
Abstract
Background
One of the human and animal models of migraine is the systemic administration of the nitric oxide donor (NO) nitroglycerin (NTG). NO can provoke migraine-like attacks in migraineurs and initiates a self-amplifying process in the trigeminal system, probably leading to central sensitization. Recent studies suggest that the endocannabinoid system is involved in nociceptive signal processing and cannabinoid receptor (CB) agonists are able to attenuate nociception in animal models of pain.
Aim
The purpose of the present study was to investigate the modulatory effects of a CB agonist anandamide (AEA) on the NTG-induced expression of transient receptor potential vanilloid type 1 (TRPV1), neuronal nitric oxide synthase (nNOS), nuclear factor kappa B (NF-κB), cyclooxygenase-2 (COX-2) and kynurenine aminotransferase-II (KAT-II) in the upper cervical spinal cord (C1–C2) of the rat, where most of the trigeminal nociceptive afferents convey.
Methods
A half hour before and one hour after NTG (10 mg/kg) or placebo injection, adult male Sprague-Dawley rats (n = 44) were treated with AEA (2 × 5 mg/kg). Four hours after placebo/NTG injection, the animals were perfused and the cervical spinal cords were removed for immunohistochemistry and Western blotting.
Results and conclusion
Our results show that NTG is able to increase TRPV1, nNOS, NF-κB and COX-2 and decrease KAT-II expression in the C1–C2 segments. On the other hand, we have found that AEA modulates the NTG-induced changes, thus it influences the activation and central sensitization process in the trigeminal system, probably via CBs.
Introduction
Migraine belongs to the group of primary headaches, and it is one of the most abundant neurological syndromes, which affects 15% of the European population (1). Despite intensive research, the exact pathomechanism of the disorder is not fully understood, but it is well known that activation and sensitization of the trigeminal system is essential during the attack (2). Continuous activation of peripheral trigeminal afferents leads firstly to peripheral (first-order) sensitization. Sustained inputs can lead to second-order, and ultimately to third-order central sensitization (3). Previous data have shown that after the onset of central sensitization during the migraine attack, the acute treatment becomes less effective (4), although this theory has been contested recently, stating that the severity of headache might be a better indicator than the symptoms of sensitization (5,6); it is generally accepted that the latter plays an essential role in the genesis of migraine (7).
One of the models of migraine is the systemic administration of nitroglycerin (NTG), generating a short-lasting headache in humans via the rapid vasodilatory effect of nitric oxide (NO) followed by a typical migraine without aura in migraine patients (8).
In rats, NTG is able to increase c-Fos-immunoreactivity in the caudal trigeminal nucleus (TNC) suggesting the activation of the second-order trigeminal neurons there (9). NO is also involved in the central sensitization process in the trigeminal system (10), probably acting via the activation of trigeminal Aδ and C fibers, since the destruction of the latter with capsaicin abolishes the effect of NTG (11).
Cannabinoid receptors (CBs) are G-protein-coupled receptors that have two subtypes: CB1 and CB2. CB1 is present in the central nervous system, liver and lung, and CB2 is expressed primarily in the immune system (12).
Animal models of pain have shown that fluctuations in the endocannabinoid levels in the nervous system are related to pain processing and antinociception (13). CB1 is present in the trigeminal ganglion (TG) and on the axon terminals of primary sensory neurons in the nociceptive areas in the spinal cord in rats (14). CB1 is able to inhibit the responses of trigeminal neurons with A- and C-fiber inputs from the dura mater (15), pointing to the importance of the endocannabinoid system in pain processing (16).
Although the psychoactive properties of cannabinoids (17) restrict their therapeutic application, the interactions between the endocannabinoid system and pain mediation is intensively studied in several laboratories.
N-Arachidonylethanolamide or anandamide (AEA) is the first discovered endocannabinoid, and is an agonist of CBs and transient receptor potential vanilloid type 1 (TRPV1), which is a nonselective cation channel activated by numerous stimuli, such as heat and vanilloids (18,19). AEA has vasodilatory actions (20) that are not mediated by CBs (21). It is well known that AEA is able to reduce NTG-induced hyperalgesia and c-Fos expression in the TNC in rats (22), which means that AEA is capable of modulating the activation of the trigeminal system.
TRPV1 is present in the spinal cord and is considered as a molecular integrator of chemical and physical stimuli that elicit pain (23). In addition, NO donors can activate TRPV1 resulting in an increase of intracellular calcium concentration in different cell types (24,25), which suggests that TRPV1 may be modulated by NO.
NO is synthesized from arginine by nitric oxide synthase, a neuronal isoform of which (nNOS) has an outmost importance in nociception and sensitization and is present in the trigeminal system (26). NO donors may trigger a self-amplifying process at the level of central projection site of the trigeminal system by increasing endogenous NO synthesis, which might be relevant in the central sensitization phenomenon (7).
Nuclear factor kappa B (NF-κB) has a crucial role in the inflammation process by controlling many genes including cytokines. Several studies have shown that proinflammatory cytokines contribute to the development of pain and hyperalgesia (27). Cyclooxygenase-2 (COX-2) is present in the dorsal horn of the spinal cord too and it has a substantial role in the processing of pain (28). Gao and Duan have found that COX-2 expression increased in the TNC after orofacial nociception (29).
Kynurenine aminotransferase-II (KAT-II) is a key enzyme in the kynurenine pathway that converts L-kynurenine (L-KYN) to kynurenic acid (KYNA), a known ionotropic glutamate receptor antagonist molecule (30) that can also block α-7-nicotinic acetylcholine receptors (31). In an NTG-induced animal model of migraine L-KYN, KYNA and KYNA analogs inhibited trigeminal activation and sensitization (32–34).
Thus modulatory effects of cannabinoids can be suggested on trigeminal activation both on peripheral and central levels. In the present paper we studied the effect of NTG injection on the TRPV1, nNOS, NF-κB, COX-2 and KAT-II expression levels in the superficial laminae of C1–C2 and its modulation by the CB agonist AEA.
Materials and methods
Animals
The procedures used in this study followed the guidelines for the Use of Animals in Research of the International Association for the Study of Pain and the directive of the European Economic Community (86/609/ECC). They were permitted by the Committee of the Animal Research of University of Szeged (I-74-12/2012) and the Scientific Ethics Committee for Animal Research of the Protection of Animals Advisory Board (XI./352/2012). Forty-four adult male Sprague-Dawley rats weighing 200–250 g were used. The animals were raised and maintained under standard laboratory conditions with tap water and regular rat chow available ad libitium on a 12-hour dark-12-hour light cycle.
Drug administration
The animals were divided into four groups (n = 6 per group for immunohistochemistry, n = 5 per group for Western blot analysis). The animals in the first group, called the placebo group, received only the vehicle solution (physiological saline) as treatment. In the second group, the rats were treated with an intraperitoneal injection of NTG (10 mg/kg bodyweight, Pohl Boskamp). In the third and fourth groups, the animals received intraperitoneal AEA (2 × 5 mg/kg bodyweight, Sigma Aldrich) a half hour before and one hour after the placebo or NTG treatment. Greco and colleagues have demonstrated that single 20 mg/kg doses of AEA before NTG administration is able to reduce NTG-induced c-Fos expression in the TNC (22). Since in our other running experiments (not reported here) we obtained results showing the efficacy of AEA at lower dosages, we used 2 × 5 mg/kg of AEA in this context using two injections because of the short half-life of the drug (35). AEA was dissolved in physiological saline. In the case of the first and second groups, animals were treated with physiological saline instead of AEA.
Immunohistochemistry
Four hours after the placebo/NTG injection, the rats were perfused transcardially with 100 ml phosphate-buffered saline (PBS, 0.1 M, pH 7.4), followed by 500 ml 4% paraformaldehyde in phosphate buffer under chloral hydrate (0.4 g/kg bodyweight) anesthesia. The C1–C2 segments of the cervical spinal cord between −5 and −11 mm from the obex were removed and postfixed overnight for immunohistochemistry in the same fixative. After cryoprotection, 30 µm cryostat sections were cut and serially collected in wells containing cold PBS. The free-floating sections were rinsed in PBS and immersed in 0.3% H2O2 in methanol or PBS for 30 minutes. After several rinses in PBS containing 1% Triton X-100, sections of C1–C2 were kept overnight at room temperature in anti-TRPV1 antibody (Santa Cruz, s.c.28759) at a dilution of 1:500, or for two nights at 4℃ in anti-nNOS antibody (EuroProxima, 2263B220-1) at a dilution of 1:5000, or for two nights at 4℃ in anti-NF-κB antibody (Abcam, ab97726) at a dilution of 1:100. The immunocytochemical reaction was visualized by the Vectastain Avidin-Biotin kit of Vector Laboratories (PK6101), and nickel ammonium sulfate-intensified 3,3’-diaminobenzidine. The specificity of the immune reaction was controlled by omitting the primary antisera.
Western blot analysis
Four hours after the placebo/NTG injection, the animals were perfused transcardially with 100 ml PBS and the dorsal horns of the C1–C2 segments were extracted. Until the measurements, the samples were stored at −80℃. The samples were sonicated in ice-cold lysis buffer containing 50 mM Tris-HCl, 150 mM NaCl, 0.1% igepal, 0.1% cholic acid, 2 µg/ml leupeptin, 2 mM phenylmethylsulphonyl fluoride, 1 µg/ml pepstatin, 2 mM ethylenediaminetetraacetic acid (EDTA) and 0.1% sodium dodecyl sulfate (SDS). The lysates were centrifuged at 12,000 revolutions per minute (RPM) for 10 minutes at 4℃ and supernatants were aliquoted and stored at −20℃ until use. Protein concentration was defined with BCA Protein Assay Kit using bovine serum albumin as a standard. Prior to loading, each sample was mixed with sample buffer, and denaturated by boiling for 3 minutes. Equal amounts of protein samples (20 µg/lane) were separated by standard SDS polyacrylamide gel electrophoresis on 10% Tris-Glycine gel and electrotransferred onto Amersham Hybond-ECL nitrocellulose membrane (0.45 µm pore size). We used the Page Ruler Prestained Protein Ladder (10–170 kDa) to define approximate molecular weights. Following the transfer, membranes were blocked for one hour at room temperature in Tris-buffered saline containing Tween 20 (TBST) and 5% nonfat dry milk powder. Then, they were incubated in TBST containing 1% nonfat dry milk and nNOS antibody (BD Biosciences, 610308, dilution: 1:2000, incubation: overnight at room temperature), or COX-2 antibody (Proteintech, 12375-1-AP, dilution: 1:1000, incubation: overnight at room temperature), or KAT-II antibody (Santa Cruz, sc-67376, dilution: 1:10,000, incubation: overnight at room temperature) or β-actin antibody (Calbiochem, CP01, dilution: 1:100 000, incubation: overnight at room temperature). The next day, the membranes were incubated in TBST containing 1% nonfat dry milk and horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody (Santa Cruz Biotechnology, sc-2030, sc-2031) for two hours at room temperature. Protein bands were visualized after incubation of membranes with the SuperSignal West Pico Chemiluminescent Substrate using Carestream Kodak BioMax Light film.
Data evaluation
All evaluations were implemented by an observer blind to the experimental groups. The detailed methods were described previously (34,36,37).
Immunohistochemistry (TRPV1, nNOS, NF-κB)
The photomicrographs of the stained sections of C1–C2 were taken using a Zeiss AxioImager microscope supplied with an AxioCam MRc Rev. 3 camera (Carl Zeiss Microscopy, Jena, Germany).
The area covered by TRPV1-immunoreactive fibers and nNOS-immunoreactive cells was determined by Image Pro Plus 6.2® image analysis software (Media Cybernetics). After image acquisition, the laminae I–II in the dorsal horn were defined manually as areas of interest and a threshold gray level was validated with the image analysis software as described in an earlier study (32,36). The program calculated the area innervated by the immunoreactive fibers and cells as the number of pixels with densities above the threshold; the data were expressed as area fractions (%) of the corresponding immunolabelled structures.
We used the unbiased optical disector method to calculate the volume densities of the NF-κB-immunoreactive cells (37).
Western blot analysis (nNOS, COX-2, KAT-II)
For densitometric analyses, films were scanned and quantified using Java ImageJ 1.47v analysis software (National Institutes of Health). The results were normalized to β-actin.
Statistical analysis
Statistical analysis of measurements were performed in SPSS Statistics software (version 20.0 for Windows, SPSS Inc) using one-way analysis of variance (ANOVA) followed by the Tukey or Tamhane post hoc test depending on variances of data, with p < 0.05 taken as statistically significant. Group values are reported as means ± SEM.
Results
NTG induced an increase in TRPV1 expression in the C1–C2, and AEA inhibited this phenomenon
On transverse sections of the C1–C2 segments, there were abundant TRPV1-positive fibers in the superficial layers of the dorsal horn. The TRPV1-immunoreactive area in the NTG-treated group was significantly higher compared to the placebo-treated group (p < 0.05). The NTG-induced increase was attenuated by treatment with AEA (p < 0.05) (Figure 1).
(a) Representative photomicrographs of the TRPV1 expression in the C1–C2 segments. (b) Changes in area fractions of TRPV1-immunoreactive fibers in superficial laminae I and II of the C1–C2 segments. In the NTG group, the area covered by TRPV1 was significantly higher than in the placebo group. AEA seemed to block this effect. Bar graph data presented here and in succeeding figures are means ± SEM. Scale bar: 100 µm, *p < 0.05. TRPV1: transient receptor potential vanilloid type 1; IR: immunoreactive; NTG: nitroglycerin; AEA: anandamide.
NTG increased nNOS expression in the C1–C2, and AEA attenuated this effect
On transverse sections of the C1–C2 segments, nNOS-immunoreactive neurons and processes with cytoplasmic staining can be observed in the superficial layers of the dorsal horn. In the NTG group, the area fraction of nNOS-immunoreactive structures was significantly higher than in the placebo-treated group (p < 0.01). AEA treatment resulted in a decrease of nNOS-immunopositive structures (p < 0.001) (Figure 2).
(a) Representative photomicrographs of nNOS expression in the C1–C2 segments. (b) Changes in the nNOS-immunoreactive area fractions. In the NTG group, the area fraction of nNOS-IR structures was increased compared to the placebo-treated group. AEA was able to attenuate this effect. Scale bar: 100 µm, **p < 0.01; ***p < 0.001. nNOS: neuronal nitric oxide synthase; IR: immunoreactive; NTG: nitroglycerin; AEA: anandamide.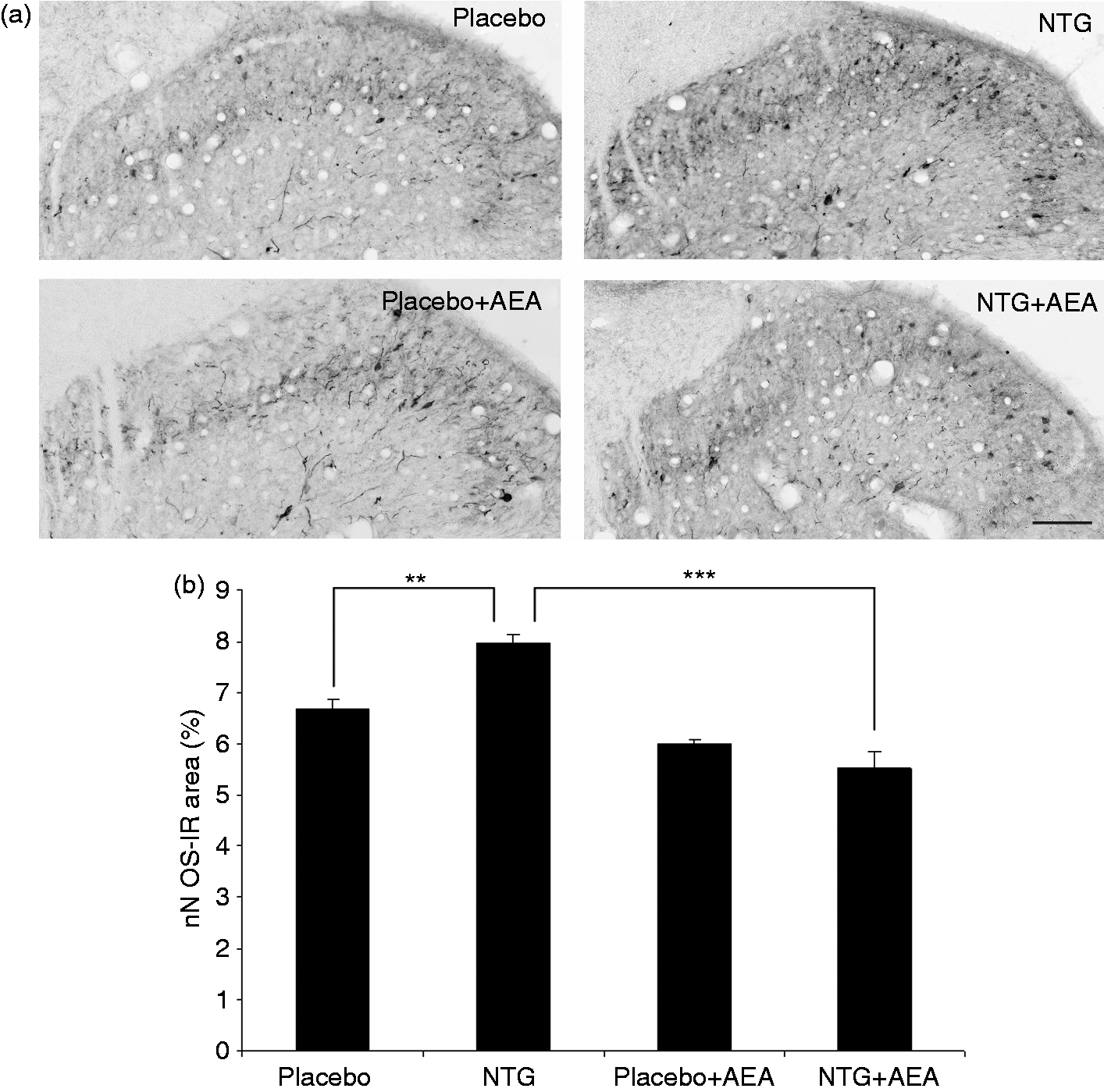
Western blot analysis of the C1–C2 region confirmed the results obtained by nNOS immunohistochemistry. A band characteristic of the nNOS protein was identified at 155 kDa. Densitometric analyses confirmed that the nNOS bands were significantly enhanced (p < 0.01) in C1–C2 after NTG administration as compared with the placebo-treated animals. This effect was blocked by the treatment with AEA (p < 0.05) (Figure 3).
(a) Western blot of nNOS and β-actin expression in the C1–C2. (b) The quantitative analysis shows that in the NTG group, the relative optical density of the nNOS-specific band was significantly higher than in the placebo group. AEA treatment weakened this effect. *p < 0.05; **p < 0.01. nNOS: neuronal nitric oxide synthase; NTG: nitroglycerin; AEA: anandamide.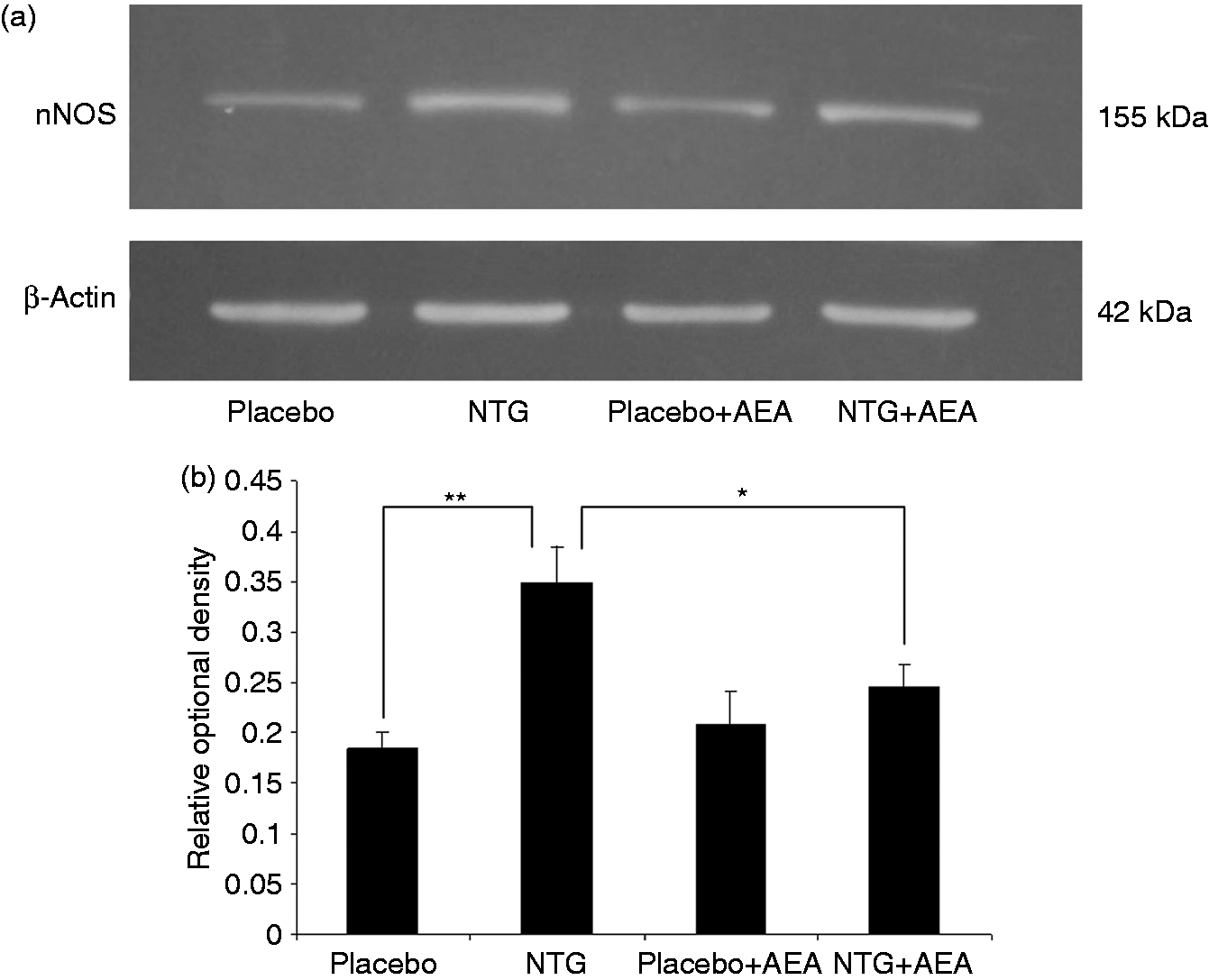
NTG treatment enhanced NF-κB expression, and AEA mitigated this phenomenon
In transverse sections of the C1–C2 region, a high number of NF-κB-positive cells can be seen in the superficial layers of the dorsal horn. In the NTG-treated animals the volume density of the NF-κB-positive cells was significantly higher than in the placebo group (p < 0.05). This value decreased in the AEA-injected group (p < 0.05) (Figure 4).
(a) Photomicrographs of the NF-κB expression in the C1–C2 segment. (b) The diagram shows that NTG treatment resulted in a significant increase in the volume density of NF-κB-immunoreactive cells. This effect was not observed in AEA-injected animals. Scale bar: 50 µm, *p < 0.05. NF-κB: nuclear factor kappa B; IR: immunoreactive; NTG: nitroglycerin; AEA: anandamide.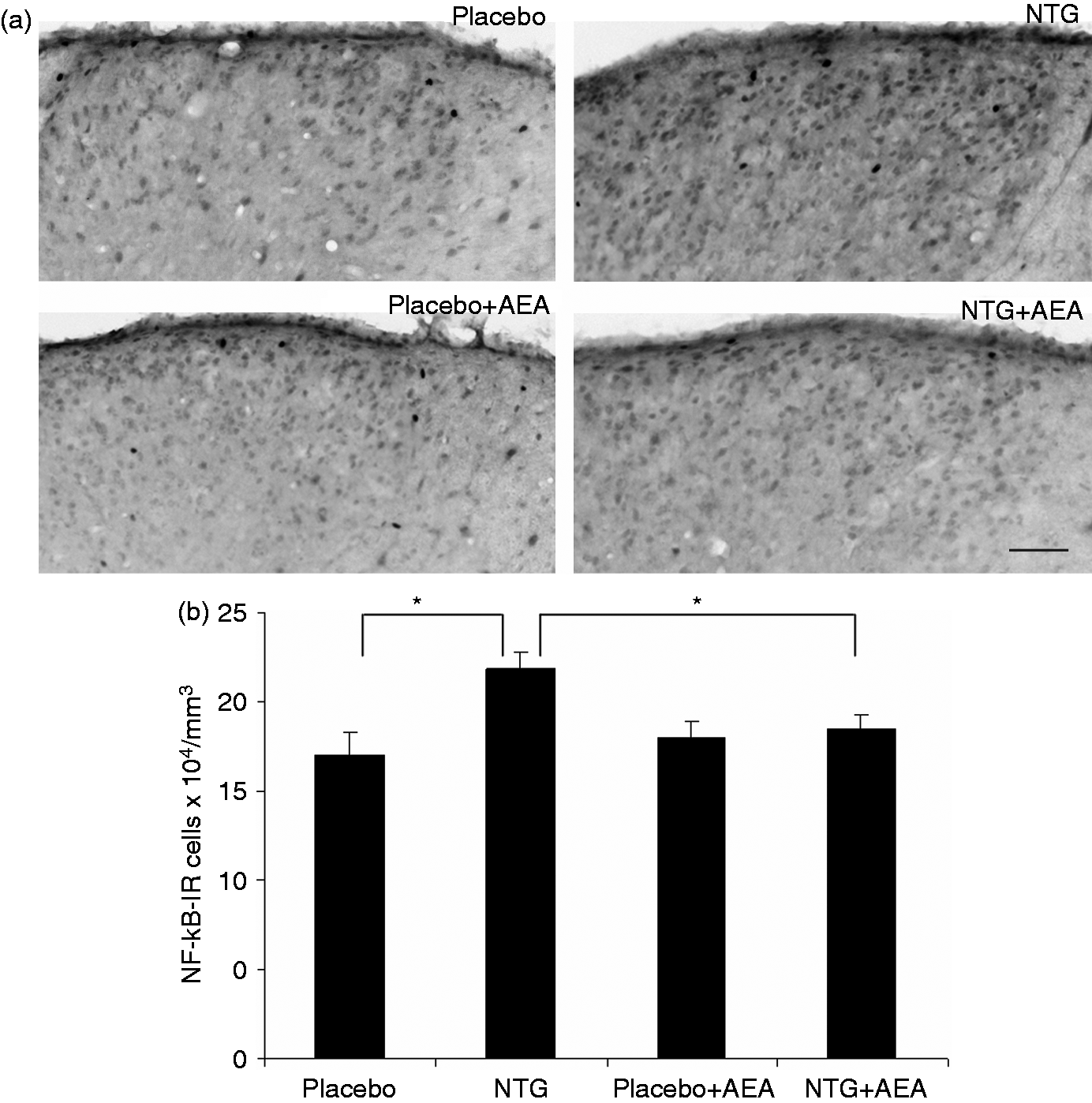
NTG enhanced expression of COX-2 enzyme, and AEA inhibited this action
A band characteristic of the COX-2 protein was identified at 68 kDa in Western blot assay. Densitometric analyses showed that the COX-2 bands were significantly enhanced (p < 0.01) in segments C1–C2 after NTG administration as compared with the placebo-treated animals. The effect of NTG was decreased by the AEA treatment (p < 0.01) (Figure 5).
(a) Western blot of COX-2 and β-actin expression in the C1–C2. (b) Densitometry of the individual bands indicated that in the NTG-treated animals the expression of COX-2 was significantly higher than in the placebo group. No such effect was seen in the AEA-injected group. **p < 0.01. COX-2: cyclooxygenase-2; NTG: nitroglycerin; AEA: anandamide.
NTG decreased KAT-II expression, which was alleviated by AEA
A band characteristic of the KAT-II protein was referred at 60 kDa in Western blot assay. Densitometric analyses confirmed that the KAT-II bands were significantly weaker (p < 0.05) in segments C1–C2 after NTG administration as compared with the placebo-treated animals. This effect was reversed by treatment with AEA (p < 0.05) (Figure 6).
(a) Western blot of KAT-II and β-actin expression in the C1–C2. (b) Quantitative data demonstrate that in the NTG group, the relative optical density of KAT-II is significantly lower than in the placebo group. AEA reduced this effect. *p < 0.05. KAT-II: kynurenine aminotransferase-II; NTG: nitroglycerin; AEA: anandamide.
Discussion
The present data indicate that NTG treatment activates the trigeminal system, and the observed changes can be interpreted as a central sensitization phenomenon. The NTG-induced alterations are reversed by the administration of AEA suggesting the involvement of CBs in this process.
It is generally accepted that the trigeminal pain processing and sensitization is an extremely complex phenomenon involving numerous cellular and molecular components but the associated pathways are not fully known. Figure 7 shows a schematic and simplified flow diagram that indicates that the connections are sometimes indirect and the signaling molecules may modulate the complex response at different target points.
Schematic illustration of the interactions among the NO-influenced molecules involved in the trigeminal pain processing and sensitization showing the possible sites of action for cannabinoids.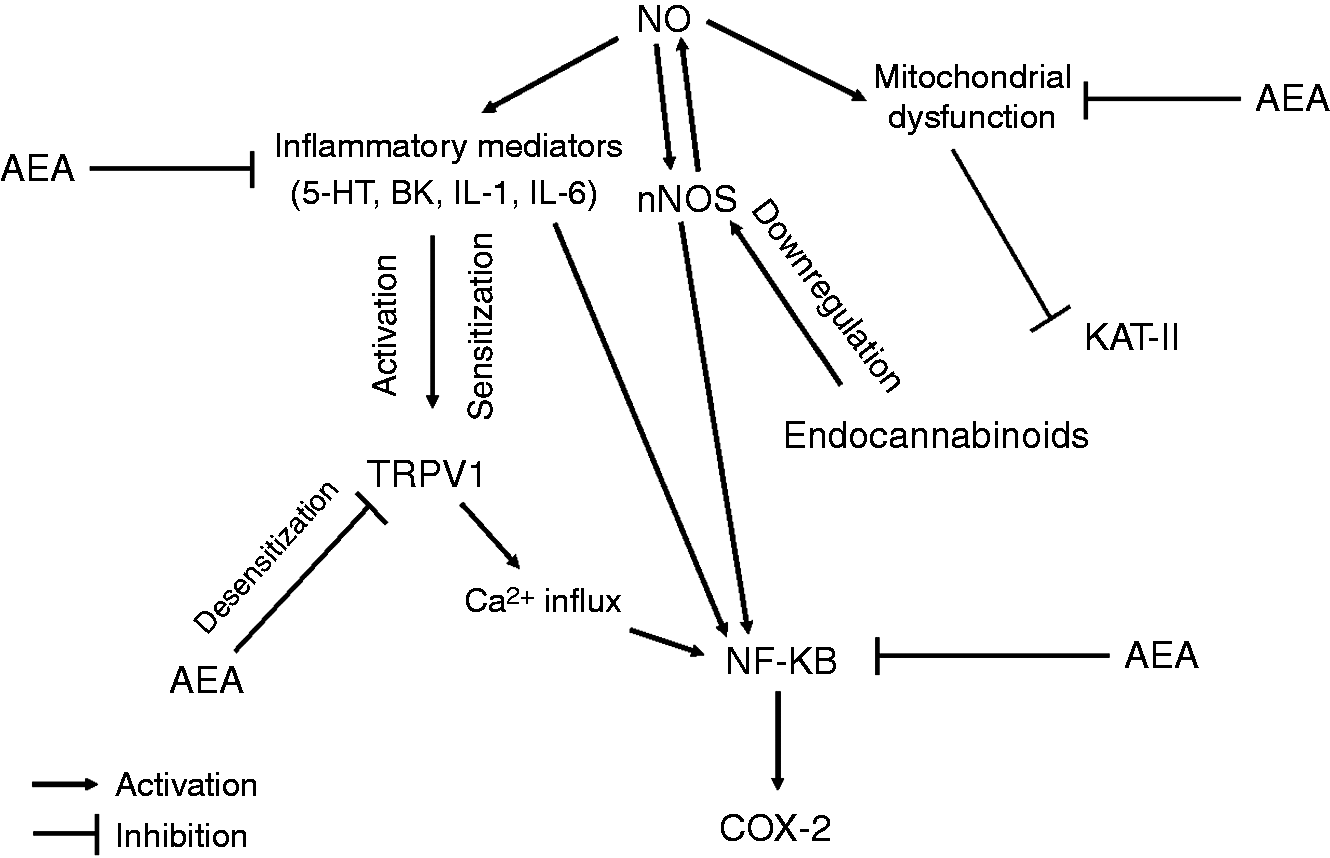
TRPV1
TRPV1 is present in the terminals of primary sensory neurons in the dorsal part of the spinal cord (38) and co-expressed with CB1 (39). Experimental data indicate that TRPV1 contributes to the peripheral sensitization, allodynia and hyperalgesia (40); central blockade of this receptor is able to attenuate central terminal sensitization (41). Our results show that NTG significantly increases TRPV1 expression in the C1–C2 segments of the rat, thus it may be the indicator of sensitization phenomena in the trigeminal system in our experimental setting.
It is well evidenced that NO donors can activate TRPV1 in several cell types (24,42), and in inflammatory pain models increased receptor expression was also reported (43,44). This indicates that the NTG effect on TRPV1 expression is indirect—it may cause neurogenic inflammation (45), and the inflammatory mediators like serotonin (5-HT) and bradykinin (BK) can activate TRPV1 by stimulating trigeminal nociceptive neurons (46). Pro-inflammatory mediators, such as tumor necrosis factor α, interleukin 1 (IL-1), interleukin 6 (IL-6) and BK enhance TRPV1 (47). In the animal model of inflammation, Complete Freud’s adjuvant increases TRPV1 messenger RNA (mRNA) expression in the dorsal root ganglion (48), this also supports the idea that inflammation upregulates TRPV1 expression. To summarize, we may assume that NTG is able to activate TRPV1 mainly via inflammatory mediators. On the other hand, there are research data indicating that systemic administration of calcitonin gene-related peptide (CGRP) increases the expression of TRPV1 in the TG of rats (49), which might also play a role in this process.
Our recent data also show that AEA, a CB receptor and TRPV1 agonist, attenuates the effect of NTG on TRPV1 changes. Activation of ionotropic CBs can result in inhibition of nociceptors and antihyperalgesia and antinociception in certain pain models (50). Intrathecal administration of AEA decreases thermal pain sensitivity and its effect can be altered with the TRPV1 antagonist capsazepine (51). AEA may also cause a desensitization of TRPV1 in skeletal muscle arterioles (52), suggesting that AEA is able to mitigate TRPV1 activity. On the other hand, AEA can inhibit neurogenic, CGRP- and NO-induced dural vasodilation, and this involves pre- and postsynaptic mechanisms (53). A recent report shows that the AEA level changes are able to modulate CGRP mRNA expression after NTG-treatment in human peripheral blood mononuclear cells (54). We do not know exactly the role of TRPV1 in the AEA-modulated sensitization process, but based on the available literature data (15,55), we hypothesize that the role of CB1 is more pronounced than TRPV1 in this context.
nNOS
It is known that nNOS is a key player in nociception (56) and its role in the sensitization cascade is intensively studied. In our present experiment administration of NTG increased the expression of nNOS in the C1–C2, which is in line with earlier results (10). The most probable explanation for this phenomenon is that NO activates small-caliber fibers in the trigeminal system and the increase of nNOS expression in the second-order trigeminal neurons induces a self-amplifying mechanism (10,11). The present results indicate that AEA is able to inhibit this effect. Several studies have shown that there is an interaction between NO and the cannabinoid system, e.g. nNOS and CB1 are co-localized in neurons in lamina II of the spinal cord (57); NTG-induced hyperalgesia is associated with a fluctuation of the endocannabinoid system in various brain areas of rats (22). Our data are in line with the findings of Hillard and colleagues, who reported that CB1 agonists inhibit KCl-induced activation of nNOS in cultured cerebellar granule cells (58). In addition, Carney and colleagues have detected that cannabinoid agonist downregulated nNOS protein and mRNA in neuronal cells (59). These data suggest that NTG is able to generate a sensitization process and AEA inhibits this effect by blocking nNOS.
NF-κB
In our experiment NTG increased NF-κB expression in the superficial layers of the dorsal horn in the C1–C2 segments. A similar effect was reported by Reuter et al., who demonstrated that NTG infusion is able to trigger the activation of NF-κB in dura mater (60). It is not clear how NTG can activate the NF-κB-pathway; it might be related to a direct neuronal effect of NTG, or an indirect effect via dural inflammation (61). Concerning the cellular mechanisms it is important to note that both TRPV1 and nNOS might play a role in this effect. It was shown that Ca2+ influx through TRPV1 may modulate the nuclear translocation and increased the activity of NF-κB (62). On the other hand the increase in nNOS expression is accompanied by increased NF-κB expression and activation (63). Furthermore, Sancho and colleagues have noticed that AEA inhibits tumor necrosis factor-α-induced activation by inhibition of a cytokine-induced cascade (64). In addition, Nakajima and colleagues have found that AEA also blocked lipopolysaccharide-induced activation, suggesting that AEA inhibits proinflammatory mediators by blocking NF-κB activation (65). Tassorelli and her group reported that parthenolide (inhibitor of NF-κB) attenuated NTG-induced c-Fos activation in TNC (66), which indicates that NF-κB may be important in the NTG-induced trigeminal activation and its inhibition is able to modulate the nociceptive process. Our data reconfirm that NTG is able to activate NF-κB, and thus can trigger neurogenic inflammation, which has a key role in sensitization phenomena. Furthermore, we detected that AEA is able to reduce this effect. Endocannabinoids might operate a negative feedback control over the proinflammatory process by suppressing the activation of transcription factors involved in the inflammatory action (67).
COX-2
NO may also cause neurogenic inflammation by increasing NF-κB levels, which may lead to the upregulation of COX-2 in inflammatory pain (68). It is well known that nonsteroidal anti-inflammatory drugs (NSAIDs), which exert their effects through the inhibition of COX-enzymes (69), are effective in the treatment of migraine and tension-type headache (70). In animal studies, it has been shown that COX-2 is involved in the NTG-induced activation and sensitization process of the trigeminal system. Pre-treatment with indomethacin (non-selective COX inhibitor) and NS398 (selective COX-2 inhibitor) reduced the NTG-induced c-Fos (11), nNOS and calmodulin-dependent protein kinase II alpha expression-changes in the TNC (71,72). Tassorelli and colleagues have demonstrated that COX-2 expression is increased in the hypothalamus and caudal brain stem after NTG injection (73), thus COX is one of the mediators of NTG-induced neuronal activation. Furthermore, NO is able to activate COX-enzymes in fibroblasts, probably by an interaction with the iron-hem center of the enzyme (74). It is important to note that AEA is one of the substrates of COX-2 producing prostaglandin and ethanolamids (75). In our study, we have found that AEA is able to inhibit the NTG-induced COX-2 increase. The cellular and molecular background of this effect remains to be determined. Our assumption is that it may be associated with a negative feedback mechanism, but it is possible that after the cleavage of AEA some metabolites may downregulate COX-2 expression. Since AEA is able to reduce cytokine-induced cascade and proinflammatory mediators, it is also possible that the reduction of the inflammatory process is able to downregulate the COX-2 expression.
KAT-II
In our experiments NTG decreased KAT-II expression in the C1–C2 segments, which may indicate that fluctuation of KAT is involved in the NTG-triggered trigeminal activation. Recent studies have shown that migraine is associated with mitochondrial dysfunction (76). It has also been demonstrated that an NO donor is able to downregulate the respiratory chain complex and cause release of the mitochondrial cytochrome C, thus causing a mitochondrial dysfunction (77). The decrease in KAT-II activity might be associated with mitochondrial damage as shown in the striatum of patients suffering in Huntington’s disease (78). This is in line with our earlier data showing that administration of mitochondrial complex II inhibitor 3-nitropropionic acid is able to reduce KAT-II activity in rats (79). On the other hand, NTG may cause neurogenic inflammation and the released pro-inflammatory cytokines can modulate the kynurenine pathway (80), which may also contribute to the change in KAT-II expression. Nevertheless, in another model of trigeminal activation (electrical stimulation of the TG), decreased KAT-immunoreactivity in dural macrophages, Schwann cells and mastocytes was found in rats (81), suggesting a prominent role of this enzyme in the process of trigeminal activation.
In the present experiment, we have detected that AEA is able to mitigate NTG-induced KAT-II expression decrease. AEA and the other endocannabinoid molecule 2-arachidonoylglicerol cause reduction of calcium-induced cytochrome C release from mitochondria and protect mitochondria from cytochrome-mediated damage, which leads to DNA fragmentation and apoptosis (82,83). Taken together, it is possible that NTG may cause mitochondrial dysfunction, and AEA is able to inhibit this effect probably by decreasing cytochrome C release.
Conclusions
In conclusion, our results show that AEA has a modulating effect on central sensitization markers in our experimental setting. These data suggest that the endocannabinoid system plays a significant role in the cellular mechanism of trigeminal sensitization and thus it may modulate the pathomechanism of migraine.
Footnotes
Article highlights
Systemic administration of nitroglycerin is able to activate and sensitize the trigeminal system in rats.
This phenomenon is reflected by alteration of the expression of biological markers (transient receptor potential vanilloid type 1 (TRPV1), neuronal nitric oxide synthase (nNOS), nuclear factor kappa B (NF-κB), cyclooxygenase-2 (COX-2) and kynurenine aminotransferase-II (KAT-II)) of this process.
Anandamide, a cannabinoid receptor agonist, is able to modulate these changes caused by nitroglycerin.
Cannabinoid receptors are involved in the activation and sensitization of the trigeminal system.
Acknowledgement
We are indebted to Mrs Valéria Vékony for histotechnical assistance.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the Hungarian Brain Research Program (grant no. KTIA_13_NAP-A-III/9); the EUROHEADPAIN (FP7-Health 2013-Innovation; grant no. 602633); the TÁMOP-4.2.2.A-11/1/KONV-2012-0052; and the European Union and the State of Hungary, co-financed by the European Social Fund in the framework of TÁMOP 4.2.4.A/2-11-1-2012-0052. Dr Árpád Párdutz was supported by the Bolyai Scholarship Programme of the Hungarian Academy of Sciences.