarettes did not significantly change the total cell number in BALF although there was a trend toward an increase (Figure 6A). However, the proportion of neutrophils increased significantly (Figure 6B) and that of macrophages significantly decreased in the BALF of cigarette smoke–exposed animals (Figure 6C). On the fifth and sixth day postexposure, the percentage of neutrophils decreased, while that of macrophages gradually increased.
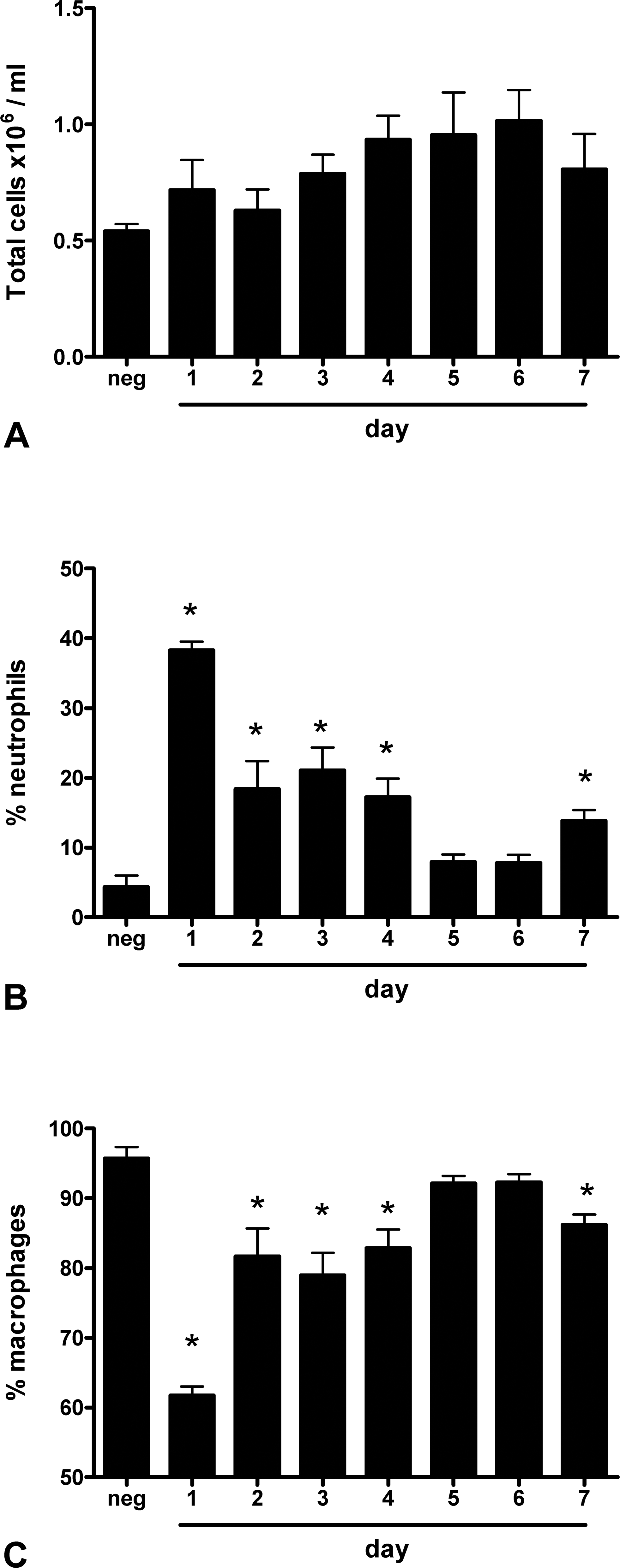
Total cell number (A), neutrophil (B) and macrophage (C) percentages in BALF 1–7 days after exposure to air (neg) or smoke from 3 + 2 cigarettes. Data are presented as means ± standard error of the mean (SEM), *p < .05, one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test.
Histology
Compared to sham controls (Figure 7A) animals exposed to smoke from 3 + 2 cigarettes for 2 consecutive days showed neutrophil accumulation 24 hr after the last cigarette, adjacent to bronchioles/arterioles and, to a lesser degree, along ADs (Figure 7B). For the first 4 days after smoke exposure, the neutrophils prevailed among inflammatory cells. A chronic inflammatory infiltrate began to emerge 4 days after the last cigarette and dominated from the fifth- till the seventh-day postexposure. As neutrophils, monocytes/macrophages mainly accumulated along bronchioles/arterioles (Figure 7C, D).
Representative photomicrographs of lung tissue of sham- (A) and cigarette smoke–exposed animals (B–D). Twenty-four hours after exposure to smoke from 3 + 2 cigarettes for 2 consecutive days (B), neutrophils accumulated along bronchioles. A chronic inflammatory infiltrate started to appear 4 days after the last cigarette (C) and prevailed over neutrophils from the fifth until the seventh (D) day postexposure. A–D: HE, magnification 200×.
Alveolar macrophage hyperplasia was visible 24 hr after the last cigarette and progressed (Figure 8B, C) continuously during the 7 days after smoke exposure to become most pronounced toward the end of the experiment. Alveolar macrophages in nonexposed animals were strongly F4/80 (data not shown) and CC-10 (Figure 9) positive, while weakly claudin-8 positive (Figure 10), and remained so throughout the experiment. In addition to pulmonary inflammation, exposure to cigarette smoke induced significant damage to select pulmonary cells. Single cell loss and small foci with naked basal membranes were present along bronchi and bronchioles 24 hr after the last cigarette in animals exposed to smoke from 3 + 2 cigarettes for 2 consecutive days (Figure 11).
Representative photomicrographs of lung tissue of sham (A) and animals exposed to smoke from 3 + 2 cigarette for 2 consecutive days (B, C). Mitotic figure (arrow) in alveolar macrophage 3 days post smoke exposure (B). Compared to sham, increased number of alveolar macrophages were present in alveolar spaces 4 days postexposure (C). A and C: HE, magnification 200×; B: Ki67, magnification 400×.
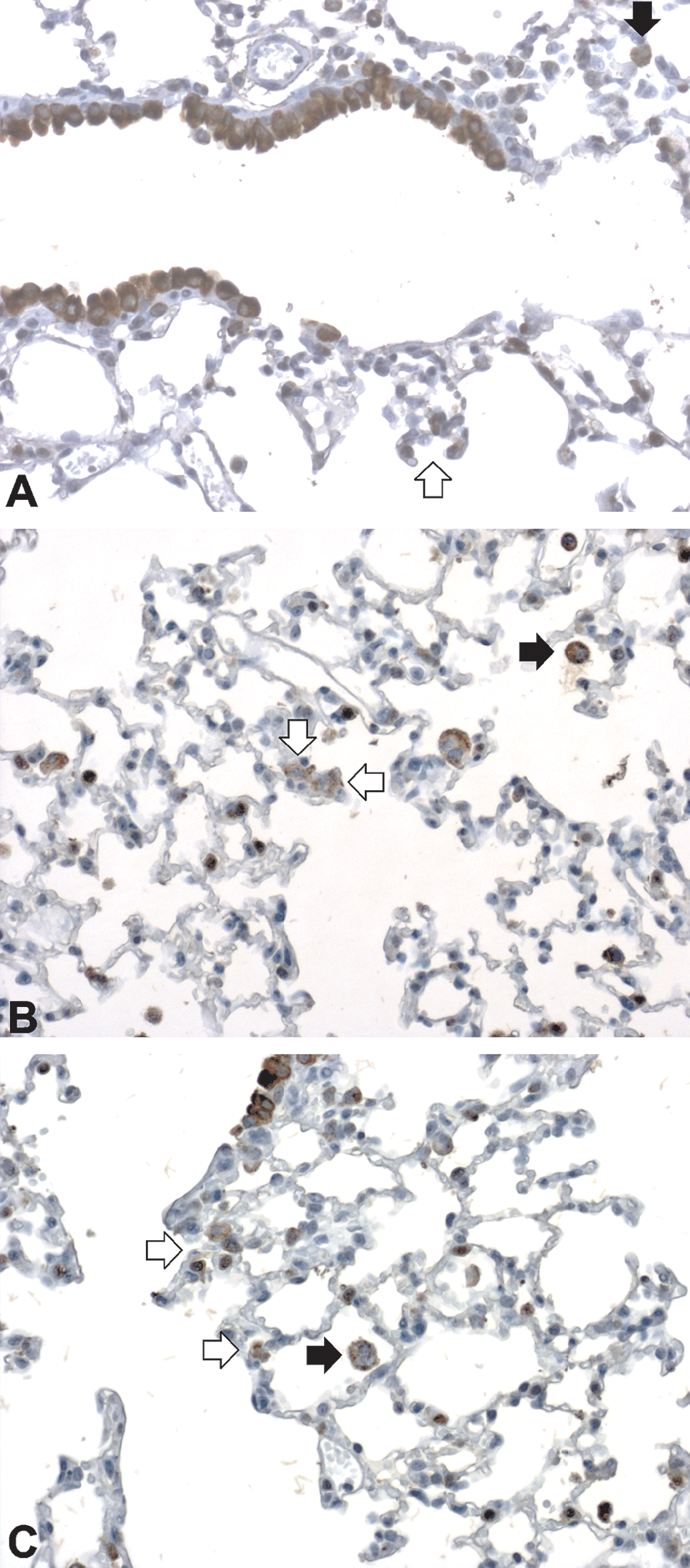
Clara cell 10 kDa protein (CC-10) expression by alveolar macrophages and alveolar epithelial cell type-II (AEC-II) in sham (A) and mice exposed to smoke from 3 + 2 cigarettes for 2 consecutive days (B, C). In normal lungs (A), apart from airway Clara cells, alveolar macrophages were strongly positive (black arrow). A small number of AEC-II showed faint staining (white arrow). Three days after the last cigarette (B), the number of AEC-II positive for CC-10 had increased (white arrow). Alveolar macrophages remained strongly positive (black arrow). At day 7 of recovery period (C), the number of CC-10 positive AEC-II had decreased (white arrow). Alveolar macrophages were CC-10 positive (black arrow). A–C: magnification 200×.
Claudin-8 expression in lungs of sham (A) and 2-day cigarette smoke (3 + 2 cigarettes)–exposed animals (B). Claudin-8 staining was present in bronchial epithelial cells of normal murine lungs (red arrow). Some alveolar epithelial cell type-II (AEC-II; white arrow) showed very weak cytoplasmic claudin-8 staining, as did some alveolar macrophages (black arrow; A). Four days after exposure to smoke from 3 + 2 cigarettes for 2 consecutive days (B), a majority of AEC-II showed weak positivity for claudin-8 (white arrow). Claudin-8 staining of alveolar macrophages remained unchanged (black arrow). A–B: magnification 200×.
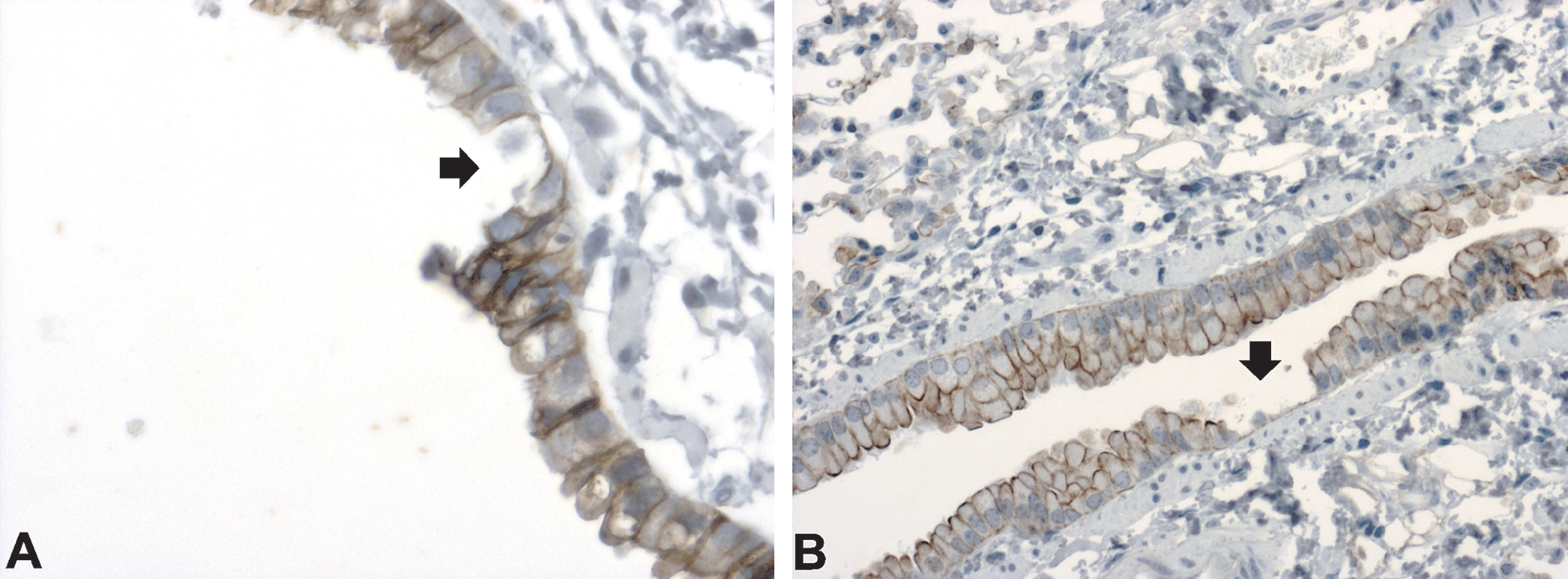
Damage to bronchial epithelial cells caused by cigarette smoke. Damage [single cell loss (A) and small foci with exposed basement membranes (B)] to epithelial cells was seen in conducting bronchioles 24 hr after exposure to smoke from 3 + 2 cigarettes for 2 consecutive days. A: claudin-3, magnification 400×; B: cytokeratin, magnification 200×.
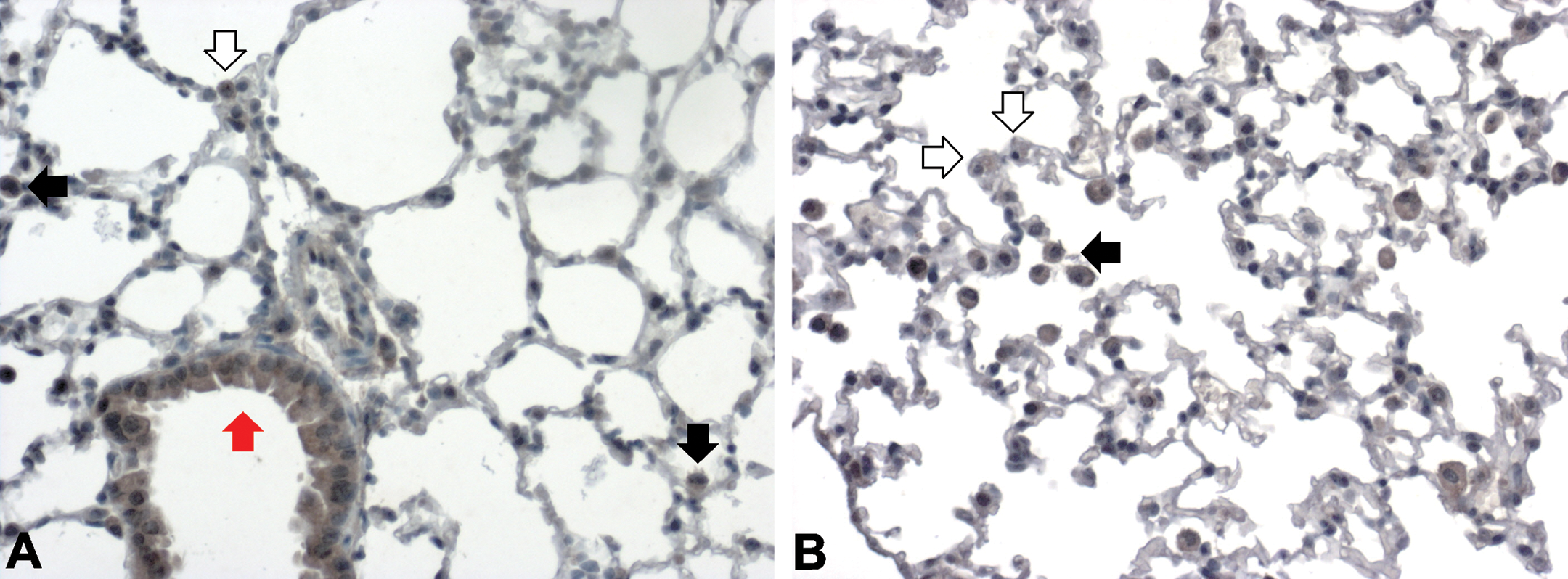
Large irregular cells, sometimes forming nests, at bifurcations of bronchioles, BADJ, and along ADs were distributed diffusely throughout the lungs (Figure 12). These cells were strongly CK (Figures 12D and 13D) and claudin-3 (Figures 12H and 13C) positive, but patchy CC-10 (Figure 12F) and claudin-8 (Figure 12J) positive, while being negative for the macrophage marker F4/80 (data not shown).
Epithelium lining terminal bronchioles and alveolar ducts in lungs of sham (A, C, E, G, I) and 2-day cigarette smoke (3 + 2 cigarettes)–exposed (B, D, F, H, J) animals 1 day after smoke exposure. Cytokeratin (D), CC-10 (F), claudin-3 (H), and claudin-8 (J) positive epithelial cells at bronchioloalveolar duct junction and bronchiole bifurcations were changed in shape and size (white arrow). De novo staining for cytokeratin (C, D), CC-10 (E, F), claudin-3 (G, H), and claudin-8 (I, J) was seen in cells lining alveolar ducts (black arrow). A, B: HE; A–J: magnification 200×.
The severity and frequency of the AD lining cell reaction slowly decreased with time (Figures 13 and 14). The improvement coincided with a switch from an acute to a chronic inflammatory reaction (Figure 6). No goblet cell metaplasia was evident, as indicated by PAS staining (data not shown). By the end of the 7-day recovery period, small foci of regenerative hyperplasia were observed within small bronchioles (Figure 15), as well as spots of bronchial metaplasia in ADs (Figure 16).
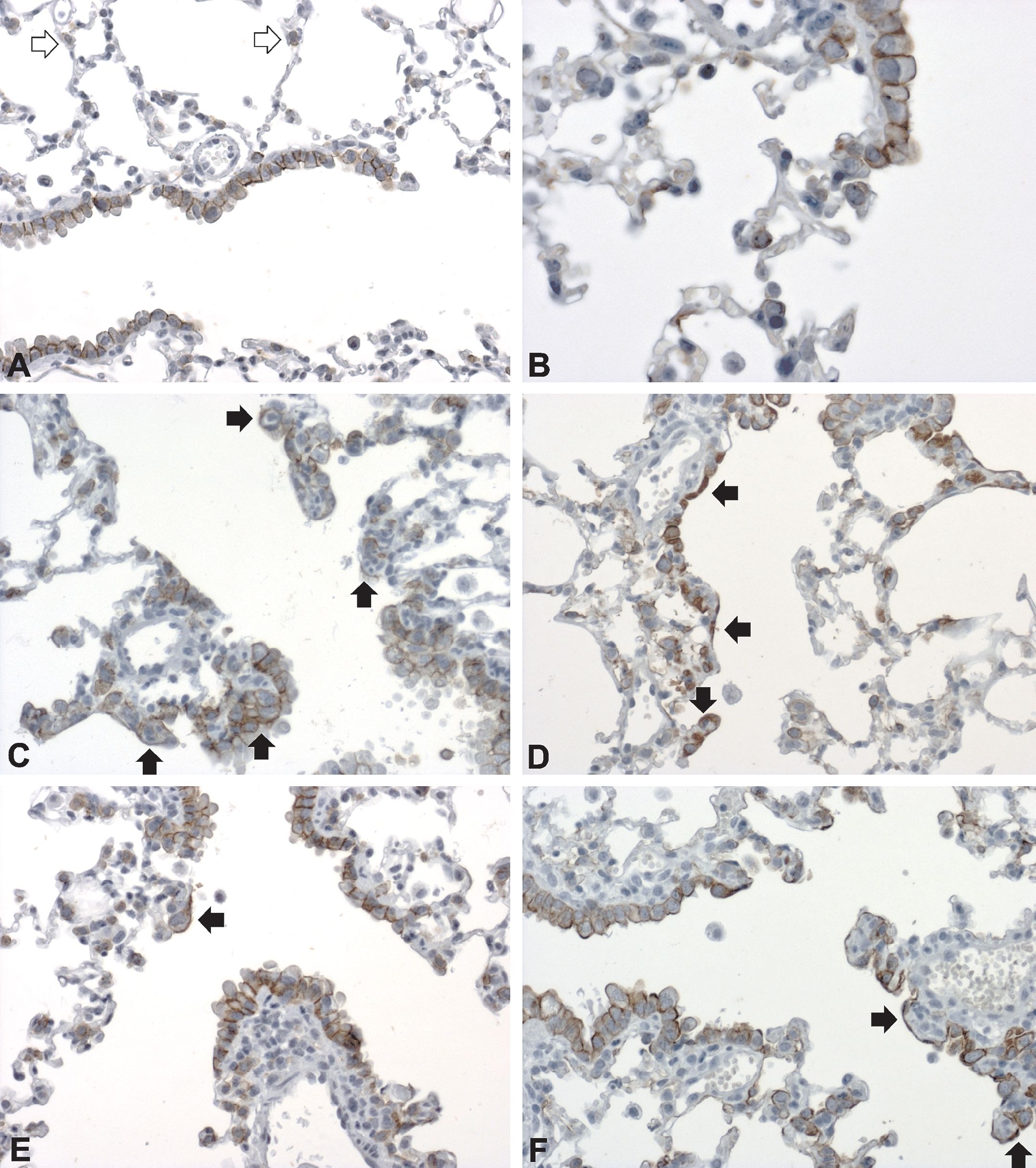
Epithelium lining terminal bronchioles and alveolar ducts in lungs of sham (A, B) and 2-day cigarette smoke (3 + 2 cigarettes) exposed (C–H) animals during the 7-day recovery period. The number of large and/or irregular cells at bifurcations of bronchioles and along alveolar ducts (black arrow) decreased from day 4 until the end of the experiment. Data shown are from 1 day (C, D), 3 days (E, F), and 7 days (G, H) after smoke exposure. Note claudin-3 positive alveolar epithelial cell type-II (AEC-II) cells (white arrow). A: claudin-3, magnification 100×; C–E: claudin-3, magnification 200×; G: claudin-3, magnification 50×; B, D, F, H: cytokeratin, magnification 200×.
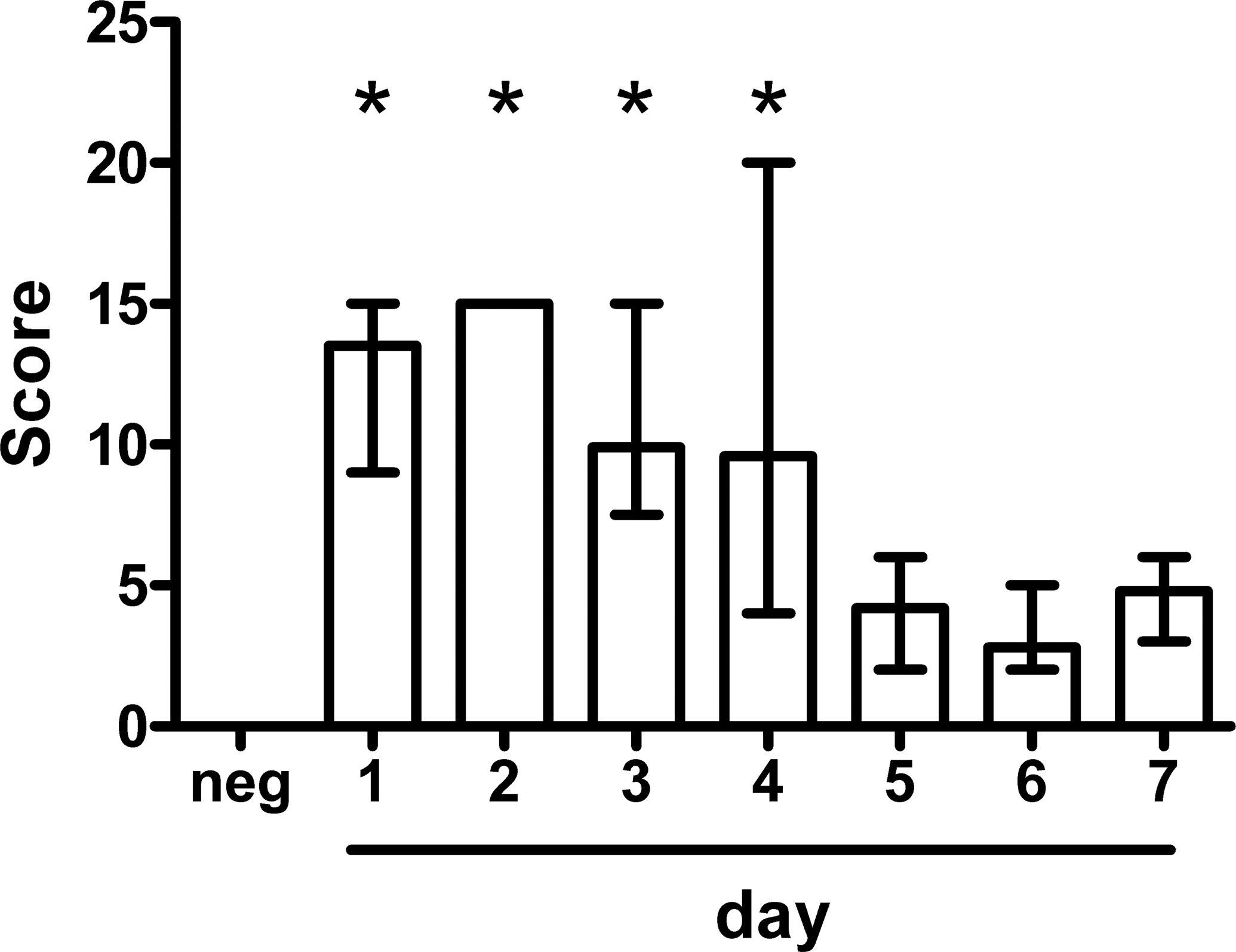
Severity and frequency of alveolar duct lining cell injury by cigarette smoke. The frequency and severity of epithelial changes along alveolar ducts decreased significantly from day 5 after exposure. Score = alveolar duct lining cell injury severity × frequency. Data are shown as medians and ranges of five animals per group. *p < .05, one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test.
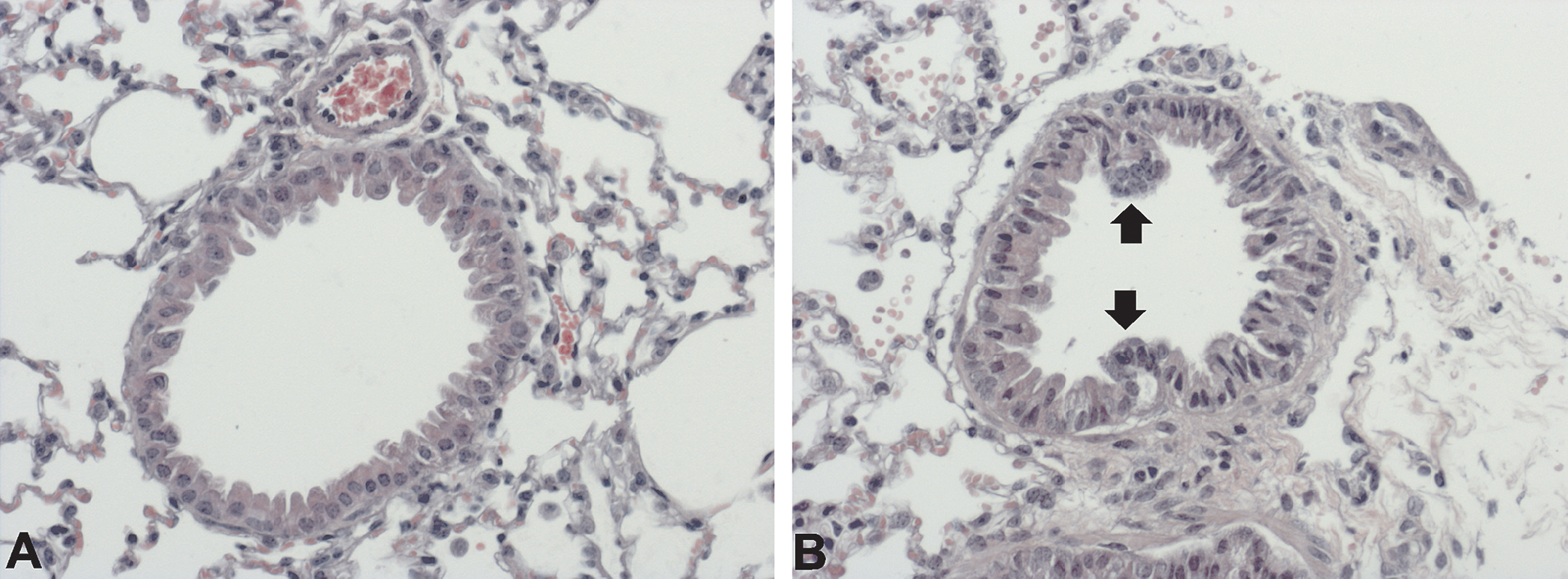
Regenerative hyperplasia of bronchial epithelium in sham (A) and animals exposed to smoke from 3 + 2 cigarettes for 2 consecutive days (B). Medium-sized bronchi are shown in sham-exposed animals (A) and minimal epithelial hyperplasia (arrow) 7 days after smoke exposure (B). A–B: HE, magnification 200×.
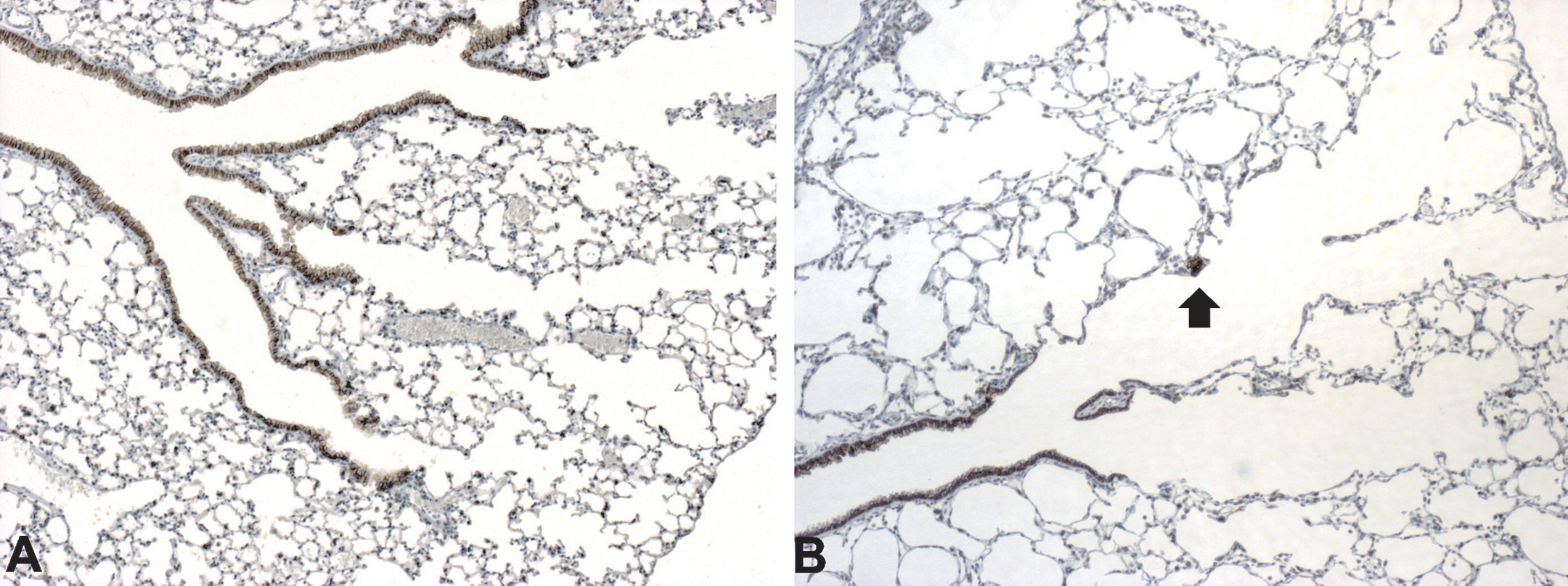
Claudin-3 staining of sham (A) and 2-day cigarette smoke (3 + 2 cigarettes)–exposed animals at day 7 postexposure (B). Small focus of bronchiolar metaplasia (arrow) in alveolar duct 7 days postexposure. A–B: claudin-3, magnification 50×.
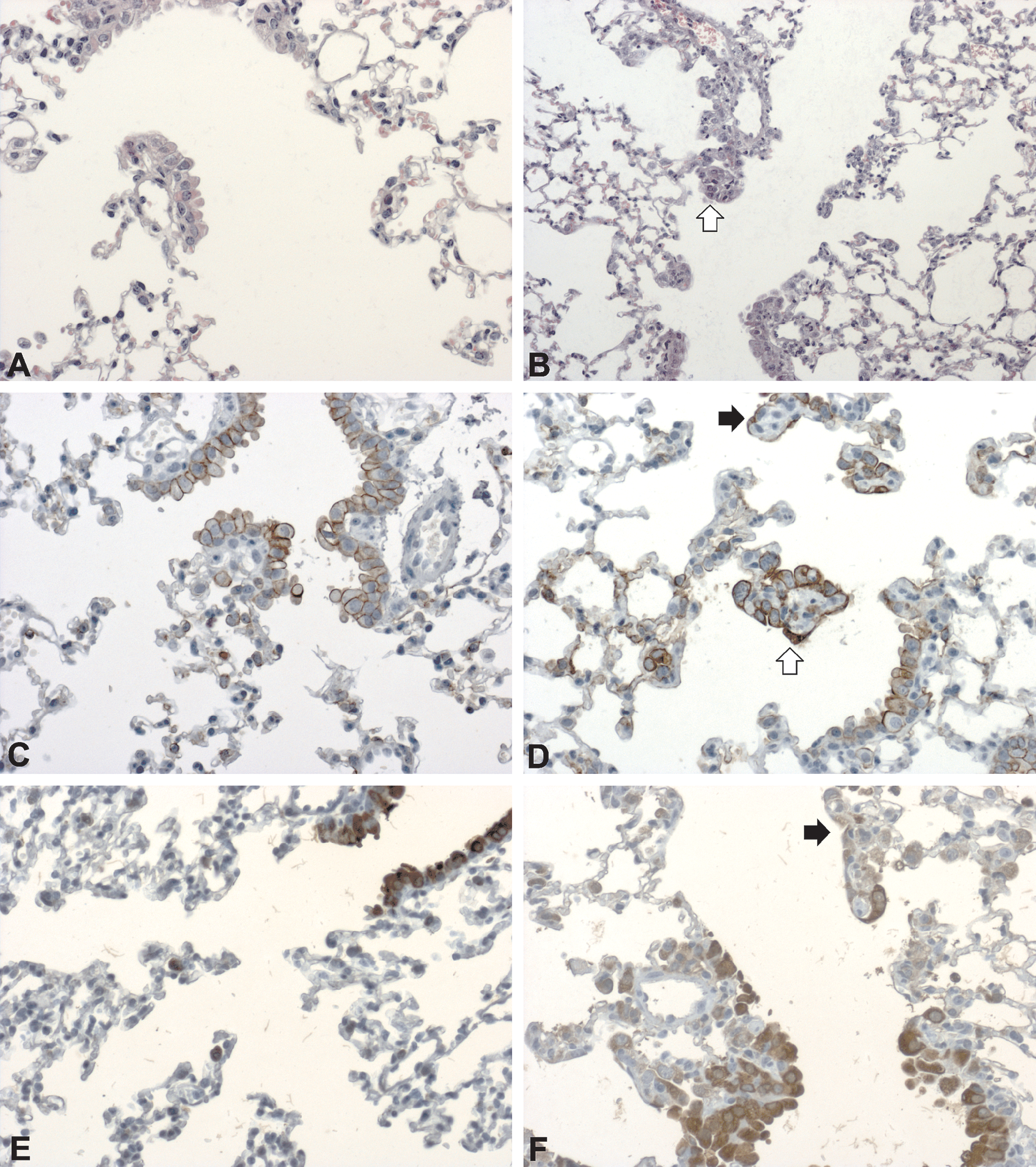
For the first 4 days after exposure to smoke from 3 + 2 cigarettes for 2 consecutive days, the majority of alveolar epithelial cells type-II (AEC-II) were enlarged (Figure 17B and C) even protruding into alveolar space (Figure 17C and 18C) and some were dividing (Figure 17D and 18D). The staining pattern for claudin-4 remained the same as for normal lungs, while the number of AEC-II expressing CC-10 increased (Figure 9), as well as the proportion of claudin-8 positive AEC-II (Figure 10B). By the end of the recovery period, that is, 7 days postexposure, the AEC-II staining pattern for all observed markers was the same as in sham-exposed lungs.
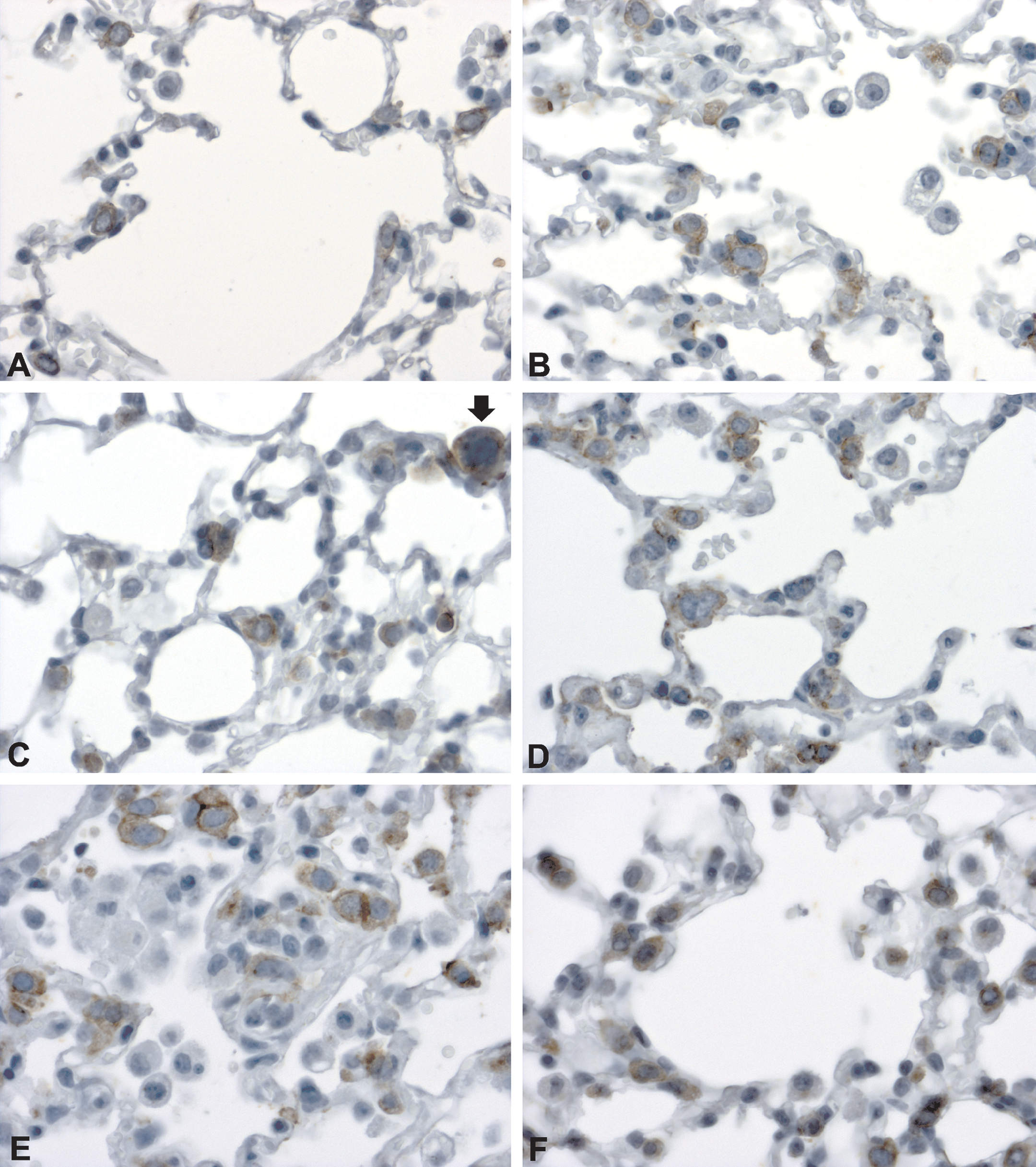
Claudin-3 staining of sham (A) and 2-day cigarette smoke (3 + 2 cigarettes)–exposed (B–F) animals during the 7-day recovery period [day 1 (B), day 2 (C), day 3 (D) day 4 (E), and day 7 (F)] after smoke exposure. Up to day 4 following smoke exposure, enlarged (arrow) alveolar epithelial cell type-II (AEC-II) were present (B, C, D, E). By the end of the experiment, AEC-II had regained their normal appearance (F). A–F: magnification 400×.
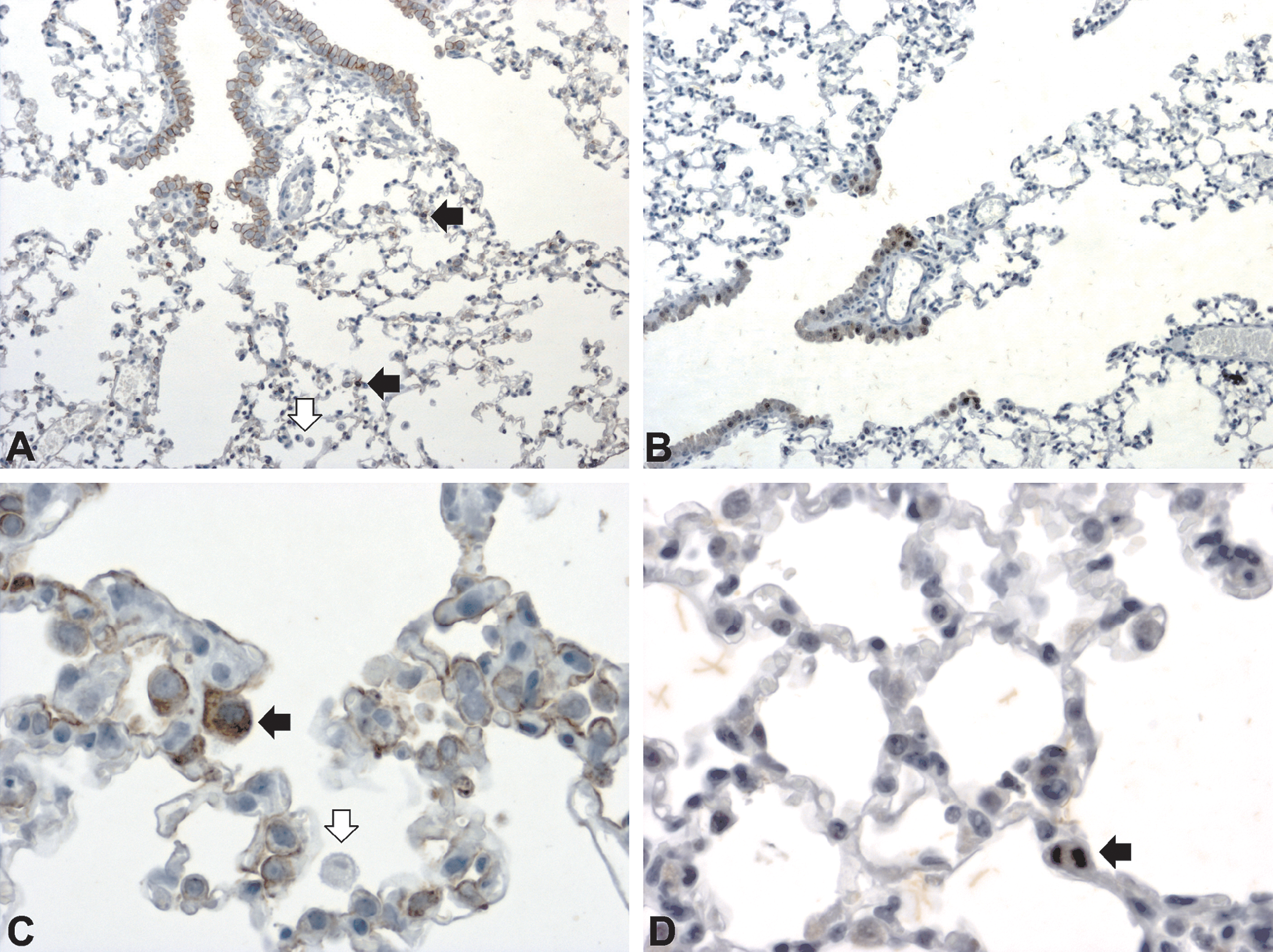
Cytokeratin (A, C) and Ki67 (B, D) expression in sham (A, B) and 3 days post exposure to smoke from 3 + 2 cigarettes for 2 consecutive days (B, D). In nonexposed lungs (A), alveolar epithelial cell type-II (AEC-II) were positive for cytokeratin (black arrow), while alveolar macrophages did not stain (white arrow). Three days postexposure cytokeratin positive AEC-II (black arrows) were enlarged and protruded into alveolar space (C), some were dividing (black arrow) (D). Note that alveolar macrophages remained cytokeratin negative (white arrow). A–B: magnification 50×; C–D: magnification 200×.
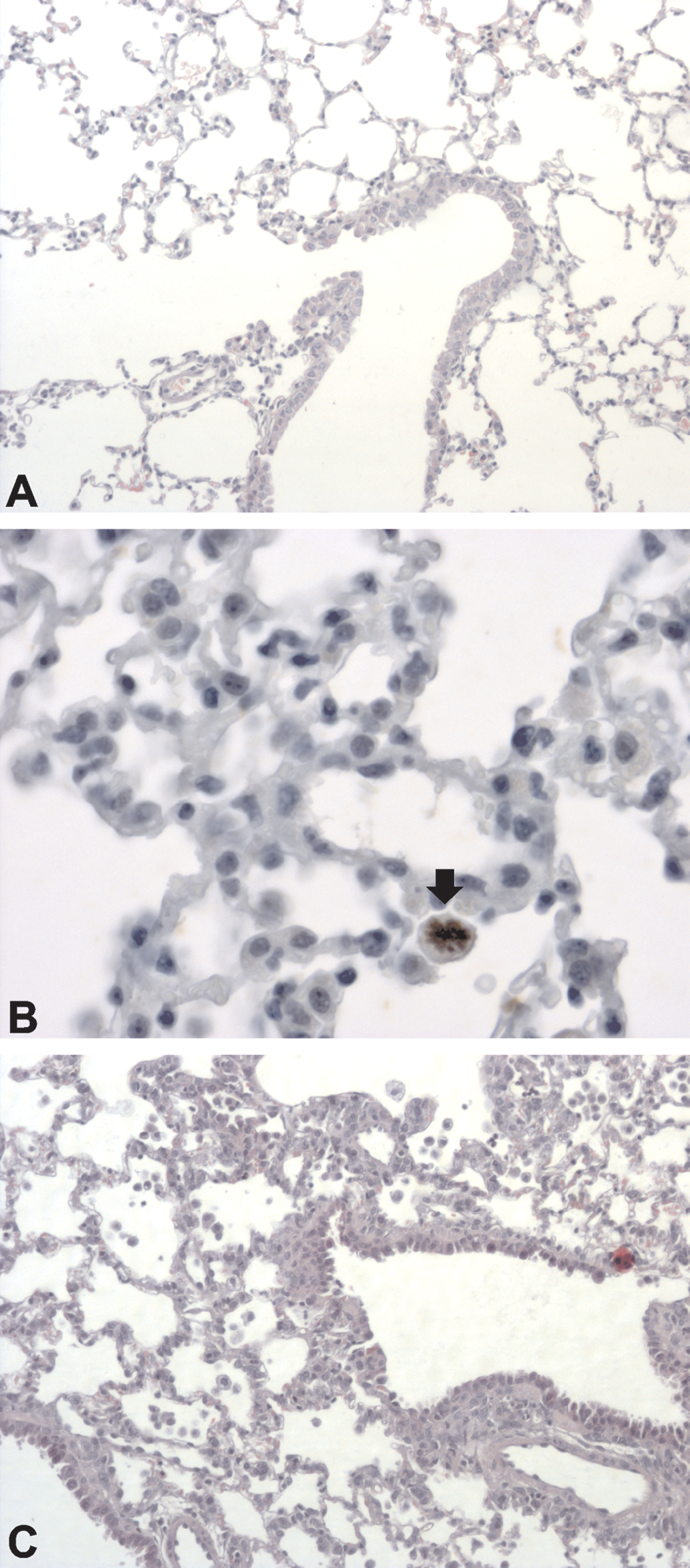
In nonsmoke-exposed lungs, nitrotyrosine was detected mainly in the cytoplasm of alveolar macrophages and single Clara cells (Figure 19A). One day after exposure to smoke from 3 + 2 cigarettes for 2 consecutive days, nitrotyrosine was present in the cytoplasm of cells lining ADs and AEC-II (Figure 19B). Nitrotyrosine staining intensity increased in alveolar macrophages (Figure 19C). By the following day, the nitrotyrosine staining pattern/intensity had returned to normal and remained so until the end of observed recovery period (Figure 19D).
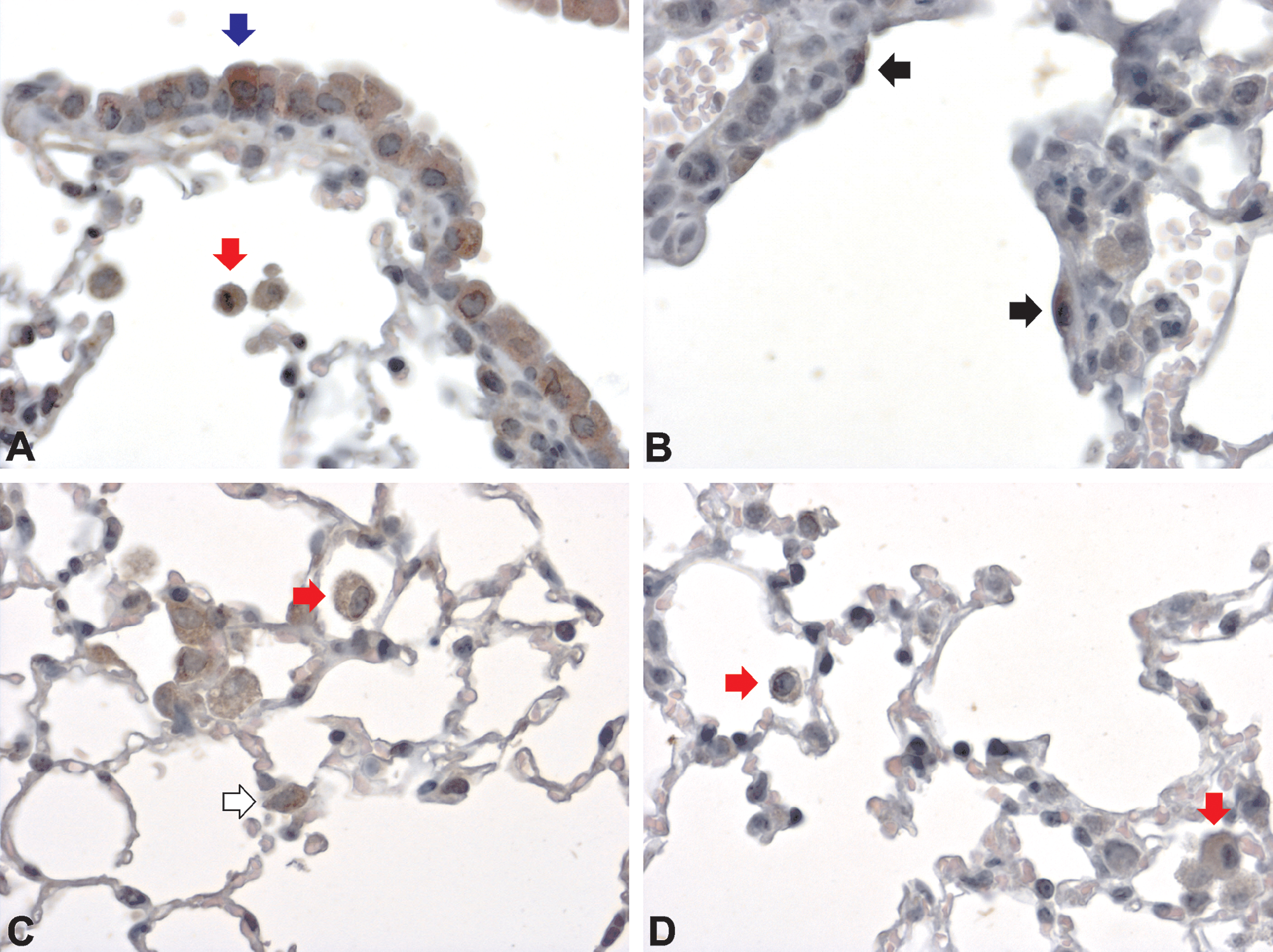
Nitrotyrosine in sham exposed lungs (A) and after smoke exposure (B-D). Nitrotyrosine in sham exposed lungs (A) was present in the cytoplasm of alveolar macrophages (red arrow) and Clara cells (blue arrow). One day after exposure to smoke from 3 + 2 cigarettes for 2 consecutive days, cells lining alveolar ducts (black arrow) (B) and AEC-II (white arrow) (C) stained positive. Alveolar macrophage staining increased in comparison to that in sham-exposed lungs (red arrow) (C). At day 7 postexposure (D), only alveolar macrophages (red arrow) stained with variable intensity. A–D: magnification 400×.
Discussion
Neutrophil accumulation in pulmonary parenchyma is one of the hallmarks of both COPD and cigarette smoke models of COPD (Bhalla et al. 2009; Di Stefano et al. 1998; Stanescu et al. 1996). In order to determine the minimal smoke exposure sufficient to induce inflammation, in the present study, we exposed BALB/c mice to smoke from one to five 3R4F cigarettes and evaluated various inflammatory parameters 4 and 24 hr postexposure. We have shown that brief exposure to smoke from a single 3R4F cigarette is sufficient to induce pulmonary neutrophilia. Four hours following exposure capillary vasodilatation and blood stasis were observed, while increased exposure to smoke induced accumulation of neutrophils in the interstitium. Twenty-four hours after exposure to smoke from a single 3R4F cigarette, neutrophils accumulated in lungs; mainly along bronchioles/ADs and adjacent vessels. Their count/percentage in BALF increased, while macrophage percentage significantly decreased. Further increasing smoke exposure to a maximum of five cigarettes did not significantly increase the 24 hr postexposure neutrophil count/percentage in BALF. Morris et al. (2008) showed that exposure to at least four 1R3F cigarettes was needed to induce an increase in the proportion of BALF neutrophils in BALB/c mice. The discrepancy between these studies could result from differences in animal gender and the type of cigarettes used.
The accumulation of neutrophils along bronchioles/arterioles and to a lesser degree along ADs is in line with the observation that 1 hr after first exposure to cigarette smoke, the concentrations of neutrophil chemoattractants, KC, MIP-2, and LIX, were strongly increased in lung homogenates.
Despite this early inflammatory reaction, smoke from a single cigarette failed to cause any epithelial damage in terms of changes in cell morphology and/or aberrant expression of markers used in immunohistochemistry. In contrast, following exposure to the smoke from two cigarettes, single cell loss along the bronchi could be detected. In addition, quite unexpectedly, this low smoke exposure level was sufficient to induce de novo expression of the tight junction protein, claudin-3, by AD epithelial cells. On the other hand, claudin-3 expression along cell membranes of bronchial epithelial cells remained unchanged following smoke exposure, as described previously (Kaarteenaho-Wiik and Soini 2009).
Prolongation of cigarette smoke exposure to five cigarettes for 2 consecutive days induced three major pulmonary changes: damage to bronchial epithelial cells, accumulation of neutrophils along bronchioles/arterioles, and hyperplasia of alveolar macrophages. Concentrations of KC and MIP-2 increased with the dose/duration of exposure to cigarette smoke as reflected in the increased intensity of neutrophilic pulmonary infiltration, detected histologically. The changes observed have been well documented in the literature on the murine short-term smoke model of COPD (D’Hulst et al. 2005). In addition, in these experimental settings, cigarette smoke induced overt intrapulmonary conducting airway epithelium cell loss.
It has been shown previously that cigarette smoke induces epithelial damage (Tarrant, Mills, and Williard 2009) in lungs with preferential damage of Clara cells (Plopper et al. 1992). In order to further highlight the airway epithelial cell reaction to cigarette smoke, antibodies against CK, claudin-3, claudin-4, and claudin-8, and CC-10 were used. Morphologically altered epithelial cells in the distal portions of the respiratory tree, that is, at bifurcations of bronchioles and BADJ, were identified as Clara cells (CK and CC-10 positive). Bolton et al. (2008) described metaplasia of Clara cells in transitional airways after exposure to LPS, which is also present in cigarette smoke. We confirmed, in line with others (Giangreco, Reynolds, and Stripp 2002; Reynolds and Malkinson 2010), the high susceptibility of cells in distal airway progenitor regions, neuroepithelial bodies, and BADJ (Giangreco et al. 2009), to toxins from cigarette smoke. These findings support the hypothesis that the onset of emphysema, characteristic for COPD, is at least partially due to the failure of repair mechanisms (Churg, Cosio, and Wright 2008; Puchelle et al. 2006).
De novo CK, CC-10, claudin-3, and claudin-8 expression by epithelial cells lining ADs could point to very early adaptive mechanisms induced by cigarette smoke exposure. Accordingly, Churg and Wright (2007) showed that ADs were the major site of lesions in a chronic murine model of smoke-induced emphysema.
In normal murine lungs, the cytoplasm of AEC-II was positive for claudin-3 and claudin-4 (data not shown), which is in agreement with observations by Kaarteenaho-Wiik and Soini (2009). Following smoke-exposure for 2 days, the majority of AEC-II were enlarged and some dividing, as observed in sections stained with claudin-3. The number of AEC-II expressing CC-10 and claudin-8 increased. There are scarce reports on claudin-8 expression in the respiratory epithelium of mammals. Expression of claudin-8 has been reported in cultured AEC-II of rats (Cohen, Lawrence, Margulies 2010) and the gills of puffer fish (Bagherie-Lachidan, Wright, and Kelly 2009). Bagherie-Lachidan proposed claudin-8 role in osmoregulation. Aberrant CC-10 expression by AEC-II was an unexpected finding. Park et al. (2006) noticed the CC-10 staining of a subpopulation of AEC-II which expressed Sox17. Sox17 is a transcription factor with a role in airway cell differentiation during both fetal and adult life. In mature lung tissue, its expression is restricted to bronchial ciliated cells. The relevance of altered claudin and CC-10 expression by AEC-II is yet to be explored.
In order to study the recovery process after exposure to smoke from 3 + 2 cigarettes for 2 consecutive days, pulmonary changes during the first 7 days after smoking cessation were observed. An acute inflammatory reaction, characterized by neutrophilic infiltration into the BALF and lung tissue, dominated for the first 4 days after the last cigarette. This was subsequently replaced by chronic monocytic inflammation. The concentration of the MCP-1 was significantly increased on the second day after smoke exposure leading to an increased accumulation of monocytes/macrophages. As a result, from the fifth day of observation, the macrophage count in BALF increased. Along with changes in BALF composition, monocytes prevailed among inflammatory cells in histological sections. Macrophages were mainly situated along bronchioles/arterioles, as indicated by the F4/80 marker. Within 7 days postexposure, their numbers diminished. In contrast, hyperplasia of alveolar macrophages was most prominent toward the end of the observation period.
In airways, no goblet cell metaplasia was evident, neither immediately after smoke exposure nor during the recovery period. Goblet cell metaplasia is characteristic of human COPD. In mice, even after prolonged smoke exposure, only minimal goblet cell metaplasia develops in lower airways. This is mainly due to different anatomical and histological characteristics in the mouse in comparison to humans (Wright, Cosio, and Churg 2008).
Changes in the epithelium lining terminal bronchioles and ADs were observed after smoke exposure, distributed diffusely throughout the lungs for the first 2 days after exposure and slowly decreased over the following days. By the end of the observation period, small foci of epithelial hyperplasia appeared, indicating repair of the bronchial mucosa. By day 7 after the last cigarette, small foci of bronchial metaplasia could be seen in ADs. Similar findings were described after exposure to naphthalene, which is found in cigarette smoke (Jensen-Taubman, Wang, and Linnoila 2010). By the end of the recovery period, the AEC-II staining pattern for all observed markers was the same as in sham-exposed lungs.
Increased levels of oxidative stress have been described in smokers and COPD patients (Churg, Cosio, and Wright 2008). In addition, oxidative stress in various lung compartments (airway epithelium, AEC-II, and alveolar macrophages) of animals exposed to cigarette smoke has been reported as well (Churg, Cosio, and Wright 2008). Aoshiba et al. (2003) observed oxidative stress mediated DNA damage in AEC-II of animals exposed to cigarette smoke, and Pechkovsky et al. (2002) showed that stimulated AEC-II and oxidative stress induced activation of alveolar macrophages. In our study, expression of nitrotyrosine was measured as an indicator of reactive nitrogen species (RNS). Cigarette smoke induced aberrant and transient nitrotyrosine expression by AEC-II and AD lining cells, as well as a sustained increase in nitrotyrosine expression by alveolar macrophages. These results support the proposal that increased nitrotyrosine expression by alveolar macrophages might be caused by cigarette smoke–induced oxidative stress in AEC-II (Pechkovsky et al. 2002). Early nitrotyrosine expression by AD lining epithelial cells may have contributed to the more prolonged aberrant expression of various epithelial markers in our study.
In summary, in this study, we have described a model of cigarette smoke–induced lung injury characterized by an early onset of bronchiolar/AD and alveolar epithelial damage. We demonstrated the high susceptibility of Clara cells situated in airway progenitor regions to toxins from cigarette smoke and the smoke-induced aberrant expression of tight junction proteins by epithelial cells lining ADs. Furthermore, AEC were affected by toxins from cigarette smoke early after exposure. Therefore, the results presented in our study suggest that this short-term murine smoke model could be used to study pulmonary epithelial changes and epithelial injury/repair relevant to COPD that have been studied predominantly in long-term models. In addition, claudin-3 and CC-10 could be used as markers of early tobacco smoke–induced epithelial injury along ADs.
Footnotes
Acknowledgments
The authors wish to thank Ivanka Pašalić, DVM, Irineja Ćubela, Slavica Skender, and Hrvoje Poduška for excellent technical assistance. The authors thank Michael J. Parnham for critical reading of the article.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by GlaxoSmithKline Research Centre Zagreb Limited.