
Abstract
Background:
Hepatic iron overload is common in patients with myelodysplastic syndromes undergoing hematopoietic cell transplantation (HCT) and may predispose to peri- and post-HCT toxicity. To better understand the mechanisms of iron overload-induced liver injury, we examined the effects of iron overload induced by ferric ammonium citrate (FAC) on oxidative stress and apoptosis signaling pathway in human hepatic cell line HH4.
Methods and Results:
Hepatic HH4 cells were exposed to FAC to force iron uptake, and cellular responses were determined. Incubation with 5 mM FAC resulted in increased intracellular iron content in a time-dependent manner. High concentration of FAC impaired cell viability and increased level of reactive oxygen species (ROS), and addition of antioxidant reagent such as glutathione or N-acetylcysteine dramatically reduced FAC-induced intracellular ROS generation. FAC overload significantly increased the phosphorylation of inhibitor of κB-α, p38 mitogen-activated protein kinase (MAPK), and nuclear factor κ light chain enhancer of activated B cells (NF-κB) p65 and promoted the nuclear translocation of NF-κB p65. Knockdown of Fas and Bid expression by small interfering RNA in iron-treated HH4 cells resulted in restoration of cell viability.
Conclusions:
We reported that FAC treatment is capable of inducing both extrinsic death receptor and intrinsic mitochondrial signaling pathway-mediated HH4 cells apoptosis through ROS-activated p38 MAPK and NF-κB pathways.
Introduction
Myelodysplastic syndromes (MDS) are clonal disorders of hematopoiesis. 1 Several therapeutic strategies, including immunosuppression, 2 hypomethylation, or allogeneic hematopoietic cell transplantation (HCT) have been developed. Preclinical and limited clinical data suggest that iron overload related to anemia, red blood cell transfusions, and possibly other factors may be associated with hepatic injury in patients undergoing HCT. 3,4 However, the exact mechanism by which iron overload might lead to liver damage in this setting is uncertain.
Iron, which is an essential form for almost all forms of life, plays a vital role in a wide variety of metabolic processes, including oxygen transport, DNA synthesis, and electron transport. 5 For example, iron is essential for nicotinamide adenine dinucleotide phosphate hydride oxidases, xanthine oxidase, lipoxygenases, cytochrome P450 enzymes, subunits of the mitochondrial electron transport chain, and the hydrogen peroxide-destroying enzyme catalase. 6 But accumulation of excessive iron usually generates free radicals via the Fenton reaction. 7 Under normal physiological conditions, reactive oxygen species (ROS) plays an important role in activating transcription factor and promoting cell proliferation and differentiation. However, excessive ROS can cause lipid peroxidation of the cell membrane and alter some cell signaling pathways in order to induce cell death. It was reported that iron overload triggered hepatocellular injury in rats via hepatic mitochondrial and microsomal lipid peroxidation. 8 Meanwhile, recent studies showed that iron overload can induce apoptosis via mitochondria-mediated caspase 3-dependent pathway in cardiac cells 9 and promote cell death through the Bcl-2/Bax pathway in dopaminergic neuronal cells. 10 In addition, previous research has indicated that excessive ROS may be an early response for intracellular iron overload, earlier than morphological changes in apoptosis. For example, Sudip Bhattacharyya et al. found that only 3 h exposure of murine hepatocytes to iron (ferrous sulfate) obviously enhanced ROS generation. 11 Despite this, the mechanisms of iron overload-induced liver injury such as ROS level and apoptosis are not very clear. The present experiments indicated that both extrinsic and intrinsic signaling pathways mediated apoptosis through ROS-activated p38 mitogen-activated protein kinase (MAPK), and nuclear factor κ light chain enhancer of activated B cells (NF-κB) pathway in ferric ammonium citrate (FAC)-induced HH4 cells.
Methods
Cell culture
The non-transformed human hepatocyte cell line HH4 was maintained as described previously. 12 Briefly, HH4 cells were maintained in Williams’ medium E (Gibco Laboratories, Grand Island, New York, USA), supplemented with 10% fetal bovine serum, 0.1% ITS, dexamethasone (0.04 μg/mL; Sigma Chemical Co., St Louis, Missouri, USA), gentamycin (50 μg/mL; Sigma Chemical Co.), HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 20mM; Gibco Laboratories), and sodium pyruvate (1 mM; Gibco Laboratories).
For specific experiments, glutathione (GSH) and N-acetylcysteine (NAC; Beyotime Institution of Biotechnology, Shanghai, China) were dissolved in phosphate-buffered saline (PBS) to prepare a 100 mM stock solution. HH4 cells were exposed to 4–8 mM GSH or 2 mM NAC for 5 h after a 5-h treatment with FAC (Sigma Chemical Co.).
Cellular iron measurement
HH4 cells were cultured in the absence or presence of 5 mM FAC for 12–72 h incubation and then collected for iron determination. Cells were rinsed twice with PBS and digested with trypsin (Gibco Laboratories). After centrifugation at 800g for 10 min, the pellet was resuspended in 400 μL of ultrapure water for a brief sonication. Protein content in an aliquot was determined using bicinchoninic acid assay (BCA) protein assay (Beyotime Institution of Biotechnology). The rest of the sample was digested in 1 mL of 1 N nitric acid for 12 h at 80°C. Total cellular iron content in HH4 was determined using a Spectra AA 220 atomic absorption spectrophotometer (Varian, Milpitas, California, USA).
Effects of FAC on cell viability
The suppression of cell viability by FAC was determined using an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sers, Yantai, China). Cells were plated at a density of 1 × 105 cells/mL in 96-well plates at 37°C in a 5% carbon dioxide atmosphere and incubated for 24 h. Cells were then exposed to 0.1–10 mM FAC and propagated for 0–72 h. After treatment, cells were incubated with 5 mg/mL MTT (20 μL/well) in medium for 4 h at 37°C. MTT-containing medium was removed, and the intracellular formazan product was dissolved in 200 μL dimethyl sulfoxide for quantification by use of double-wavelength spectrophotometry (595 nm and 655 nm) with a microplate reader (Bio-Rad, Hercules, California, USA).
Detection of intracellular ROS
Intracellular ROS was measured by an ROS assay kit (Beyotime Institution of Biotechnology). Dichloro-dihydro-fluorescein diacetate (DCFH-DA) is cleaved into DCFH (nonfluorescent form) by intracellular esterase. Then DCFH is oxidized by intracellular ROS in viable cells to produce a fluorescent dichlorofluorescein (DCF). HH4 cells were exposed to serum-free medium containing 10 μM DCFH-DA after being treated with 5 mM FAC for 0–72 h. For ROS scavenging assays, cells were treated with 4–8 mM GSH or 2 mM NAC, respectively, for 5 h following a 5-h exposure to FAC. After 1 h incubation with 10 μM DCFH-DA, cells were washed three times with serum-free medium, and fluorescence intensity was measured by a fluorescence-activated cell sorting (FACS) Calibur flow cytometer (BD Biosciences, Franklin Lakes, New Jersey, USA) with excitation and emission wavelengths of 488 and 525 nm, respectively.
Western blot analysis
Cells were lysed with radioimmunoprecipitation assay (1% Triton X-100, 150 mM sodium chloride, 25 mM Tris pH 7.4, 5 mM ethylenediaminetetraacetic acid, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 5 mM tetrasodium pyrophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 2 mM phenylmethanesulfonyl fluoride, and 0.076 U/mL aprotinin). After centrifugation at 13,800g for 15 min, the supernatants were collected. Protein content was determined using BCA protein assay. Equal amounts of protein were subjected to SDS-polyacrylamide gel electrophoresis. Proteins were electrophoretically transferred to polyvinylidene fluoride membranes (Bio-Rad) and blocked with PBS/0.05% Tween-20 (PBS-T)-containing 5% nonfat dry milk or bovine serum albumin at room temperature (RT) for 1 h. Membranes were incubated with primary antibody at a 1:1000 dilution in PBS-T at 4°C overnight, washed four times with PBS-T, and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (Beyotime Institution of Biotechnology) in PBS-T at RT for 1 h. Protein was detected using an enhanced chemiluminescence (ECL) kit (Tiangen, Beijing, China).
Real-time PCR
Total RNA was extracted using RNA pure tissue kit (Cwbio, Beijing, China) according to the manufacturer’s instruction. Real-time reverse transcription polymerase chain reaction (RT-PCR) amplification was performed using an Ultra SYBR mixture kit (Cwbio, Beijing, China) on the C1000 Touch Thermal Cycler (Bio-Rad), and the results were analyzed using CFX Manager software (Bio-Rad). Target gene expression levels were quantified using the formula
Forward and reverse primer sequences, annealing temperature, and product size for different genes.

Detection of apoptotic cells
HH4 cells were incubated for 12–72 h with or without 5 mM FAC. The cells were then washed twice with cold PBS and resuspended in 1× binding buffer. The cells were incubated with a mixture-containing 5 μL of annexin V–fluorescein isothiocyanate and 10 μL of propidium iodide (BD Biosciences Pharmingen, San Diego, California, USA) at RT in the dark for 15 min. The cells were then analyzed by flow cytometry using FACS Calibur flow cytometer.
RNA interference
For gene silencing assays, the small interfering RNA of the Fas (siFas) or Bid (siBid) gene and negative control siRNA were synthesized by Invitrogen Corporation (Shanghai, China). The sense and antisense strands of siRNAs were: Fas, 5′-CCCUUGCACCAAAUGUGAATT-3′ (sense), 5′-UUCACAUUUGGUGCAAGGGTT-3′ (antisense); Bid, 5′-GGGAAGAAUAGAGGCAGAUTT-3′ (sense), 5′-AUCUGCCUCUAUUCUUCCCTT-3′ (antisense). Hepatocytes were transiently transfected with oligofectamine according to the manufacturer’s protocol in the presence or absence of 27.3 nmol siRNA duplex. Quantitative real-time RT-PCR assay of messenger RNA (mRNA) expression level of Fas or Bid was performed to identify inhibitory effect of siRNA in HH4 cell. After 48 h transfection with indicated siRNA, cells were treated with 5 mM FAC for 24 h. Cell viability assay was performed with MTT chromometry.
Statistical analysis
Results were analyzed by one-way analysis of variance (ANOVA) or two-sample unpaired t-test using Prism 5 program. Differences in results were considered significant when p ≤ 0.05. Results are given as mean ± SEM of at least three independent experiments.
Results
FAC causes cellular iron increase in a time-dependent manner
To investigate the overall effect of FAC on cellular metabolism, we firstly determined the kinetics of cellular iron accumulation. HH4 cells were treated with 5 mM FAC for different periods of time (12–72 h). As shown in Figure 1, the total iron content assimilated by HH4 cells increased in a time-dependent manner. After 72 h of FAC exposure, cellular iron content was about sixfold higher than in the control group, suggesting that FAC exposure upregulated the total cellular iron content in HH4 cells.

FAC exposure increased iron content in HH4 cells. HH4 cells were treated with 5 mM FAC for 12–72 h. Cells were harvested and iron content was determined by spectrophotometer. Results are shown as mean ± SEM of three independent experiments. *p < 0.001: Statistically significant differences compared with the Veh group (no treatment) using one-way ANOVA test. FAC: ferric ammonium citrate; ANOVA: analysis of variance.
FAC-induced iron overload suppresses HH4 cell viability
Following exposure to FAC at 5–10 mM, numbers of HH4 cells declined by 48 h (Figure S1). In addition, determination of the level of mitochondrial succinodehydrogenase (SDH) activity, as assessed by the MTT assay, showed a significant decrease in HH4 viability after 12–72 h of exposure to FAC at 5–10 mM (Figure 2).

Cell viability was measured with MTT assay after FAC exposure in HH4 cells. HH4 cells were treated with FAC (0.1, 1, 5, or 10 mM) for 0–72 h. Data were showed as relative cells viability to Veh group (no treatment). Results are shown as mean ± SEM of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001: Statistically significant differences compared with the Veh group (no treatment) using one-way ANOVA test. FAC: ferric ammonium citrate; ANOVA: analysis of variance; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
FAC-triggered generation of ROS is attenuated by antioxidants
As shown in Figure 3(a), mean fluorescence intensities reflecting ROS levels in the cytoplasm were markedly increased following 5 mM or 10 mM FAC exposure, and the highest values of mean fluorescence intensities were observed when cells were treated with 5 mM FAC at 48 h. However, additional GSH and NAC in treated HH4 cells reduced cellular ROS generation. Meanwhile, HH4 cells treated with 8 mM GSH resulted in more dominant effect than with 4 mM GSH or 2 mM NAC (Figure S2 and Figure 3(b)).

Intracellular ROS production in HH4 cells induced by FAC. (a) The levels of intracellular ROS in HH4 cells were measured by oxidized DCF. (b) Attenuation of ROS generation by GSH and NAC in iron-overload HH4 cells. HH4 cells were treated with 4 mM GSH, 8 mM GSH, or 2 mM NAC for 5 h, after a 5-h exposure to FAC. GSH and NAC significantly decreased the amount of FAC-induced ROS. Results are shown as mean ± SEM of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001: Statistically significant differences compared with the Veh group (no treatment) (a) or 5 mM FAC group (b) using one-way ANOVA test or the two-sample unpaired t-test. ROS: reactive oxygen species; GSH: glutathione; DCF: dichlorofluorescein; NAC: N-acetylcysteine; FAC: ferric ammonium citrate; ANOVA: analysis of variance.
FAC exposure activates MAPK and NF-κB signaling pathways
Previous studies suggested that excessive ROS induced activation of p38 MAPK in hematopoietic stem cells 13 and NF-κB signaling pathway 14 in human neuroblastoma cells. We next examined the effect of FAC exposure on ROS-related MAPK signal pathways and NF-κB activation in HH4 cells. As shown in Figure 4(a), total p38 MAPK and NF-κB protein levels kept unchanged before and after 5 mM FAC treatment. However, phospho (p)-p38 MAPK, p-IκB-α, and p-NF-κB protein levels dramatically increased, suggesting that p38 MAPK, IκB-α, and NF-κB were activated in HH4 following FAC exposure. In addition, the effect of FAC overload on nuclear translocation of NF-κB was examined. The results showed that FAC exposure significantly promoted nuclear translocation of NF-κB (Figure 4(b)).

Involvement of the p38 MAPK and NF-κB pathways in FAC-treated HH4 cells. (a) Protein expression of p-p38 MAPK, p38 MAPK, p-IκB-α, p-NF-κB, and NF-κB in HH4 treated with 5 mM FAC for 0–72 h by Western blot (Cell Signaling Technology, Boston, Massachusetts, USA). (b) Nuclear translocation of NF-κB p65 in HH4 cells treated with 5 mM FAC for 24 h. p-IκB-α: phospho inhibitor of κB-α; MAPK: mitogen-activated protein kinase; p-NF-κB: phosphio-nuclear factor κ light chain enhancer of activated B cells; FAC: ferric ammonium citrate.
FAC-induced iron overload activates apoptotic signaling pathway
Next, we observed that the apoptosis rate of HH4 cells treated with 5 mM FAC for 12–72 h was significantly increased comparing with the control (Figure 5(a)). In order to further investigate the effect of iron loading on apoptosis, we next examined the expression of fas, caspase-8, cytc and caspase 3 in HH4 cells treated with FAC for 0–72 h. As shown in Figure 5(b), there was upregulation of Fas and Caspase 8 at 12 and 24 h, and a more delayed increase in cytochrome c (Cyt c) and caspase 3 expression. Furthermore, Western blots showed significant, time-dependent changes in protein levels of Fas-associated protein with death domain (FADD), caspase 8, Bid, and caspase 9 in HH4 cells treated with 5 mM FAC (Figure 5(c)). In order to verify whether Fas-related extrinsic pathway and mitochondrial intrinsic pathway are involved in HH4 cells apoptosis induced by FAC, cells were pretreated with siRNA for Fas or Bid, which are the key molecules for extrinsic pathway and intrinsic pathway, respectively. 15 These blocking experiments clearly enhanced cell viability of FAC-exposed hepatocytes, compared with the control groups (Figure S3 and Figure 5(d)).

FAC-induced iron overload promoted HH4 death via activation of the extrinsic (Fas) and intrinsic (mitochondrial) apoptotic pathway. HH4 cells were treated with 5 mM FAC for various times. (a) Flow cytometric analysis of apoptosis rate on 5 mM FAC-treated HH4 cells. Cells were incubated with 5 mM FAC for 0–72 h. The apoptosis feature of HH4 cells was determined by annexin V staining. Results are shown as mean ± SEM of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001: Statistically significant differences compared with the Veh group (no treatment) using one-way ANOVA test. (b) Change of Fas, Caspase 8, Cyt c and Caspase 3 gene expression was determined by RT-PCR. The expression of genes was classified as upregulated if expression levels were >0.5-fold and as downregulated if levels were <0.5-fold, relative to control HH4 cells. Expression of β-actin was used as internal control. (c) Protein expression of FADD and caspase 8 and activation of Bid, caspase 9 were analyzed by Western blot (Cell Signaling Technology, Boston, Massachusetts, USA). (d) Inhibitory effects of siRNA for Fas or Bid on cell viability caused by FAC overload. Cells (105 cells/well) were pretreated with 27.3 nM siRNA for Fas or Bid for 48 h. Then HH4 cells were treated with 5 mM FAC for 24 h, and cell viability was measured by MTT. Results are shown as mean ± SEM of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001: Statistically significant differences compared with the Veh group (no treatment) using one-way ANOVA test. Statistically significant differences between siFas and siBid groups are compared using the two-sample unpaired t-test. siRNA: small interfering RNA; SCR: scrambled siRNA; siFas: Fas siRNA; siBid: Bid siRNA; FAC: ferric ammonium citrate; ANOVA: analysis of variance; RT-PCR: reverse transcription polymerase chain reaction; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; FADD: Fas-associated protein with death domain.
Effects of GSH on ROS-related signaling and apoptotic pathways
To evaluate whether antioxidant reagent can reduce intracellular oxidant stress, HH4 cells were incubated with 8 mM GSH after treatment with 5 mM FAC. As shown in Figure 6(a), protein levels of p-p38 MAPK and p-NF-κB decreased in GSH-treated HH4 cells compared with cells not exposed to GSH. In addition, mRNA levels of caspase 8, Cyt c, and caspase 3 and the apoptosis rate in GSH-treated cells also decreased markedly (Figure 6(b) and (c)). Furthermore, we also investigated the effect of NF-κB activation on expression level of Fas gene. Results showed interruption of the NF-κB pathway by pharmacologic NF-κB inhibitor Bay 11-7082 markedly attenuated Fas gene expression in HH4 cells (Figure 6(d)).

The addition of GSH attenuated the occurrence of apoptosis-related events (a). (b) Effects of GSH on expression of p-p38 MAPK, p38 MAPK, p-NF-κB, NF-κB, and apoptosis-related genes. Cells were treated with 5 mM FAC for 5 h and subsequently further incubated with 8 mM GSH for 5 h. The expression of p-p38 MAPK (a), p38 MAPK (a), p-NF-κB (a), NF-κB (a), caspase 8 (b), Cyt c (b), and caspase 3 (b) were analyzed by Western blot or RT-PCR, using tubulin or actin as control, respectively. (c) FAC-induced early apoptosis were weakened via GSH treatment. Cells were treated with 5 mM FAC for 5 h and subsequently further incubated with 8 mM GSH for 5 h. (d) The addition of NF-κB inhibitor Bay 11-7082 (Beyotime Institution of Biotechnology, Shanghai, China) attenuated expression level of Fas gene in HH4 cells. Results are shown as mean ± SEM of three independent experiments. *p < 0.05; **p < 0.01: Statistically significant differences compared with the control groups using unpaired Student’s t-test. GSH: glutathione; p-IκB-α: phospho inhibitor of κB-α; MAPK: mitogen-activated protein kinase; p-NF-κB: phosphio-nuclear factor κ light chain enhancer of activated B cells; FAC: ferric ammonium citrate; RT-PCR: reverse transcription polymerase chain reaction.
Discussion
At present, HCT is considered to be the only therapeutic modality for MDS patients. 16 However, patients commonly have an increase in nontransferrin-bound iron and show hepatic iron overload already before HCT or after transplant conditioning. 12,17 Although the liver functions as an iron storage organ, excess iron is toxic to the liver. Iron can stimulate the formation of ROS via the Fenton reaction, 18 and progressive accumulation of ROS may damage mitochondrial and nuclear DNA through lipid peroxidation. 19
In the present study, long-term exposure of HH4 hepatocytes to FAC resulted in iron accumulation and a time- and concentration-dependent decrease in mitochondrial succinate dehydrogenase activity. Our data showed dramatic increase of ROS production in FAC-treated HH4 cells, which was also confirmed in human MG-63 cells and hematopoietic cells/mesenchymal stem cells by others. 20,21 Furthermore, we showed that ROS production caused by iron accumulation in our model was strikingly reduced in the presence of antioxidants GSH or NAC.
It is well known that oxidative stress can trigger the early activation of redox-sensitive signaling cascades (such as MAPKs and NF-κB). Moreover, MAPKs play an important role in cell proliferation, differentiation, development, transformation, and apoptosis. 22 –24 Meanwhile, NF-κB is one of the best characterized transcription factors, which could bind to a 10-bp site in the κ light chain enhancer (also called κB), 25 and its activation requires phosphorylation and degradation of inhibitory protein IκB. 26 In previous reports, Yvonne Nolan et al. had showed that increasing levels of ROS due to oxidative stress can elicit the phosphorylation of p38 MAPK in rat cortex and hippocampal tissue. 27 Another study has suggested that activation of p38 MAPK could induce the activity of NF-κB in C2C12 cells by reducing IκBα levels, inducing NF-κB DNA-binding activity and potentiating the transactivating activity of NF-κB p65. 28 In agreement with those studies, our results showed that ROS caused by FAC exposure triggered a sustained activation of p38 MAPK, IκB-α, and NF-κB, which can be attenuated by antioxidant reagent GSH (Figures 4 and 6(a)).
It is found that under different pathophysiological conditions, oxidative stress-activated MAPKs and NF-κB can induce different pathways of cellular apoptosis. 11,29 Normally, apoptosis, the programmed cell death, is mediated via two major pathways, initiated by death receptors or via mitochondrial stress. 30 Our experiments showed upregulation of mRNA and protein levels of Fas, FADD, caspase 8, and caspase 3 genes, supporting involvement of the death receptor pathway in HH4 hepatocytes exposed to treatment with FAC. In addition, our results also demonstrated that FAC-induced apoptosis in HH4 involved activation of Bid, forming a 15-kDa cleaved active form, and then translocation of truncated Bid leads to the release of cyt c and downstream activation of the caspase 9/caspase 3 cascade (Figure 5(b) and (c)), indicating that typically mitochondria pathway was also involved in the apoptosis process. To further characterize the FAC-induced apoptosis, we employed gene silencing of relevant apoptosis-related genes. The results of inhibition of Fas and Bid expression by RNA interference suggested that FAC exposure induced apoptosis via both the extrinsic (Fas-initiated) pathway and intrinsic (mitochondrial) pathway (Figure 5(d)). There is evidence that NF-κB activation can induce upregulation of proapoptotic genes (such as TNFα, TNFR1 and FasL), leading to etoposide-induced apoptosis by endogenous and exogenous apoptotic signaling pathways in T lymphocytes. 31,32 Our data proved that Fas gene expression decreased when HH4 cells were treated with NF-κB inhibitor (Figure 6(d)).
In conclusion, this work suggests that FAC exposure can increase the level of ROS and induce apoptosis in HH4 cells by activating both p38 MAPK and NF-κB pathways. While the disturbance of Fas and Bid expression by RNA interference demonstrated that both death receptor pathway and mitochondrial apoptotic pathway play an important role in FAC-induced HH4 cell apoptosis. Simultaneously, both GSH and NAC act as an antioxidant and antiapoptotic agent and provide protection to iron overload HH4 cells by inhibiting activation of p38 MAPK and NF-κB and downregulating expression of apoptosis-related gene. The probable mechanism of FAC-induced cytotoxicity to HH4 has been outlined in Figure 7. Our results provide a theoretical basis for drug therapy of iron-associated liver damage.
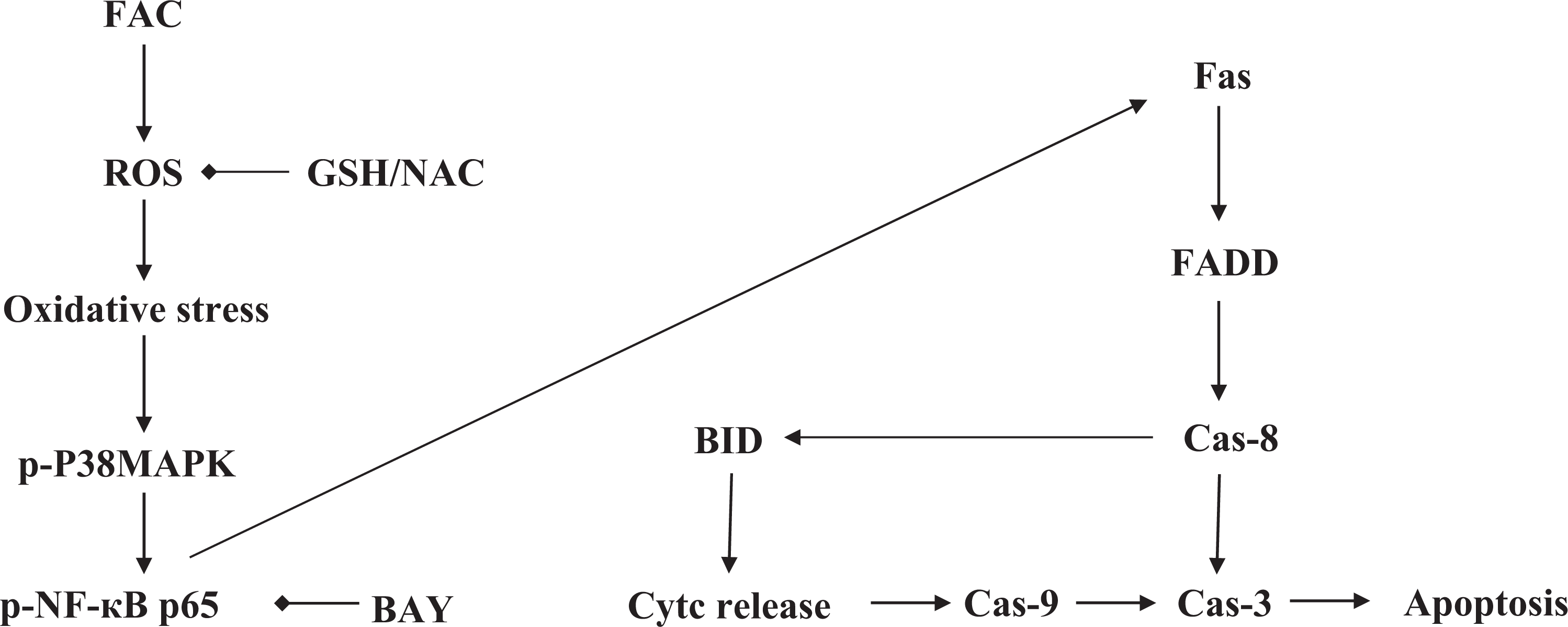
Schematic representation of possible signal transduction pathways in HH4 with FAC treatment. FAC: ferric ammonium citrate.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Science Foundation of China (grant nos. 81470294 and 31400691), the Natural Science Foundation of Jiangsu Province (grant no. BK20140169), Jiangsu Province Recruiting Plan for High-level, Innovative and Entrepreneurial Talents, the Fundamental Research Funds for the Central Universities (grant no. JUSRP51319B), Jiangsu Province “Six Summit Talent” Foundation (grant no. 2013-SWYY-019), and the 111 Project (grant no. 111-2-06).