
Abstract
Industrial solvents pose a significant threat to the humankind. The mechanisms of their toxicity still remain in debate. Trichloroethylene (TCE) is a widespread industrial solvent responsible for severe liver dysfunction, cutaneous toxicity in occupationally exposed humans. We utilized an in vitro system of human epidermal keratinocyte (HaCaT) cells in this study to avoid complex cell and extracellular interactions. We report the cytotoxicity of organic solvent TCE in HaCaT and its reversal by a natural flavanone, naringenin (Nar). The cytotoxicity was attributed to the rapid intracellular free calcium (Ca2+) release, which might lead to the elevation of protein kinase C along with robust free radical generation, instability due to energy depletion, and sensitization of intracellular stress signal transducer nuclear factor κB. These effects were actually seen to induce significant amount of genomic DNA fragmentation. Furthermore, all these effects of TCE were effectively reversed by the treatment of Nar, a natural flavanone. Our studies identify intracellular Ca as a unique target used by organic solvents in the cytotoxicity and highlight the Ca2+ ion stabilizer properties of Nar.
Introduction
Industrialization had posed serious threats to human health via synthetic chemicals like trichloroethylene (TCE). This solvent has been widely used as a cleaning and degreasing agent in ink, varnishes, and several commercial and industrial applications. 1 –3 Cutaneous toxicity and inflammation are the most common health hazards for organic solvent users, 4,5 which is primarily due to massive inflammation and defatting, that is, dissolution of skin lipids and cytotoxicity. 6
Several studies have been undertaken to explain the mechanisms of the toxicants by which they affect animals and humans. Literature accumulated till date suggests generation of intracellular reactive oxygen species (ROS) and reactive nitrogen intermediates (RNIs) as the most common pathway. 7 The outcome of the interaction of toxicants with the cells is tightly regulated by the intracellular signal transduction. One of such mechanisms is the activation of protein kinase C (PKC), a family of serine/threonine protein kinases well known for their tumor-promoting abilities. 8 PKCs have versatile biological functions, including differentiation and cell cycle progression as well as apoptosis. For example, PKCα and PKCβ have been found to interfere with the formation of the Fas death-inducing signaling complex (DISC) in Jurkat T cells by blocking Fas-associated protein with death domain recruitment and subsequent caspase-8 activation. 9 Inhibition of DISC formation by PKC activation has also been observed in HeLa cells during tumor necrosis factor–related apoptosis-inducing ligand-induced apoptosis, 10 apart from cellular apoptosis of cardiomyocytes due to free radical generation by the increase in the calcium (Ca) flux induced by glucose. 11
Increase in the intracellular Ca levels is the signature of upregulation of various downstream targets involved in toxicity and cell death. 12 Free Ca2+ leads to the dissipation of mitochondrial membrane potential (MMP) and apoptosis. 13 – 16
ROS generation is one of the first and basic responses of the cells toward any kind of endogenous or environmental stress. 17 ROS production and degradation involve fine equilibrium, and during toxic insult, the equilibrium shifts toward massive ROS production. This sustained ROS generation activates several downstream molecular targets involved in cellular toxicity.
In recent past, many active compounds of plant origin have shown promising potential, especially in the field of pharmaceutical sciences and medicine due to their capability to prevent a number of chronic and degenerative diseases including cancer. 18,19 Several studies, both in vitro and in vivo, have shown that flavonoids possess several biological and pharmacological activities, 20 including anticancer, 21 antioxidant, 22 antimutagenic, antiviral, antimicrobial, anti-inflammatory, and immunomodulatory affects. The exact cellular and molecular mechanisms of flavonoids are not very well understood. One among them is the ability of flavonoid to act as antioxidants, direct radical scavengers, metal ion chelators, carcinogen inactivators, modulators of gene expression, and DNA repair. 23
Naringenin (Nar) is a readily available flavanone present in grapefruit and consumed in high quantity through dietary sources. It elicits wide array of bioactive effects 24 on living system, which includes antioxidant, 25 anti-inflammatory, and immunomodulatory effects. 26 The role of Nar in cellular signal transduction and interception of various molecular targets involved in cell death has not been ascertained.
In this study, we have demonstrated useful observations underlying TCE-induced toxicity in human epidermal keratinocytes (HaCaT) and its reversal by dietary natural compound Nar.
Materials and methods
Cell culture
HaCaT cells were procured from National Centre for Cell Science Central Cell Repository, Pune, Maharashtra, India. Cells were maintained in 1:1 Dulbecco’s modified Eagle’s medium (DMEM):Ham F12 media with 10% fetal bovine serum (FBS) and 1.5% antibiotic/antimycotic solution containing 0.2% sodium bicarbonate at 37°C under a humidified atmosphere of 5% carbon dioxide (CO2)/95% air. Cells were passaged twice weekly using a standard trypsin–ethylenediaminetetraacetic acid (EDTA) protocol. Prior to commencement of experiments, cells were subcultured into six-well plates (or 96-well plates for the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay) for various assays.
Chemicals
TCE was obtained from Merck Ltd, Germany. Neutral red dye, low-melting point agarose, ethidium bromide (EtBr), Triton X-100, N-acetyl cysteine (NAC) were purchased from Sigma Chemical Co. Ltd (St Louis, Missouri, USA). Normal melting agarose, Nar (N5893; Sigma), EDTA disodium salt, and MTT dye were purchased from Hi Media Laboratories (Mumbai, Maharashtra, India). Phosphate-buffered saline (Ca2+, magnesium ions (Mg2+) free; PBS), DMEM, Ham F12 media, trypsin–EDTA, FBS, antibiotic, and antimycotic solution (10,000 U/mL penicillin, 10 mg/mL streptomycin, 25 µg/mL amphotericin-B) were purchased from Gibco, Grand Island, NY, USA. Fluorescent probe 5,5′,6,6′-0-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1), propidium iodide (PI), and 2,7-dichlorofluorescein diacetate were purchased from Sigma. All other chemicals were obtained locally in India and were of analytical reagent grade.
Treatment and exposure of TCE
HaCaT cells were plated at a density of 104 cells/mL in a 96-well plate for the cytotoxicity assays (MTT and neutral red uptake) and 24-well plate for other parameters (propidium iodide cell cycle study, comet assay, apoptosis, MMP, and immunocytochemistry), respectively, and allowed to attach for 24 h before the addition of TCE dissolved in serum-free media. A stock solution of TCE (50 µM) was prepared and diluted to different concentrations (0.5–32 µM). 27 Cells were then exposed to the above concentrations for 24 h and cytotoxicity assays were performed. The molecular parameters were performed according to experimental design and the doses used were 16 µM for TCE and 10 µM for Nar, as these doses were found to be less than inhibitory concentrations.
Cell viability assays
MTT reduction
The already described method of Mosmann 28 was used. In a 96-well plate , 1 × 104 cells were seeded and treated with different doses of TCE and Nar. For TCE half-maximal inhibitory concentration (IC50) determination in HaCaT cells, cells were treated with different doses of TCE ranging from 0.5 µM to 32 µM as previously done. After the 24 h incubation, 20 µL of MTT (5 mg/mL in PBS) was added to each well and incubated for 5 h. Then the media was aspirated and 200 µL of dimethyl sulfoxide (DMSO) was added in each well to solubilize formazon crystals and the change in absorbance was read at 570 nm on a Bio-Rad enzyme-linked immunosorbent assay (ELISA) plate reader (Hercules, California, USA). The IC50 value of TCE at 24 h was calculated from MTT assay using the formula followed by Reed and Muench in their study. 29
Protective effect of Nar in pre- and posttreatment was determined by MTT assay. Cells were seeded in a 96-well plate. Both pretreatment and posttreatment of the Nar (10 µM) were assessed against toxic response due to increasing dose of TCE.
Estimation of reduced GSH
Estimation of reduced glutathione (GSH) was done by the method described by Sedlak and Linsay. 30 Briefly, cells were lysed and spun at 5000g, and the supernatant was deproteinized with an equal volume of TCA (10%) and allowed to stand at 4°C for 2 h. The contents were then centrifuged at 1500 r/min for 15 min, and the supernatant was collected. Finally, 75 µL buffers were added to the microtiter plates and mixed with 20 µL samples (supernatant). Then 25 µL 5,5′-dithiobis-(2-nitrobenzoic acid) solutions were added and finally 100–150 µL methanol was mixed. Absorbance of the chromogen (yellow) was read at 412 nm in a Bio-Rad ELISA plate reader. The result was expressed as microgram GSH per gram protein using molar extinction coefficient of 13,600.
Catalase activity
Catalase activity was measured according to the method described by Sinha. 31 To an Eppendorf tube, 50 µL of reaction buffer was poured. Then, 20 µL of distilled water was added along with 5 µL of post mitochondrial supernatant (PMS). After shaking the sample, 25 µL of hydrogen peroxide (H2O2) was mixed and incubated for 2 min at 37°C. The sample was vortexed and added with 100 µL of dichromate acid reagent (0.1 M potassium dichromate in glacial acetic acid). The samples were heated in a water bath at 100°C for about 10–15 min until the color of the sample was greenish/faint greenish yellow. The contents were spun at 3000 rpm and an aliquot 150–200 µL of supernatant was transferred into microtiter plate with flat bottom. Finally, the samples were read at 570 nm on a Bio-Rad ELISA plate reader and converted into the activity using molar extinction of catalase as 43.6 expressed as micromoles of H2O2 consumed per minute per milligram protein.
SOD activity
Superoxide dismutase (SOD) activity was carried out by the method described by Marklund and Marklund. 32 The assay system consisted of 2.875 mL tris(hydroxymethyl)aminomethane (Tris)–hydrochloric acid (HCl) buffer (50 mM, pH 8.5), pyrogallol (24 mM in 10 mM HCl), and 100 µL of cell lysate in a total volume of 3 mL. The enzyme activity was measured at 420 nm and was expressed as units per milligram protein on a Perkin Elmer spectrophotometer (Waltham, Massachusetts, USA). One unit of enzyme is defined as the enzyme activity that inhibits autoxidation of pyrogallol by 50%.
LDH release
Lactate dehydrogenase (LDH) activity was analyzed according to the method followed by Kitazawa et al.. 33 After the cells were treated in 24-well plates according to treatment regimen, both the supernatant and cell lysate were evaluated for LDH release and the data were optimized. Briefly, 10 µL of extracellular supernatant and the supernatant got after the lysis of cell pellet were added to 200 µL of 0.08 M Tris buffer with pH 7.2 containing 0.2 M sodium chloride (NaCl), 0.2 mM nicotinamide adenine dinucleotide reduced, and 1.6 mM sodium pyruvate. LDH activity was measured continuously by monitoring the decrease in the rate of absorbance at 490 nm using the Bio-Rad ELISA microplate reader. Changes in absorbance were used to calculate the LDH release by the cells in response to TCE toxicity and extent of protection by Nar. Data were expressed as percentage change in comparison with the control values.
Intracellular ROS
A fluorometric assay was used to determine intracellular levels of ROS by Wan et al. 34 Dichloro-dihydro-fluorescein diacetate (DCFH-DA) is a nonpolar compound that is converted into a membrane-impermeable nonfluorescent polar derivative (DCFH) by cellular esterase after incorporation into cells. The trapped DCFH is rapidly oxidized to florescent 2,7-dichlorofluorescein (DCF) by intracellular peroxides. HaCaT cells were plated in 24-well plates and treated according to treatment regimen. After the designated time, cells were washed with PBS and incubated with 20 µM DCFC-DA for 1 h in a carbon dioxide (CO2) incubator at 37°C and 5% CO2. H2O2 was used as positive control and NAC was used as negative control as well as to ascertain whether the activation of various downstream molecular targets is due to production of intracellular ROS. Pretreatment of 25 µL of 1,2-bis(O-aminophenoxy)ethane-N,N,N′,N′-tetracetic acetomethyl ester (BAPTA-AM) in the TCE exposure was also included as one group to ascertain the effect of Ca2+ ions on intracellular ROS production. After 1 h incubation, the reaction mixture was then replaced by 200 µL of PBS and fluorescence intensity was measured in a fluorometer at excitation and emission wavelengths of 485 nm and 528 nm, respectively.
Nitrite estimation
The nitrite concentration in the culture supernatant was used as a measure of nitric oxide (NO) production according to Wilms et al.. 35 After TCE administration, the generation of NO in the cell culture supernatants was determined by measuring nitrite accumulation in the medium using Griess reagent (1% sulfanilamide and 0.1% N-(1-naphthyl)-ethylenediamine dihydrochloride in 5% phosphoric acid (Sigma). Then, 100 μl of culture supernatant and 100 μl Griess reagent were mixed and incubated for 5 min. The absorption was estimated in an automated plate reader (on Bio-Rad ELISA plate reader) at 550 nm. Sodium nitrite was used to generate a standard curve for quantification. Results were obtained from three separate measurements.
Intracellular Ca levels
Intracellular Ca levels were measured according to the method of Masaki et al. 36 HaCaT cells were seeded at the concentration of 1 × 106 cells/well in 24-well plates and treated according to the treatment protocol. After the treatment, cells were washed with PBS and trypsinized and centrifuged at 800 r/min. Pellet was resuspended in PBS-containing 5 µM Fura 2-AM fluorescent tag and incubated for 30 min at 37°C and 5% CO2 incubator. After incubation, the fluorescence intensity was measured in an LSB50 fluorometer at excitation and emission wavelengths of 340 and 510 nm, respectively.
Mitochondrial membrane potential
The loss of MMP is a hallmark for apoptosis. It is an early event preceding phosphatidylserine externalization and coinciding with caspase activation. It is measured according to the method described by Smiley et al.. 37 Cells were seeded in 24-well plates and treated according to treatment plan. After the desired time, cells were washed with PBS and trypsinized and suspended in 10 µM JC-1 fluorescent dye for 1 h in a CO2 incubator at 37°C and 5% CO2. After completion of incubation, cells were centrifuged and pellets were resuspended in 2 mL PBS and immediately read in an LSB50 Perkin Elmer fluorometer at excitation and emission wavelengths of 488 and 525 nm, respectively.
PKC estimation
PKC activity was evaluated by a commercially available kit of Enzo Life Sciences (Farmingdale, New York, USA; catalog number: ADI-EKS-420A) according to manufacturer’s instructions and suggestion from an article by Shih et al.. 38
Briefly, the HaCaT cells after treatments with TCE and Nar were scrapped out of flask and washed with Ca2+ and Mg2+ free PBS and suspended in cell lysating buffer (0.6 M KCl, 5 mM ethylene glycol tetraacetic acid, 5 mM EDTA, 1% Triton X-100, and 1 mM phenylmethanesulfonylfluoride in PBS).
The cellular fractions so obtained were added to the PKC substrate microtiter plate after soaking the plate with kinase assay dilution buffer. Reaction was initiated by adding adenosine triphosphate (ATP) and incubating the plate on a gentle shaker at 30°C for 90 min. The reaction was stopped by aspirating the contents and then phosphospecific substrate antibody was added provided in the kit. Further incubation was done at room temperature for 60 min. After aspiration, several washings with wash buffer was done and finally anti-rabbit immunoglobulin G:horseradish peroxidase (HRP) conjugate was added and incubated for 30 min at room temperature. The whole reaction was stopped by adding 3,3′,5,5′-tetramethylbenzidine substrate and acidic stop solution. The absorbance was read at 450 nm on a Bio-Rad ELISA plate reader. The standard of active PKC was used for the calculations. The PKC activity in cell lysate was calculated using the following formula:

Annexin V/Cy3 apoptosis assay using fluorescent microscopy
Cells were treated with TCE and Nar according to experimental protocol and harvested with trypsin–EDTA and washed with PBS twice and centrifuged at 800g and suspended in 1 mL PBS at concentration of 2 × 106/mL.
Then, 50 µL cells were incubated with 50 µL double-staining solution on poly-
Briefly, 50 µL cell suspension was placed in the circle drawn on the poly-
Cell cycle analysis
Log-phase HaCaT cells were seeded in 24-well plates and treated according to treatment regime. The cells were harvested, washed with PBS, and fixed with 95% ethanol at −20°C overnight. After an additional washing step, cells were incubated with RNAase A (20 μg/mL) at 37°C for 30 min, stained with PI (100 μg/mL) for 10 min, and analyzed by flow cytometry.
Single-cell gel electrophoresis
The cells exposed according to treatment protocol were washed with serum-free medium and harvested with 0.06% trypsin. The cells were resuspended in culture medium supplemented with 10% FBS, and slides were prepared by the method described by Bajpayee et al. 39 Two slides were prepared from each well (one well/concentration) and kept overnight at 4°C in lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10) with 1% Triton X-100 (added just before use). The slides were subjected to DNA unwinding for 20 min and freshly prepared electrophoresis buffer (1 mM EDTA sodium salt and 300 mM sodium hydroxide). The excess alkali was neutralized with Tris buffer (400 mM, pH 7.4), and slides were stained with 20 ug/mL EtBr and stored at 4°C in a humidified slide box until scoring. Slides were scored at a final magnification of 400X using an image analysis system (Komet 4.0, ANDOR technology, Belfast, UK) attached to a microscope (DMLB, Leica, Germany) equipped with a fluorescence attachment of a charge-coupled device camera. The comet parameters used to measure DNA damage in the cells were Olive tail moment (OTM) and tail DNA (TD) (%). Images from 50 random cells (25 from each replicate slide) were analyzed for each experiment per the guidelines (Tice RR). 40 The experiment (and not the cell) was used as the experimental unit for data analysis.
ICC of NF-κB and COX-2 proteins
Cells were seeded in 24-well plates at density 20,000 cells/well and incubated overnight. Thereafter cells were treated with TCE and Nar according to the experimental protocol. Medium was carefully discarded and the cells were subsequently washed twice with PBS (pH 7.4). Optimal fixation of cells was achieved by 4% paraformaldehyde for 30 min and after washing twice with PBS (pH 7.4)-containing 1 N HCl on ice for 10 min and 2 N HCl for 10 min at room temperature. After brief rinsing in PBS (pH 6.0), cells were incubated in methanol containing 0.3% H2O2 for 15 min, followed by incubation in blocking buffer (1.5% normal goat serum, 0.5% bovine serum albumin (BSA), and 0.1% Triton X-100) for 30 min. Finally, cells were incubated with anti-NFκBp65 antibody (1:100) and anti-cytochrome c oxidase-2 (COX-2) antibody (1:150) in 1% BSA at 4°C overnight. Cells were washed twice with PBS (pH 7.4) and treated with Ultra vision ONE Detection System HRP polymer and 3,3′-diaminobenzidine (DAB) chromogen (cat. no. TL-015-HDJ) according to manufacturer’s instructions. Cells were washed twice with PBS (pH 7.4), and peroxide complex was visualized with DAB (3,30-diaminobenzidine tetra hydrochloride) under a microscope. Brown cells were photographed with an Olympus CX2 inverted microscope.
Protein estimation
Estimation of protein content per the need of the experiment was done after an already described method of Bradford. 41
Statistical analysis
All the data were expressed as mean ± SEM of three independent experiments. Nonparametric one-way analysis of variance (ANOVA) with post hoc analysis to resolve the significant differences between the treatment groups was employed. We employed ANOVA to know the significance difference between the means of groups and further post hoc test signifies which group differs. Tukey–Kramer test was used as post hoc analysis. Any significant difference with p < 0.05 was considered significant.
Results
Effect of TCE and Nar on cell viability of HaCaT cells
MTT assay done after 24 h of TCE exposure showed decrease in cell viability at various doses from 0.5 µM to 32 µM. It was observed that TCE causes significant decrease in the cell viability from 0.5 µM and it reaches 50% decrease at a concentration of 16 µM and further viability decrease to less than 30% at 32 µM concentration (Figure 1(a)).
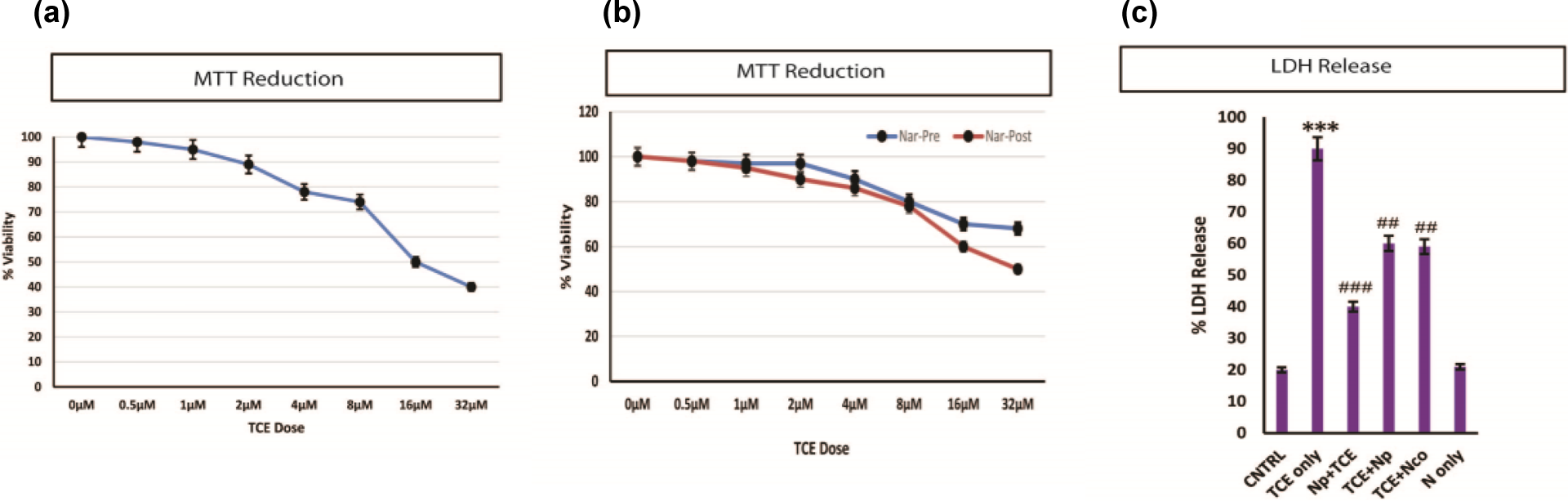
Cytotoxic evaluation of TCE. (a) Decrease in the viability of the HaCaT cells is shown after a linear dose challenge of TCE starting from 0.5 µM to 32 µM. (b) Pretreatment of Nar effectively reversed the cellular viability decline in comparison to post treatment. (c) The LDH release due to TCE exposure and protective effect of Nar is shown. ***p < 0.001: control vs. TCE; ## p < 0.01: TCE vs. others; ### p < 0.001: TCE vs. others. TCE: trichloroethylene; Nar: naringenin; LDH: lactate dehydrogenase.
The pre- and posttreatment protective effects of Nar were also assessed by MTT, demonstrating marked protection of epidermal cells against varied concentrations of TCE in the pretreatment of Nar (Np + TCE) at 10 µM dose. However, posttreatment also showed some resuscitating effects of Nar, but it was not as effective when compared with the pretreatment of Nar (Figure 1(b)). These findings were further substantiated by the estimation of LDH release (Figure 1(c)). LDH activity was observed to be significantly increased in TCE exposed cells (p < 0.001) and Nar pretreatment effectively reversed LDH (p < 0.001) release.
Effect of TCE and Nar on redox enzymes
Figure 2(a) shows the marked depletion in GSH level after TCE exposure (p < 0.001). The pretreatment (p < 0.001) protocol was more successful in restoring the GSH level than the post- and co-treatment (p < 0.05). Significant decrease in the activity of catalase was also observed due to TCE exposure (p < 0.001), and Nar pretreatment showed significant restoration (p < 0.001; Figure 2(b)). SOD activity was also found to be decreased in the TCE exposed cells (p < 0.001), which was normalized after Nar (p < 0.05; Figure 2(c)).

(a) Decrease in the levels of GSH (p < 0.001) post TCE treatment and the pretreatment of Nar (10 µM) was more successful in restoring the change to the normal level in comparison with post- and co-treatment. (b) Significant decrease in the catalase activity of HaCaT cells after TCE treatment and subsequently significant induction of the activity was done through pretreatment of Nar. (c) Another important free radical scavenging enzyme, that is, SOD. There was significant decrease SOD activity following TCE treatment and Nar treatments were able to significantly induce its activity in the HaCaT cells. ***p < 0.001: control vs. TCE; # p < 0.05: TCE vs. others. ## p < 0.01: TCE vs. others. ### p < 0.001: TCE vs. others. GSH: reduced glutathione; TCE: trichloroethylene; Nar: naringenin; SOD: superoxide dismutase.
Effect of TCE and Nar on reactive intermediates and signaling
DCFC-DA was used to measure intracellular ROS, and it was found that there was a massive generation of intracellular ROS due to TCE exposure (p < 0.001; Figure 3(a)). A similar kind of the ROS generation was noticed after H2O2 treatment also (Figure 3(a)). Along with ROS, RNIs (p < 0.001) also increased significantly (Figure 3(b)). In the same set of experiments, we pretreated cells with the BAPTA-AM (Ca2+ chelator) and a challenge by TCE failed to increase the ROS generation. These results pulsed us toward the role of Ca2+ in the TCE-mediated cytotoxicity and ROS generation in HacaT cells. In order to strengthen the claim, we studied the intracellular Ca2+ release in the cells following TCE treatment. Surprisingly, TCE led to a massive Ca2+ burst in the cells (p < 0.001; Figure 3(d)). In another set of experiments, it was noticed that H2O2 also leads to increasing ROS generation with increasing dose of H2O2, but this elevation in the intracellular ROS was not dependent on Ca2+ flux as shown in Figure 3(c). Abrupt increase in the intracellular Ca2+ can lead to the signal transduction activation by protein kinases, therefore, we evaluated PKC activity following TCE challenge and found that there was significant increase in its activity due to TCE exposure (p < 0.001; Figure 3(e)). The signal transduction cascade initiated by the TCE was well dampened by the Nar pretreatment. The posttreatment of Nar was also effective but to a lower extent. Therefore, Nar pretreatment might prevent the Ca2+ burst and associated toxicity after TCE administration in HaCaT cells.
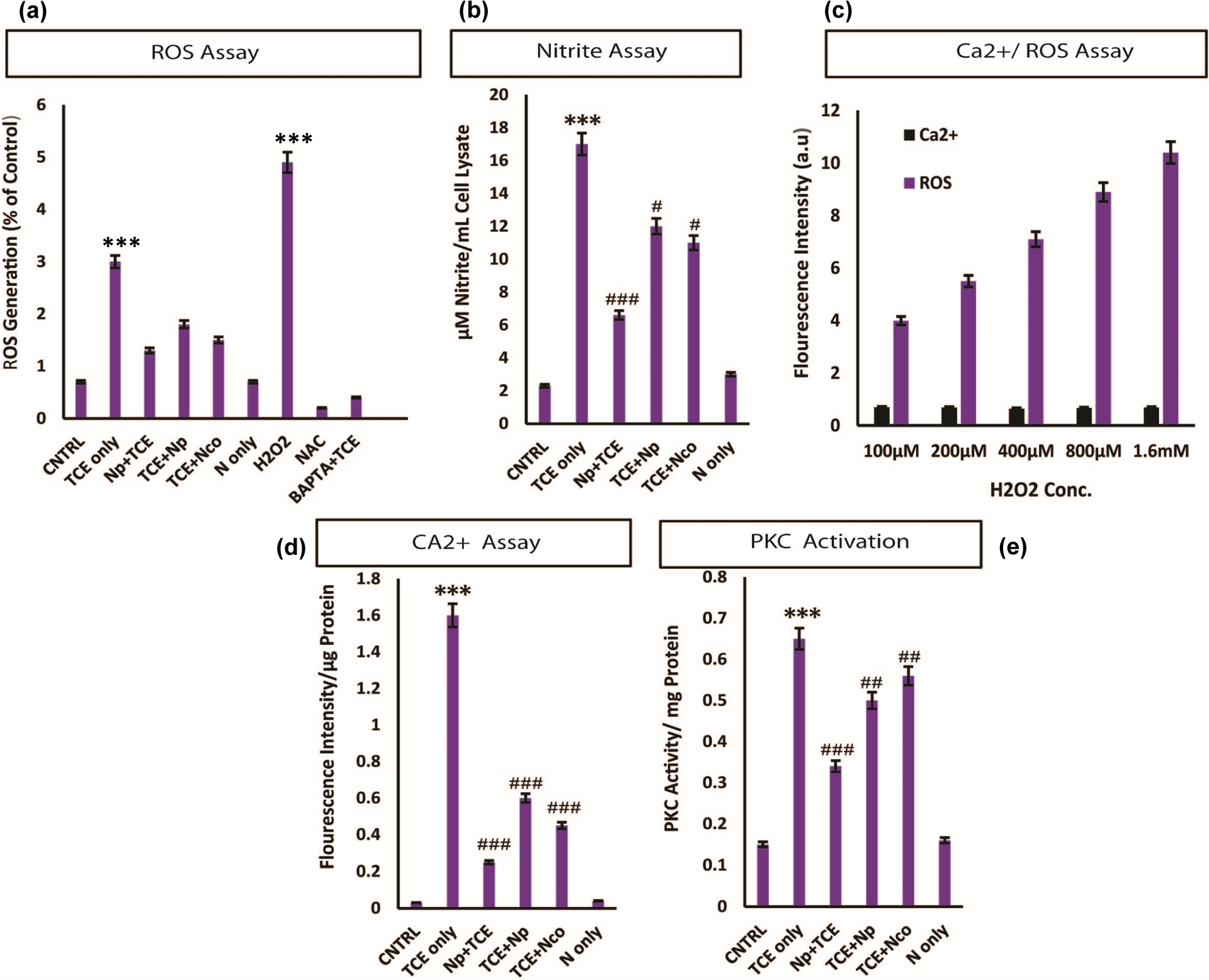
ROS generation in HaCaT cells is Ca dependent. Significant increase in the ROS was observed due to TCE (a) exposure, which was quenched by the BAPTA-AM pretreatment. NAC was used as negative control and H2O2 was used as positive control. TCE exposure also lead to a sudden increase in the RNIs (b). (c) HaCaT cells were challenged with linear increasing dose of H2O2 leading to the linear rise in free radical generation with no effect on the intracellular free Ca2+. (d) TCE treatment leads to the rapid calcium flux. TCE exposure leads to the PKC activation. ***p < 0.001: control vs. TCE; # p < 0.05; ## p < 0.01; ### p < 0.001: when compared with TCE; NS: non significant. ROS: reactive oxygen species; TCE: trichloroethylene; BAPTA-AM: 1,2-bis(O-aminophenoxy)ethane-N,N,N′,N′-tetracetic acetomethyl ester; NAC: N-acetyl cysteine; H2O2: hydrogen peroxide.
Effect of TCE and Nar on COX-2 and NF-κB expression
Expression of NF-κB and COX-2 proteins were analyzed by the immunocytochemical method. We observed a massive increase in the COX-2 expression following TCE treatment, suggesting an inflammatory response in the cells (Figure 4(a)). The initiation of the signal transduction also leads to the activation of the transcription factor NF-κB (Figure 4(b)), which can itself initiate the death cascade. However, Nar was effective in downregulating the signal, which can be a direct effect on the NF-κB or because of the reduced cell stress. Particularly, the pretreatment of Nar showed significant protection but posttreatment and co-treatment regimen was not as successful when compared with the pretreatment of Nar against TCE-induced COX-2 expression.
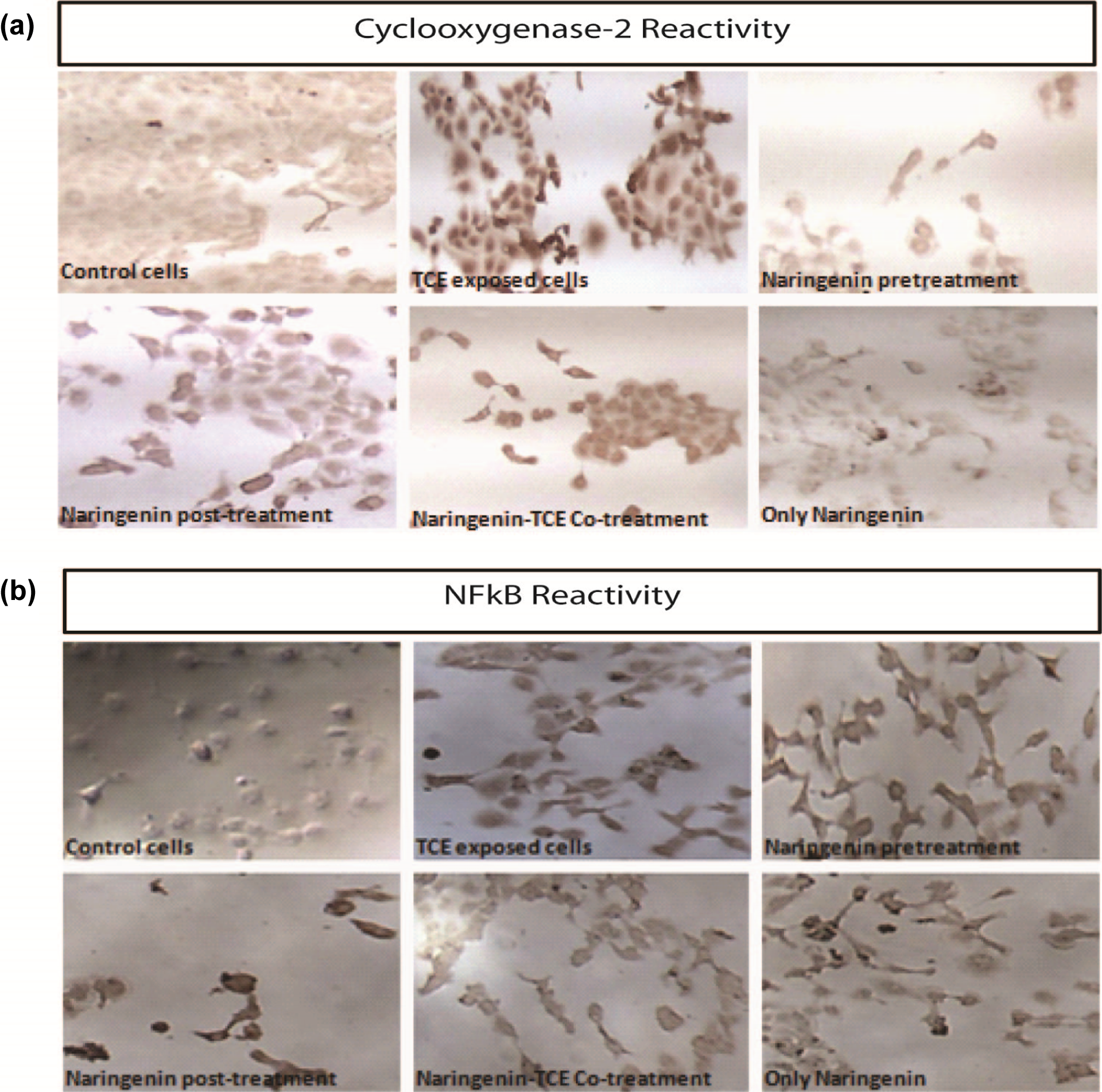
(a) Representative microphotographs of immunocytochemical localization of COX-2 proteins in HaCaT cells exposed to TCE and Nar treatment regimen. COX-2: cyclooxygenase-2; TCE: trichloroethylene; Nar: naringenin.
Effect of TCE and Nar on the HaCaT cell cycle
PI was used as a chelating agent to analyze the cell cycle mechanism using fluorescence-activated cell sorting (FACS). Figure 5 shows that less than 10% cell population was sub-diploid in the control cells, while majority of the cells were in S phase and G phase. TCE-exposed cells showed significantly higher number of cellular population (>38%), which has no growth (arrested cells). Nar pretreatment rescued the cells from TCE-induced growth arrest by 27% and posttreatment by 17% and co-treatment by 16%.

Propidium iodide was used to study healthy and apoptotic cell population by FACS. The figure shows less than 10% cell population undergoing apoptosis and cell death in normal control cells, TCE-exposed cells shows significantly higher number of cellular population undergoing apoptosis (>38%). Nar pretreatment rescued the cells from TCE induced cell death by 27% and posttreatment by 17% and co-treatment by 16%. FACS: fluorescence-activated cell sorting; TCE: trichloroethylene; Nar: naringenin.
Effect of TCE and Nar on MMP and cell death
TCE caused marked depolarization of mitochondrial membranes designated ΔΨm, which was evident from the graph showing quantitative ratio of red to green fluorescence. The TCE-exposed cells showed significant decreased ratio of red to green fluorescence (p < 0.001), which was successfully intercepted and restored by Nar treatments. The pretreatment was comparatively more efficient (p < 0.001) than post and co-treatment (Figure 6(a)) regimen.
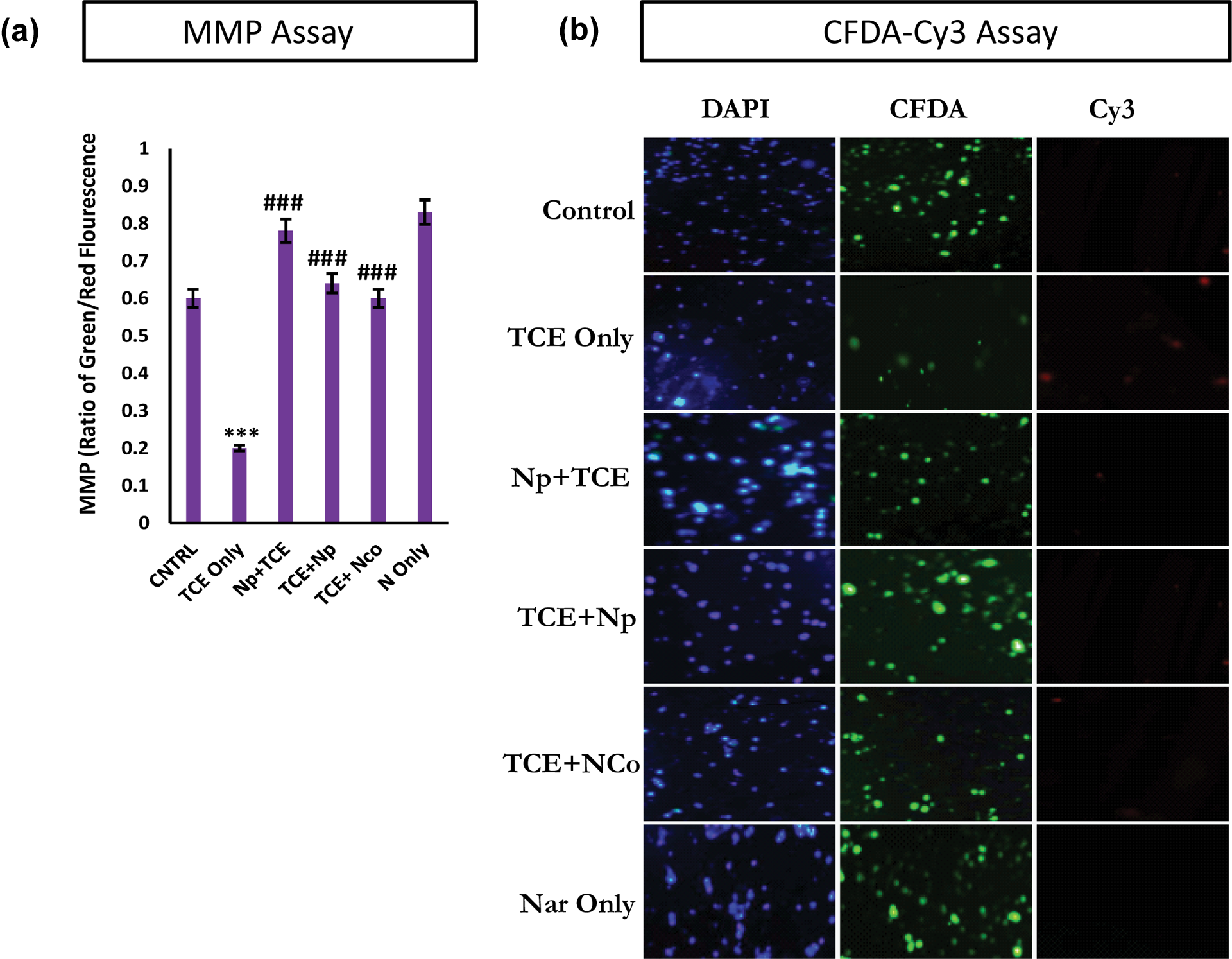
(a) The ratio of red to green (MMP). Significant decrease was observed in the TCE-treated group and was restored with the Nar treatments. (b) The FCDA-Cy3 apoptosis assay. TCE-treated cells show weak FCDA fluorescence but strong Cy3 signal showing significant amount of apoptosis induction in the HaCaT cells. Nar pretreatment was able to significantly prevent the apoptosis in comparison to post- and co-treatment against TCE toxicity. MMP: mitochondrial membrane potential; TCE: trichloroethylene; Nar: naringenin;
Annexin-Cy3 (AnnCy3) is a group of homologous protein that binds to phosphatidylserine that is present in the outer part of the plasma membrane of cells. This interaction is observed as red fluorescence and designates cell demise. 6-CFDA is used to measure viability. It is converted to 6-CF due to hydrolyzing action of esterases. This appears as green fluorescence. There was marked increase in the red fluorescence in cells due to TCE exposure. The Nar treated groups were able to decrease the red fluorescence and subsequently increased the green fluorescence. The pretreatment of Nar was a more successful preventive strategy than the post and co-protection of Nar (Figure 6(b)).
Effect of TCE and Nar on the integrity of genomic DNA
Single-cell gel electrophoresis was used to ascertain the DNA damage because of TCE exposure. Comet assay pictograph, tail length (TL), TD, and OTM parameters were used to assess the degree of DNA damage and genotoxicity. Control cells showed normal intact cell membrane, while TCE-exposed cells showed the clear disintegration of cell membrane and comet tail formation by the damaged DNA. The TL, TD, and OTM show significantly higher values when compared with the control cells. Ethyl methyl sulfone was used as positive control and DMSO as negative or vehicle control. Nar treatments were used to protect the DNA from TCE-induced damage. Pretreatment of Nar was found to significantly protect cells from TCE-induced DNA damage as observed from the pictograph and the vital parameters to assess the extent of DNA damage, namely, TL, TD, and OTM, were all significantly lowered in the Nar treatment groups, showing maximum efficacy in pretreatment group. TL, TD, and OTM were found to be significantly higher in TCE-exposed cells (p < 0.001) when compared with the control cells (Figure 7).

Single-cell gel electrophoresis assay for DNA .The data were analyzed using KOMET 6.0 software. Data in the tabular form represent the TL of the comet under different treatment schemes. TL: tail length.
Discussion
With increasing industrialization, wide array of chemicals have crept in to our normal life. 42 Several studies have highlighted the target organ-specific toxicity of industrial chemicals, especially solvents. 43 TCE can lead to toxic alterations by modulating the cellular environment, cellular receptors, or the nucleic acid integrity. This study shows the cytotoxic events involved in the cellular demise after TCE treatment and its effective prevention by Nar.
Previous findings have suggested the cytotoxicity of HepG2 cells by exposure to TCE 44 through the introduction of the cellular free radical stress and genomic DNA fragmentation. In human keratinocytes, TCE has been demonstrated to diminish the ATP synthesis regulated by the loss of the MMP. 45 It appears that large amount of ROS production is the main cause of TCE-induced cellular death. This study is able to conclude through various parameters that TCE-induced cell death follows precise pathway of increase in Ca2+ and PKC activation leading to upregulation of NF-κB and COX-2 and finally leading to DNA damage-induced cell death.
It was observed that TCE decreases the cellular viability of the HaCaT cells, which was evidenced by the mitochondrial MTT reduction. Subsequently, depletion of major antioxidative enzymes was also observed due to TCE exposure. Nar treatments were able to restore these levels.
Numerous studies have suggested high levels of intracellular Ca2+ can activate numerous molecular receptors and targets involved in the cell death. 46,47 Apart from these, cellular oxidative stress has also been associated with the rise of free Ca2+. 48 We were interested to know whether the free radical generation (ROS) induced by the TCE exposure was Ca2+ dependent and mediated by the activation of PKC. The increased ROS generation was seen to be dependent on the flux of free Ca2+ and was actually inhibited by the pretreatment of the Ca2+ chelator, BAPTA-AM. When the cells were challenged with H2O2, there was no significant increase of intracellular Ca2+ levels although intracellular ROS increased in dose-dependent manner. So it was evident that TCE leads to the increase in free Ca2+ inside the cells. Several studies suggest the role of PKC in the rise of the free Ca2+ ions under the influence of the environmental toxicants and consequent oxidative stress resulting in cell death. This assumption was tested, and it was found that the rise in the intracellular free Ca2+ after TCE exposure led to the activation of PKC. All these results suggest alteration of the cellular environment or activation of specific receptors by TCE to increase the intracellular free Ca2+, which leads to the acute free radical production. However, the pretreatment with Nar effectively reduced the free Ca2+, thereby preventing free radical generation.
The generation of free radicals of oxygen or nitrogen directly affects the balance of the cellular antioxidants and may lead to the degradation of the genomic DNA. Our results support the assumption and HaCaT cells displayed reduced enzymatic as well as nonenzymatic antioxidants after TCE treatment. Moreover, the TCE exposure elevated the cellular nitrite content, which can potentiate the toxicity by depletion of the GSH-dependent redox buffering. In addition, elevated nitrite can lead to the nitration of essential proteins rendering them inactive along with the shuttling of the cytoplasmic Bax to mitochondria, thereby changing the permeability of mitochondrial membranes.
We also checked the expression of COX-2 and NF-κB to ascertain the involvement of these proteins in the toxicity of TCE and observed that there was strong reactivity to COX-2 expression, suggesting that toxicity of TCE inflicts strong inflammatory signal transduction pathway. The expression of NF-κB was noticeably higher in the TCE-exposed cells and posttreatment cells. It should be noted that NF-κB leads to the transcriptional activation of stress-responsive elements that can lead to cellular demise. Nar pretreatment was observed to be an efficient strategy to decrease the protein expression in the cells. The increased expression of COX-2 and NF-κB due to TCE exposure further reveals the inflammatory mechanism of TCE-mediated toxicity in HaCaT cells.
We used a mitochondrial probe, JC-1, a dual fluorescence cationic dye to evaluate the change in membrane permeability and loss of potential (MMP) following TCE. The results confirmed the membrane fluidity change and loss of MMP following TCE exposure in HaCaT cells. MMP is involved in various viability-associated molecular pathways like cellular respiration and programmed cell death. 49,50 Loss of MMP triggers the decline of the cellular ATP production and secretion of several proapoptotic factors like cytochrome-C into the cytoplasm and trigger downstream targets of apoptosis/necrosis.
Finally, in order to validate whether exposure to TCE was able to propagate apoptosis, AnnCy3 cytotag was used to evaluate pre-genomic events of apoptosis. TCE-exposed cells showed significantly increasing the number of cells undergoing apoptosis revealed by significantly increased annexin V positive, 6-CFDA-positive cells, and eliminating the possibility of necrosis (annexin V positive and 6-CFDA negative). When the cells after TCE exposure were sorted for DNA content, a significantly higher number of nuclei (48%) displayed a sub-diploid peak, characteristic of apoptotic cells. However, unexposed cells showed only 10% of sub-diploid genome. These results pointed to the fragmentation of the diploid genome following the TCE exposure. We employed very sensitive single-cell gel electrophoresis programs to detect DNA fragmentation in TCE exposed cells.
We recorded a significant amount of DNA damage evidenced by increased OTM and decreased head DNA in the TCE-treated cells, which was compared with the positive control ethyl methyl sulfone. Therefore, Ca burst after solvent exposure to naive cells might be considered as a general physiological mechanism behind the generation of cellular stress and finally apoptosis.
Conclusion
In summary (Figure 12), TCE-mediated cellular toxicity HaCaT cells involves upregulation of multiple molecular pathways. The free radicals generated were pioneer signal in regulating other downstream targets of inflammation and cell death. We are reporting for the first time the involvement of Ca2+ burst and subsequent activation of PKC in the toxicity of TCE and also that there is pronounced upregulation of NF-κB and inflammatory enzyme COX-2.
The study warrants the need for validation of the cellular interaction of TCE and many other organic solvents in order to establish a base for understanding the patterns of toxicities induced by these chemicals and highlights a natural molecule that can be used to limit the adverse effects on cells with similar cellular interaction and consequences.
Footnotes
Acknowledgments
We are thankful to the University Grants Commission of the Government of India, New Delhi, for providing a Senior Research Fellowship to FA during the tenure of the study. Dr Sultana also acknowledges kind help of Dr Alok Dhawan Scientist F Indian Institute of Toxicological Research for allowing FA to perform comet assay in his laboratory.
Conflict of interest
The authors declared no conflicts of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.