
Abstract
A short-term 5-day cigarette smoke exposure study was conducted in Fischer 344 rats to identify smoke-induced lung protein changes. Groups of 10 male and 10 female rats at 5 weeks of age were randomly assigned to one of four exposure groups. Animals received filtered air (control) or 75, 200, or 400 mg total particulate matter (TPM)/m3 of diluted Kentucky reference 3R4F cigarette smoke. Nose-only exposures were conducted for 3 hours/day for 5 consecutive days. Mean body weights were significantly reduced only in male rats exposed to 400 mg TPM/m3. Body weight gains were significantly reduced in 200- and 400-mg TPM/m3–exposed males and in all smoke-exposed females compared with controls. Alveolar histiocytosis increased slightly in all smoke exposed-females and 200- and 400-mg TPM/m3–exposed males. Cyclooxygenase-2 staining increased at 400 mg TPM/m3. Matrix metalloproteinase–12 staining of alveolar macrophages and bronchiolar epithelia increased in smoke-exposed animals, especially 400-mg TPM/m3–exposed females. Protein kinase C–α staining increased in macrophages at 200- and 400-mg TPM/m3 doses. c-Jun NH2-terminal kinases staining decreased in smoke-exposed tissues. The identified changed proteins play roles in inflammation, transformation, proliferation, stress activation, and apoptosis.
Introduction
Long-term animal studies for assessment of toxicity and carcinogenesis are not ideal models partly because of the length of time it takes to induce grossly visible tumors. Moreover, significant increases in the numbers of malignant tumors in the respiratory tract have not been seen in rats, mice, dogs, hamsters, or nonhuman primates exposed for long periods to very high concentrations of smoke (Coggins 1998, 2001). Various attempts have been made to develop a novel animal model for smoke-induced lung cancer (Coggins 2001). A new approach has been suggested that focuses on induction of toxicological changes relevant to disease, such as cell proliferation, chronic inflammation, and inhibition of apoptosis (Battershill 2005). Prolonged smoking can cause lung inflammation and destroy alveoli, causing chronic obstructive pulmonary disease (COPD; Fujita and Nakanishi 2007). A number of benign nonpulmonary and pulmonary diseases characterized by chronic inflammation increases the risk of cancer at the affected site. A large body of evidence also suggests that smoking-induced pulmonary inflammation may play an important role in increasing lung cancer risk in smokers (Smith, Perfetti, and King 2006). Animal models displaying tumorigenic responses following exposure to either whole smoke or smoke fractions show elevated rates of cellular proliferation. A relationship between pulmonary inflammation and lung cancer is mechanistically plausible because inflammatory cells secrete activated oxygen species, inflammatory mediators, and proteolytic enzymes that can both damage DNA and lead to increases in reparative cell proliferation rates (Smith, Perfetti, and King 2006).
Biomarkers of effect as well as evaluation of putative carcinogenic mechanisms in rats and mice exposed to tobacco smoke allow detection of early events and their modification by tobacco smoke and are an alternative to performing more and more long-term animal studies (Witschi 2007). Assessment of toxicological changes and candidate biomarkers in short-term animal studies could provide a new approach to evaluate the mechanism(s) of toxin action as well as new assays for product testing. The purpose of the present study was to evaluate any pathological changes as well as changes in selected lung protein amount and staining patterns in nose-only–exposed Fischer 344 rats to mainstream smoke generated from 3R4F Kentucky reference cigarettes at concentrations of 75, 200, or 400 mg total particulate matter (TPM)/m3 for 5 consecutive days. A negative control of filtered air with 0 mg TPM/m3 was compared with the cigarette smoke exposure groups. In this article, reference to smoke treatment of rats refers to cigarette smoke exposure. We sought to evaluate the utility of short-term rodent studies for assessment of cigarette smoke inhalation–induced protein changes. We hypothesized that tissue from cigarette smoke–exposed rats possessed elevated levels of inflammatory mediators, tumor biomarkers, and other proteins associated with lung disease compared with unexposed animals. We expected that proteins associated with inflammation and carcinogenesis would increase upon cigarette smoke exposure. Specifically, we hypothesized that cigarette smoke exposure would induce changes in the following lung proteins: epidermal growth factor-receptor (EGF-R), transforming growth factor–beta (TGF-β, nuclear factor kappa B (NF-κB), cyclooxygenase II (COX-2), matrix metalloproteinase (MMP)–12, c-Jun NH2-terminal kinases (JNK), and protein kinase C (PKC)–α.
Materials and Methods
Test Article
The test article was the mainstream smoke from 3R4F Kentucky reference cigarettes purchased from the University of Kentucky, Lexington. All cigarettes were stored at approximately 4°C (0°C–10°C) in a secure location in their original cartons until use. All cigarettes were conditioned prior to use by storage in an enclosure for at least 48 hours at 22°C ± 1°C and 60% ± 3% humidity.
Animals and Animal Maintenance
Receipt, housing, group assignment, and environment
The study protocol was approved by the Institutional Animal Care and Use Committee at the Illinois Institute of Technology Research Institute (Chicago, IL, USA). Forty-five male and 45 female Fischer 344 rats at 5 weeks of age were received from Charles River Laboratories (Kingston, NY, USA). Animals received water and Certified Rodent Chow 5002 (PMI Nutrition International, Inc., Brentwood, MO, USA) ad libitum, except during exposure periods. Blood samples were collected from the first two surviving male rats per treatment group on study day 5 and analyzed for carboxyhemoglobin (COHb), nicotine, and cotinine. The carboxyhemoglobin levels were measured using an IL-482 CO-Oximeter (Instrumentation Laboratory, Inc., Lexington, MA, USA). The serum nicotine and cotinine levels were measured using a previously described radioimmunoassay procedure (Van Vunakis, Gjika, and Langone 1987).
During nonexposure hours, the animals were housed individually in suspended stainless-steel wire mesh cages equipped with an automatic drinking water supply system and food trays. Following a 9-day quarantine and body weight measurement, suitable animals (90) were randomly assigned to control or treatment groups using a computerized randomization program (RANSD.EXE) constrained by body weight. A total of four housing chambers were used: one for the air control group and three for smoke-exposed groups. Temperature, relative humidity, and air flow were monitored continuously; temperature was maintained between 18°C and 26°C and the relative humidity between 30% and 70%. An automatic 12-hour light-dark cycle was maintained in the exposure and housing chamber laboratories. Three weekdays prior to the first exposure, the animals were conditioned to placement and restraint in the nose-only exposure tubes to reduce stress during the exposure phase. Rats were acclimated in the exposure tubes for periods of 60, 120, and 180 minutes on the three weekdays prior to the first exposure.
Experimental Design
The experimental design is summarized in Table 1 . Groups of 10 male and 10 female rats were assigned to one of four exposure groups. Depending on the group, animals received either filtered air (air control) or 75, 200, or 400 mg TPM/m3 of diluted cigarette smoke generated from the 3R4F Kentucky reference research cigarette on the CONDOR® smoking machines (Borgwaldt-KC, Richmond, VA, USA). Exposures were conducted for 3 hours per day for 5 consecutive days. Scheduled necropsies were conducted on study day 5 of 10 rats/gender/group immediately after the final exposure. Finally, the surveillance animals consisted of the first 2 surviving male rats per exposure group from which carboxyhemoglobin, nicotine, and cotinine were measured on study day 5.
Experimental design.
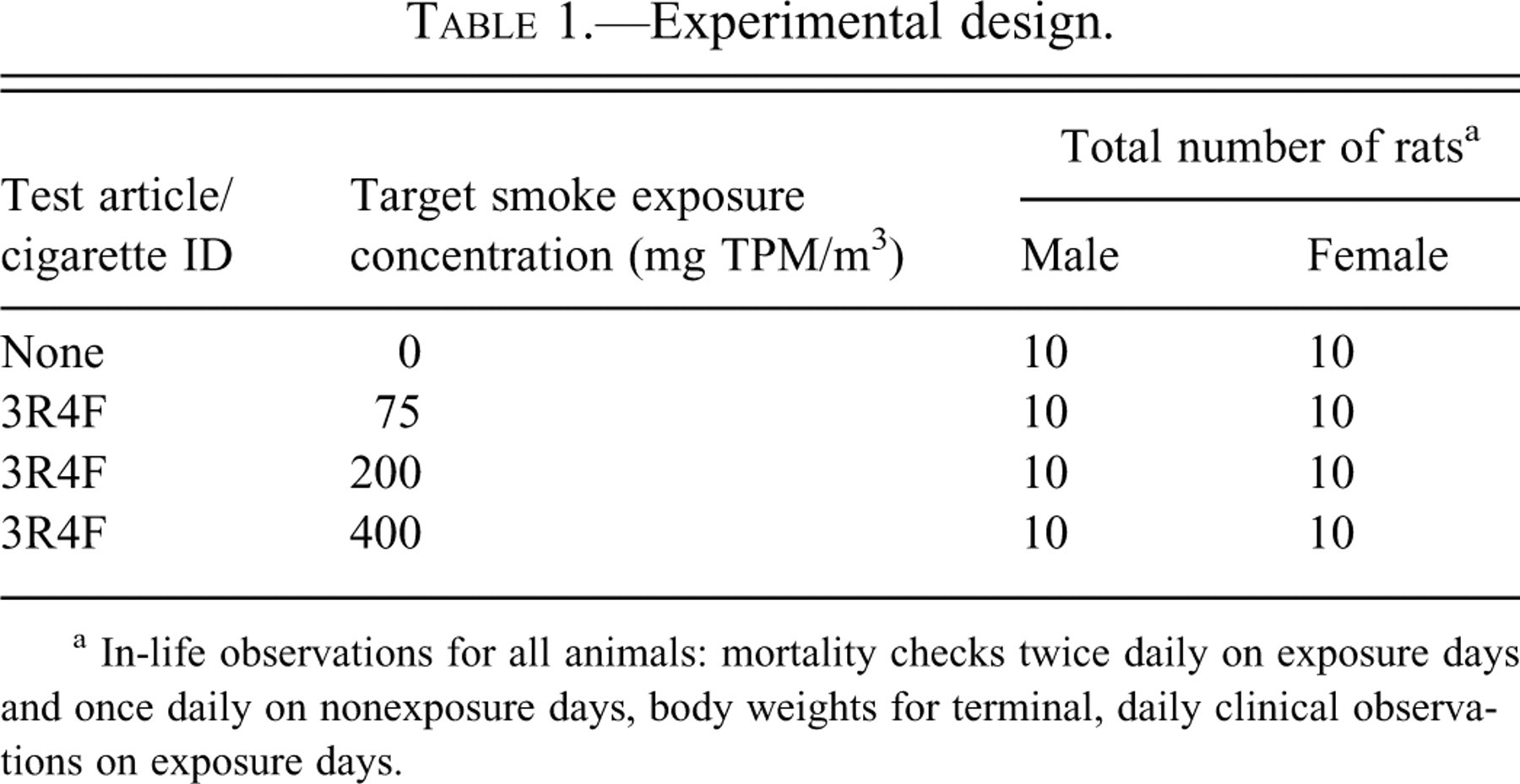
a In-life observations for all animals: mortality checks twice daily on exposure days and once daily on nonexposure days, body weights for terminal, daily clinical observations on exposure days.
Inhalation Exposure Laboratory Conditions
Cigarette smoke exposures were conducted in a laboratory equipped with 64-port nose-only inhalation exposure chambers (Lab Products Inc., Seaford, DE, USA) contained in acrylic enclosures. During the inhalation exposures, the rats were restrained in nose-only exposure animal holding tubes (CH Technologies [USA] Inc., Westwood, NJ). Animal tube loading, unloading, tube insertion, and removal from the exposure chamber manifold were performed according to standard procedures designed to minimize stress to study rats. Following exposure, the holders were removed from the chamber. The rats were removed from the holders and returned to their home cages. No food or water was provided during exposures.
Test Atmosphere Generation
The cigarette smoke test atmospheres were generated using 30-port Condor® smoking machines with a 4-piston smoke pump. Each exposure chamber had a dedicated smoking machine. Conditioned cigarettes were smoked in basic conformity with ISO Standards 3308 and 4387. The smoking machines were supplied with pressure-regulated, filtered, conditioned air. Vacuum exhaust for the smoke machines was provided by a dedicated regenerative ring blower and exhausted outside the building. The mainstream smoke was diluted in two steps with breathable quality air, with first a 1:60 dilution immediately at the exit of the smoke pump and then a second dilution set to the levels required to attain the target smoke concentration. In this study, the smoke parameters were set to approximately 9 puffs per cigarette (ranging from 8.9–9.1 puffs for all cigarettes), with a 35-mm butt length and a puff volume of 35 mL over a 2-second puffing period taken once each minute.
Test Atmosphere Monitoring
During each inhalation exposure period, the TPM concentration for cigarette smoke–exposed groups was determined at least once per hour during the 3-hour exposure period in each smoke chamber (once from filtered air) using a gravimetric filter-collection method. Carbon monoxide (CO) levels were monitored continuously in the smoke exposure and air control chambers with an infrared gas analyzer (Model ZRH; California Analytical Instruments, Inc., Orange, CA, USA). The instrument was calibrated during the test atmosphere development, and the calibration was checked prior to exposure with a zero air standard and an appropriate CO standard. CO concentration in each chamber was averaged over 20-minute periods, and the daily mean was calculated from these values. Aerosol particle size distribution was monitored once in each exposure chamber (excluding air control) using a Quartz Crystal Microbalance (QCM)–based 10-stage cascade impactor (California Measurements, Inc., Sierra Madre, CA, USA) at a sampling rate of 0.24 L/min. The mass median aerodynamic diameter (MMAD) and geometric standard deviation (GSD) were calculated from the mass accumulated on each stage of the QCM.
Exposure Biomarkers
During the exposure week, two male rats per treatment group were designated as sentinels and bled for determination of COHb, nicotine, and cotinine levels. As soon as possible (within minutes) after being removed from the exposure chamber, the animals were anesthetized with 70% CO2/30% O2, bled from the retro-orbital sinus using a tube with EDTA, and the level of COHb determined using an IL-482 CO oximeter. The serum nicotine and cotinine levels were measured using radioimmunoassay (Van Vunakis, Gjika, and Langone 1987).
Toxicology Endpoints
Body weights and clinical observations
The animals were observed twice daily for mortality or moribundity: once at the start of the exposure day and once at the end of the exposure day. Weights of all animals received were determined within 1 day following arrival. Weights of all animals were measured 8 days following receipt for randomization to facilitate test subject selection. All rats were weighed prior to exposure, on exposure day 1 and day 5.
Mortem and Postmortem Procedures
Euthanization and necropsy
Necropsies were conducted on study day 5 after the final exposure. Rats were euthanized by an overdose of sodium pentobarbital with subsequent exsanguination. Complete gross postmortem examinations were performed on all animals. Necropsy included examination of the external surface of all orifices, the external surfaces of the brain and spinal cord, and the organs and tissues of the cranial, thoracic, abdominal and pelvic cavities, and neck. The weights of the lungs and heart/aorta were recorded. Organ weight/body ratios were calculated using the nonfasted body weight obtained the day of necropsy.
Tissue processing/histopathological evaluations
The lungs were collected from all animals. The left lung was infused with neutral buffered formalin and immersed in neutral buffered formalin. After 24 hours, the left lung was placed in 70% ethanol and then processed for pathology and immunohistochemistry. Histopathology evaluations were performed on lung tissues collected at exposure day 5. Lung lobe with attached mainstem bronchi was left whole and placed in the cassette. The lung was arranged longitudinally within the cassette. Lung tissues were embedded in paraffin, 5-μm sections were obtained, and tissues were processed by routine histological methods, stained with hematoxylin and eosin, and evaluated microscopically by a board-certified veterinary pathologist.
Immunohistochemistry
Additional immunohistochemistry was performed on the lung as follows for the following proteins: EGF-R, TGF-B, NF-κB, COX-2, MMP-12, JNK, and PKC-α. We include methods for immunostaining only for the proteins that changed on smoke exposure that are included in the Results section. For MMP-12 and JNK staining, lung sections were deparraffinized and hydrated to distilled water and then soaked in 1X Tris wash buffer for 15 minutes. Then 3% hydrogen peroxide (Fisher, Pittsburgh, PA, USA ) was applied for 5 minutes followed by two rinses in 1X Tris wash buffer, then avidin block (Vector, Laboratories, Burlingame, CA, USA) was applied for 15 minutes. Following two rinses in 1X Tris wash buffer, a biotin block (Vector Laboratories) was applied for 15 minutes, then a serum-free protein block (DAKO, Carpinteria, CA, USA) was applied for 15 minutes; sections were then incubated in MMP-12 rabbit polyclonal antibody (ab66157; Abcam, Cambridge, MA, USA) at 1:750 for 2 hours. For JNK staining, pan JNK rabbit polyclonal antibody (AF1387; R and D Systems, Minneapolis, MN), detecting endogenous p46 and p54 JNK, was applied for 60 minutes at a 1:1,000 dilution. Following four rinses in wash buffer, secondary antibody (anti-rabbit IgG; Vector) was applied for 20 minutes. After four rinses in wash buffer, the avidin biotin complex (ABC) label from the Vectastain Kit (Vector) was applied for 30 minutes. Then sections were rinsed four times in wash buffer and diaminobenzidine (DAB; DAKO) was applied for 5 minutes. Following a rinse in distilled water, sections were counterstained in Mayer’s hematoxylin (Rowley Biochemical Institute, Danvers, MA, USA) for 2 minutes, rinsed in tap water, blued in 1X Tris wash buffer for 30 seconds, rinsed again in tap water, and dehydrated through a series of alcohols and xylene prior to coverslipping.
For PKC-α and COX-2 staining, lung sections were deparraffinized and hydrated to distilled water and then soaked in 1X Tris wash buffer for 5 minutes. Slides were treated with 1:20 preheated citrate buffer (InVitrogen, Carlsbad, CA, USA) for 8 minutes under pressure and then cooled for 10 minutes. Following 2- to 3-minute distilled water rinses, slides were rinsed with 1X Tris wash buffer for 5 minutes. Then 3% hydrogen peroxide was applied for 15 minutes followed by two rinses in 1X Tris wash buffer; Avidin block was then applied for 15 minutes. Following two rinses in 1X Tris wash buffer, biotin block was applied for 15 minutes. After two rinses in wash buffer, serum-free protein block was applied for 15 minutes; sections were then incubated in PKC-α mouse monoclonal antibody (sc-8393) specific for phosphorylated PKC-α(S657) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:50 dilution for 2 hours at room temperature. For COX-2 staining, normal goat block (Vectastain Kit) was applied for 30 minutes prior to incubation in rabbit anti–COX-2 antibody (160106; Cayman Chemical Co., Ann Arbor, MI, USA) for 60 minutes at a 1:200 dilution at room temperature. Following two rinses in wash buffer, appropriate secondary antibody (Vectastain Kit) was applied for 30 minutes. After two rinses in wash buffer, the ABC label complex from the Vectastain Kit was applied for 30 minutes. Then sections were rinsed two times in wash buffer, and DAB was applied for 5 minutes. Following a rinse in distilled water, sections were counterstained in Mayer’s hematoxylin for 2 minutes, rinsed in tap water, blued in 1X Tris wash buffer for 30 seconds, rinsed again in tap water, and dehydrated through a series of alcohols and xylene prior to coverslipping.
Statistical Analysis
Data were analyzed using Microsoft Excel software. Differences between samples were tested using a two-sided Student’s t test. p values less than .01 were considered statistically significant. Data are reported as mean values and standard deviation.
Results
Test Atmosphere
The gravimetrically determined mean TPM concentrations closely approximated the target levels set at 75, 200, and 400 mg/m3 because the average gravimetrically determined values were 73, 199, and 392 mg/m3 in the 3R4F research cigarettes, respectively. The mean MMAD of the smoke particles ranged from 0.59 to 0.63 μm, with a GSD range of 2.06 to 2.14. The mean CO concentration ranged from 0, 92, 234, and 420 ppm for control, 75, 200, and 400 mg TPM/m3, respectively. Mean nicotine levels were 0.00, 2.82, 11.13, and 24.08 mg/m3 for control, 75, 200, and 400 mg TPM/m3 respectively (Table 2 ).
Summary of aerosol mass concentration, particle size distribution, and carbon monoxide concentration in cigarette smoke.
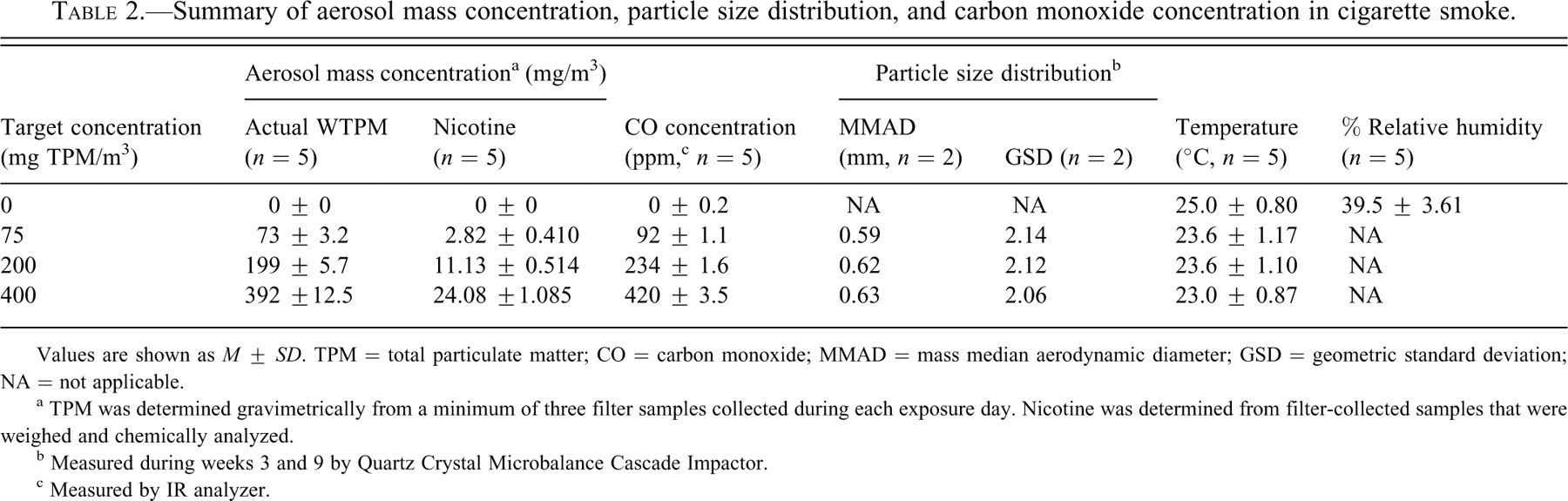
Values are shown as M ± SD. TPM = total particulate matter; CO = carbon monoxide; MMAD = mass median aerodynamic diameter; GSD = geometric standard deviation; NA = not applicable.
a TPM was determined gravimetrically from a minimum of three filter samples collected during each exposure day. Nicotine was determined from filter-collected samples that were weighed and chemically analyzed.
b Measured during weeks 3 and 9 by Quartz Crystal Microbalance Cascade Impactor.
c Measured by IR analyzer.
Biomarkers of Exposure
The mean COHb, nicotine, and cotinine levels of male rats exposed to cigarette smoke was increased when compared with the air control. COHb was increased in a dose-dependent manner in male rats as follows: control = 0.1 g/dL COHb, 75 mg TPM/m3 = 1.6 g/dL COHb, 200 mg TPM/m3 = 3.0 g/dL COHb, and 400 mg TPM/m3 = 4.8 g/dL COHb (Table 3 ). Nicotine was increased in a dose-dependent manner in male rats as follows: control = 0.6 ng/mL, 75 mg TPM/m3 = 113.6 ng/m:, 200 mg TPM/m3 = 213.1 ng/mL, and 400 mg TPM/m3 = 507.6 ng/mL (Table 4 ). Cotinine was increased in a dose-dependent manner in male rats as follows: control = 0.3 ng/mL, 75 mg TPM/m3 = 192.1 ng/mL, 200 mg TPM/m3 = 332.8 ng/mL, and 400 mg TPM/m3 = 355.6 ng/mL (Table 4).
Summary of animal blood oximetry data: Terminal male.
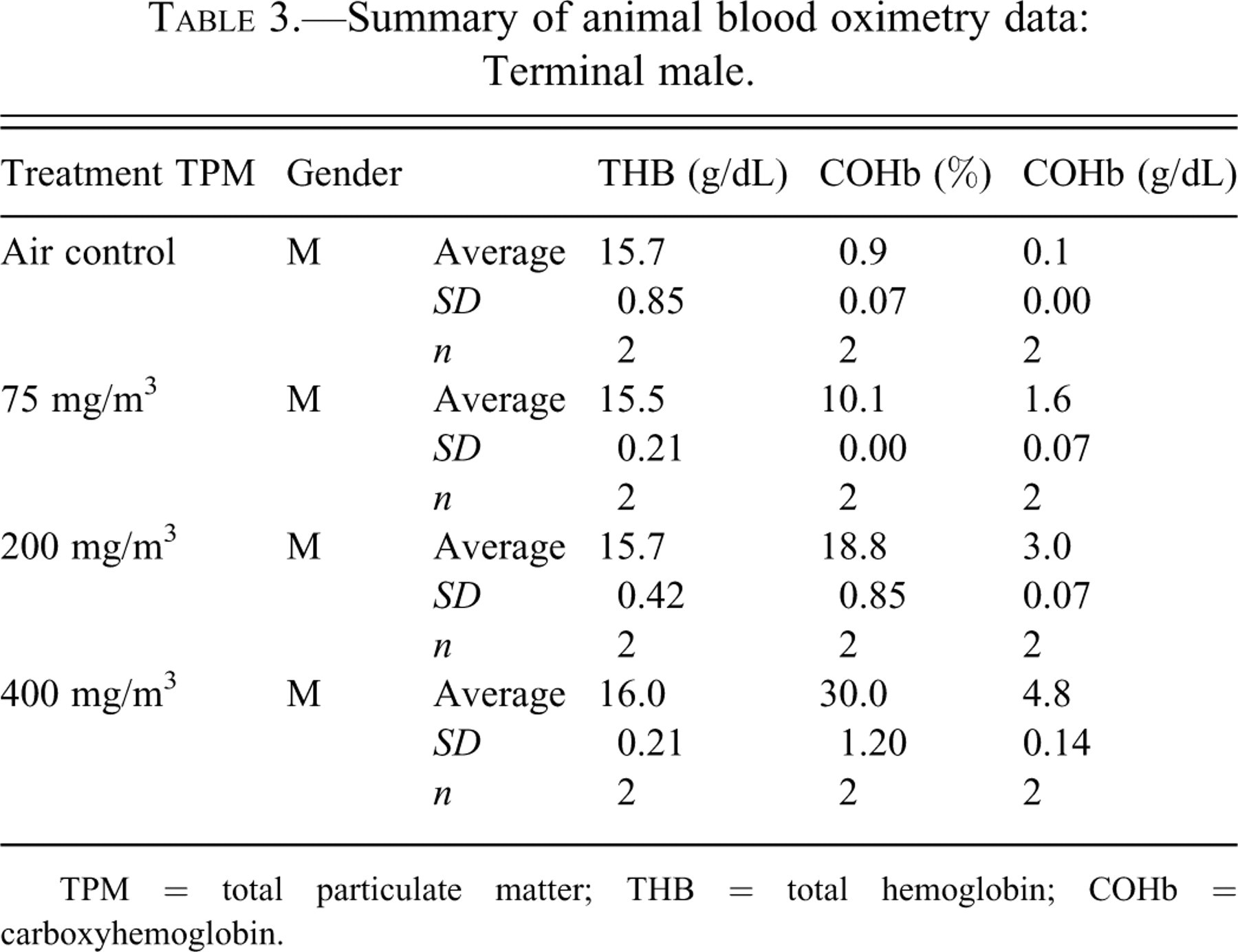
TPM = total particulate matter; THB = total hemoglobin; COHb = carboxyhemoglobin.
Summary of animal serum nicotine and cotinine data: Terminal male.
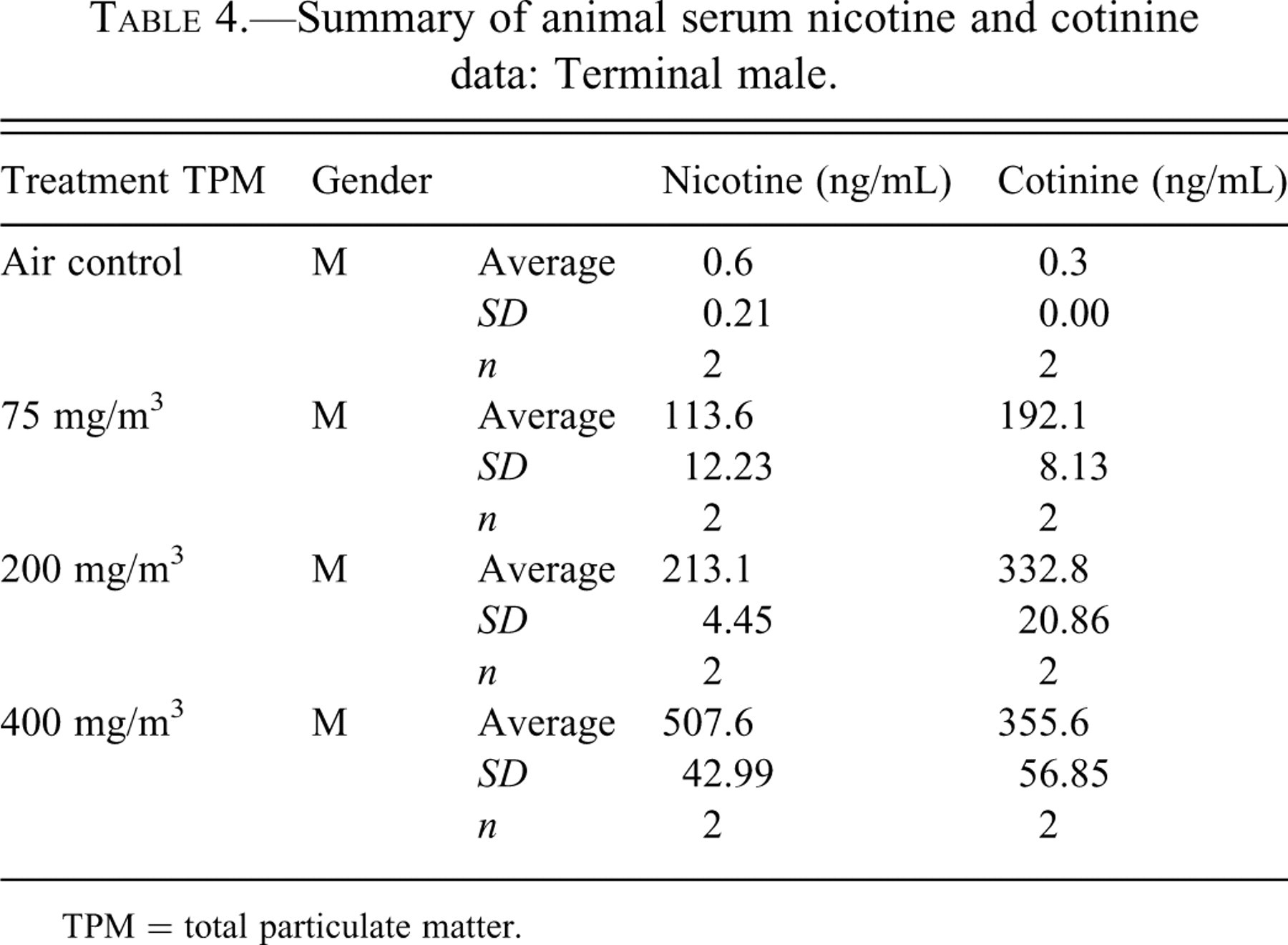
TPM = total particulate matter.
Gross Observations
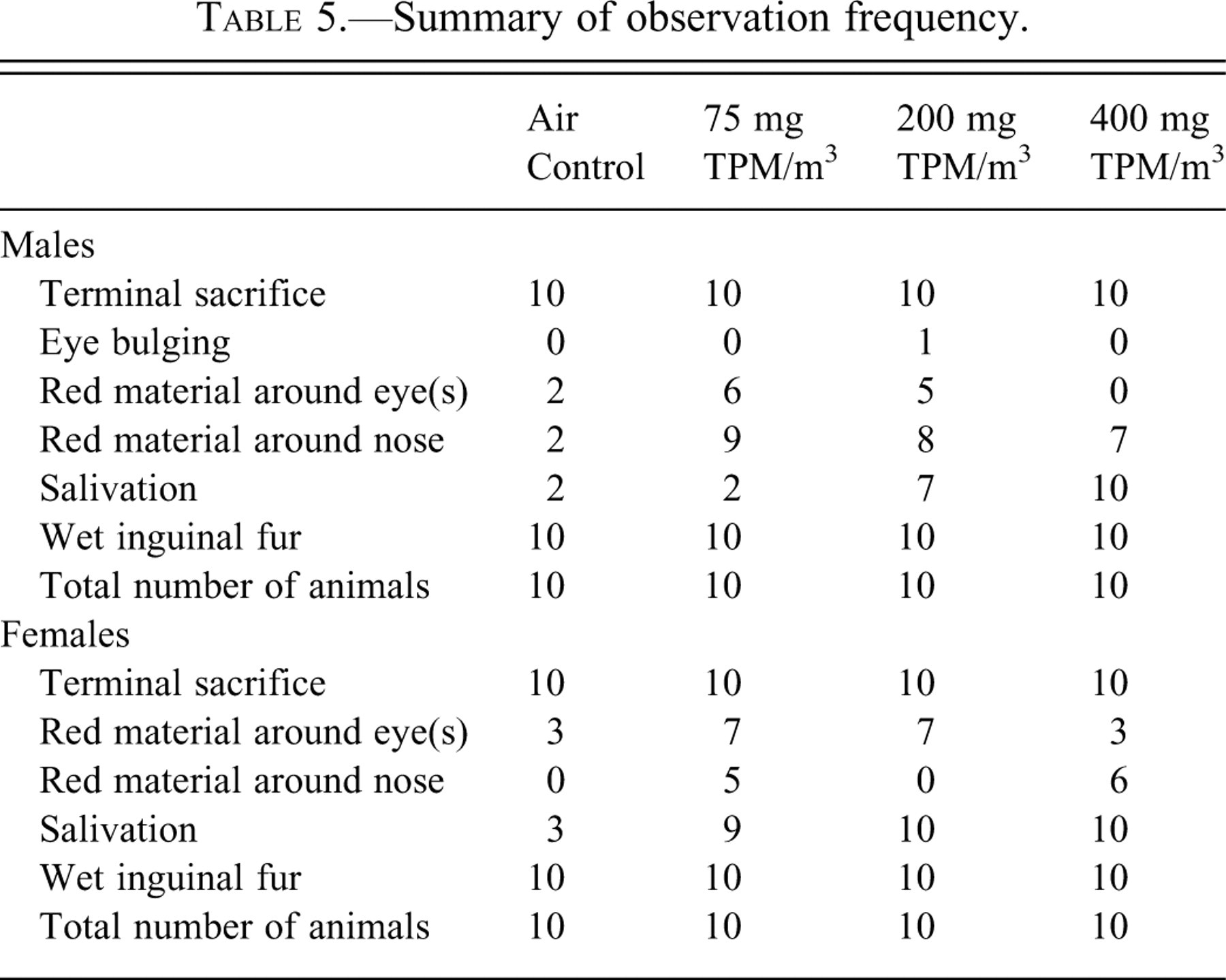
At the median concentrations of 75 and 200 mg TPM/m3, male and female rats displayed an increased amount of red material around eyes and noses compared with controls (Table 5 ). Increased salivation was seen at the 200- and 400-mg TPM/m3 doses (Table 5). No gross lesions were observed in any of the animal organs at necropsy.
Summary of observation frequency.
Body and Organ Weights
The mean body weights of male rats exposed to 400-mg TPM/m3 cigarette smoke were significantly decreased compared with their air control counterparts, while none of the female rats displayed a difference in body weights (Table 6 ). Body weight gains were significantly reduced in males at the 200- and 400-mg TPM/m3 doses compared with the controls, while body weight gains were significantly reduced in females at all smoke concentrations (Table 7 ). Male rats displayed differences in absolute organ weights, which consisted of increased heart/aorta weights in 75- and 200-mg TPM/m3 exposures when compared with the air control group (Table 8 ). Both male and female rats displayed differences in absolute organ weights, which consisted of decreased lung weights in 75- and 200-mg TPM/m3 exposure groups when compared with the air control group (Table 8).
Summary of body weights.
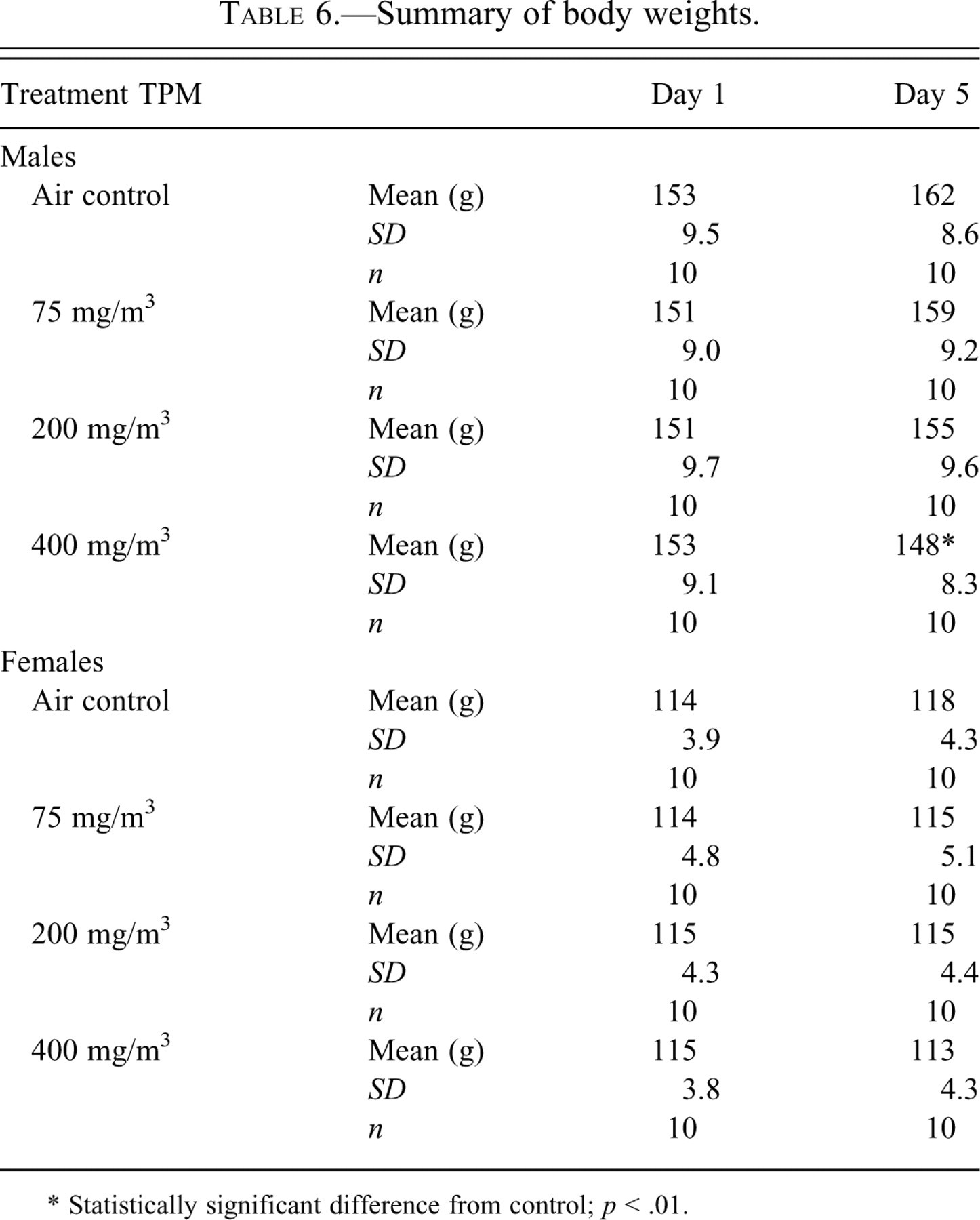
* Statistically significant difference from control; p < .01.
Summary of body weight gains.
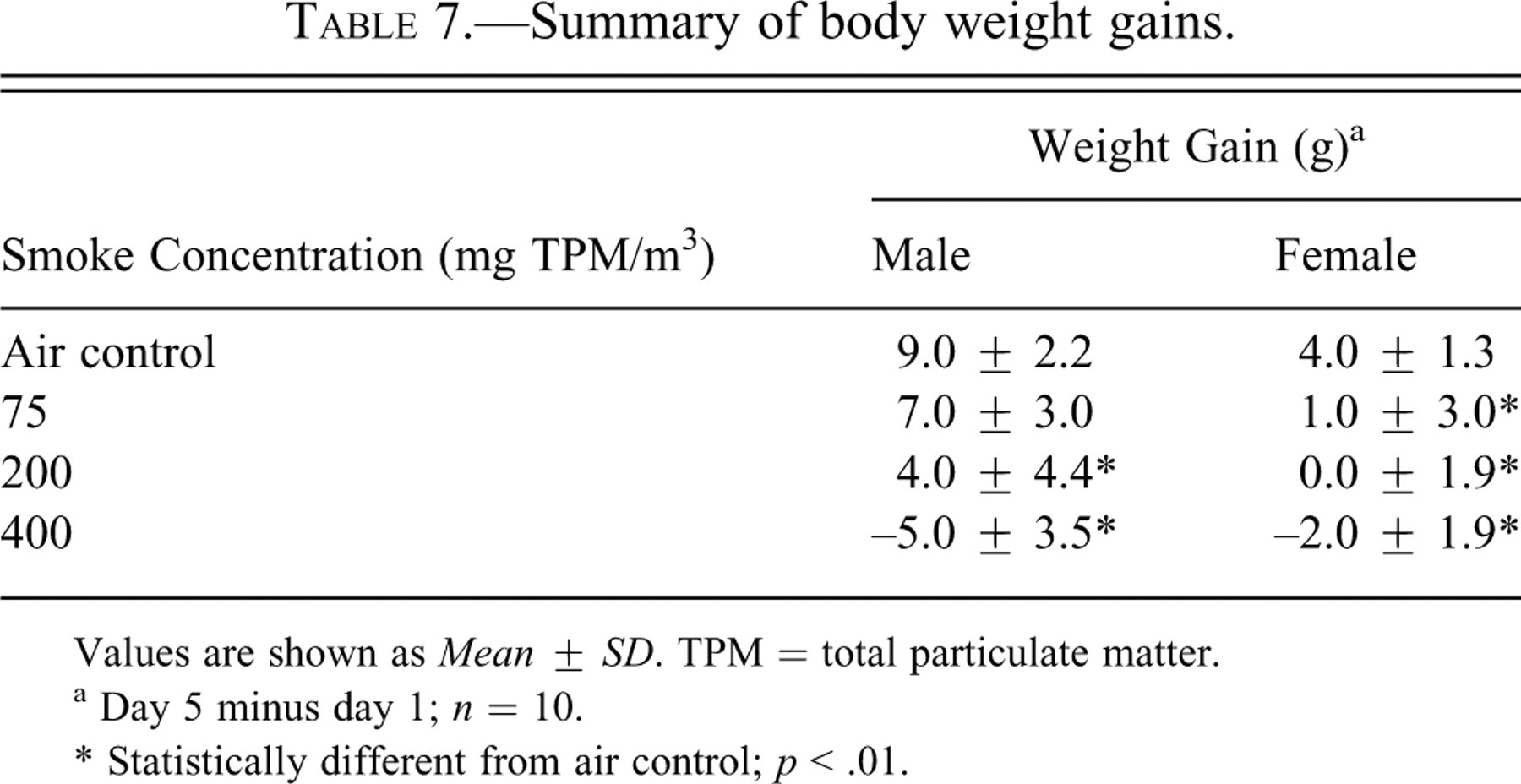
Values are shown as Mean ± SD. TPM = total particulate matter.
a Day 5 minus day 1; n = 10.
* Statistically different from air control; p < .01.
Summary of absolute weights.
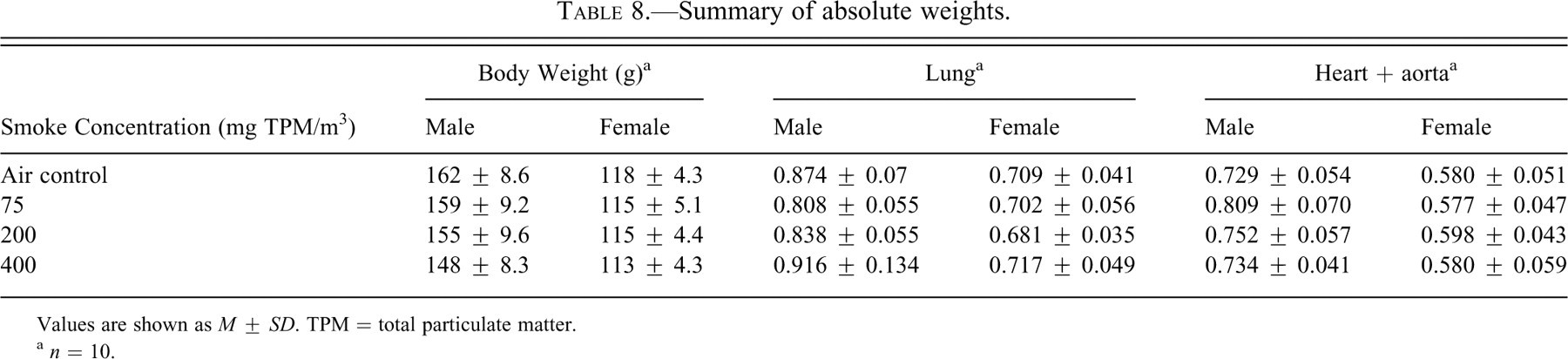
Values are shown as M ± SD. TPM = total particulate matter.
a n = 10.
Pathology
Bronchial epithelia from an unexposed male showed no hyperplasia (Figure 1A ). Bronchial epithelial hyperplasia was rare and observed in only one male rat exposed to 400 mg TPM/m3 (Figure 1B). Lung inflammation is defined by small clusters of inflammatory cells within alveoli consisting mainly of alveolar macrophages and lymphocytes. Some clusters of inflammatory cells were seen in 400-mg TPM/m3–exposed male lung (Figure 1D) compared with unexposed male lung (Figure 1C). A slight increase in alveolar histiocytosis was observed in all smoke-exposed females and 200- and 400-mg TPM/m3–exposed males. Figure 1F shows a slight increase in alveolar macrophages in 400-mg TPM/m3–exposed female lung compared with unexposed female lung (Figure 1E). Increased alveolar histiocytosis was scattered throughout the tissue and not prevalent in all areas examined.
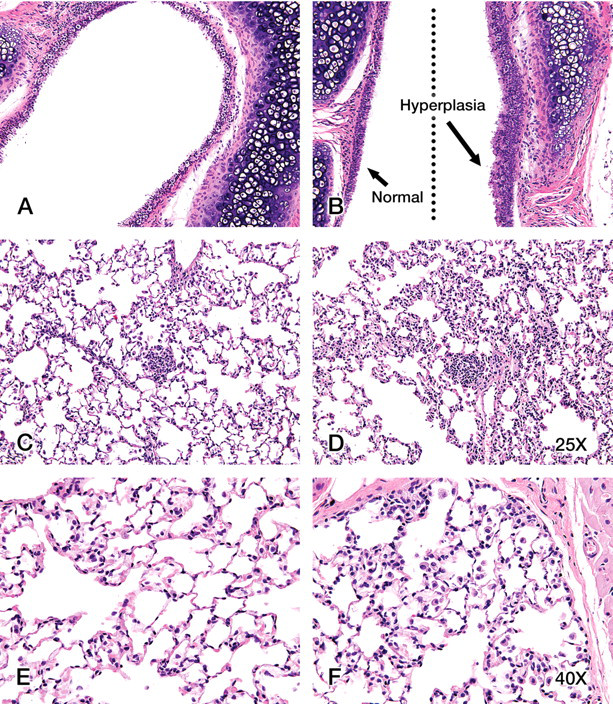
(A) Unexposed male lung with a typical epithelial lining. Bronchial epithelial hyperplasia was observed in only one animal, a male exposed to 400 mg TPM/m3 (B). Note that one epithelial border is hyperplastic (arrow) while the other epithelial border in this lung tissue appears near normal. Cross sections of the lung from an unexposed male (C) compared with a 400-mg TPM/m3–exposed male (D) show that the treated lung exhibits rare interstitial clusters of inflammatory cells (typically alveolar macrophages). Images of the control female (E) lung and 400-mg TPM/m3–exposed female (F) lung show the slight increase in alveolar macrophages found with smoke inhalation. A–D = 25×; E, F = 40×.
Immunohistochemistry
Changes in staining patterns and amount of EGF-R, TGF-β, and NF-κB were not observed between control and smoke-exposed animals in this short-term study (data not shown). Proteins that we evaluated that showed changes in staining patterns and amounts in response to smoke treatment were PKC-α, JNK, COX-2, and MMP-12.
Representative example images of interstitial PKC-α staining are shown in Figure 2 . Alveolar macrophages stained positively for PKC-α, and there was an increase in staining and an increase in the number of positive staining macrophages in the 200- and 400-mg TPM/m3 groups. Control female (Figure 2A) and male (Figure 2E) displayed minimal staining of alveolar macrophages for PKC-α, while staining was similar in the 75-mg TPM/m3–exposed male lung (Figure 2F) but nearly absent from the female lung (Figure 2B). Staining was increased in the 200-mg TPM/m3 female (Figure 2C) but limited in the male (Figure 2G), while staining increased in the 400-mg TPM/m3–exposed female (Figure 2D) and male (Figure 2H).
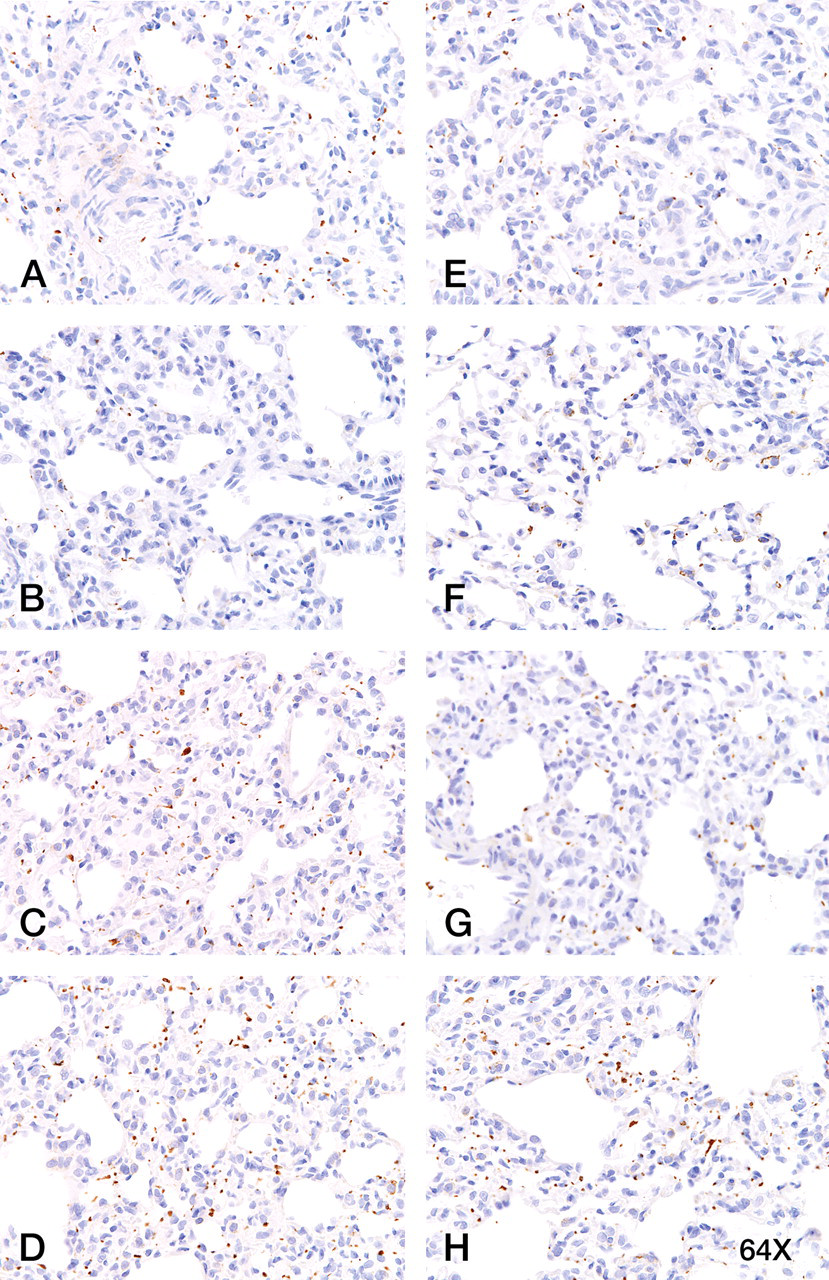
Control female (A) and male (E) lung displayed minimal interstitial staining for activated protein kinase C (PKC)–α (intravascular platelets). PKC-α staining was decreased at the 75-mg TPM/m3 dose in the female (B), but staining was similar to the unexposed tissues in the 75-and 200-mg TPM/m3–exposed male (F, G). In the 200-mg TPM/m3–exposed female (C) and the 400-mg TPM/m3–exposed female (D) and male (H) lung, there was an increase in PKC-α staining intensity in macrophages and in the number of positive staining macrophages. Magnification = 64×.
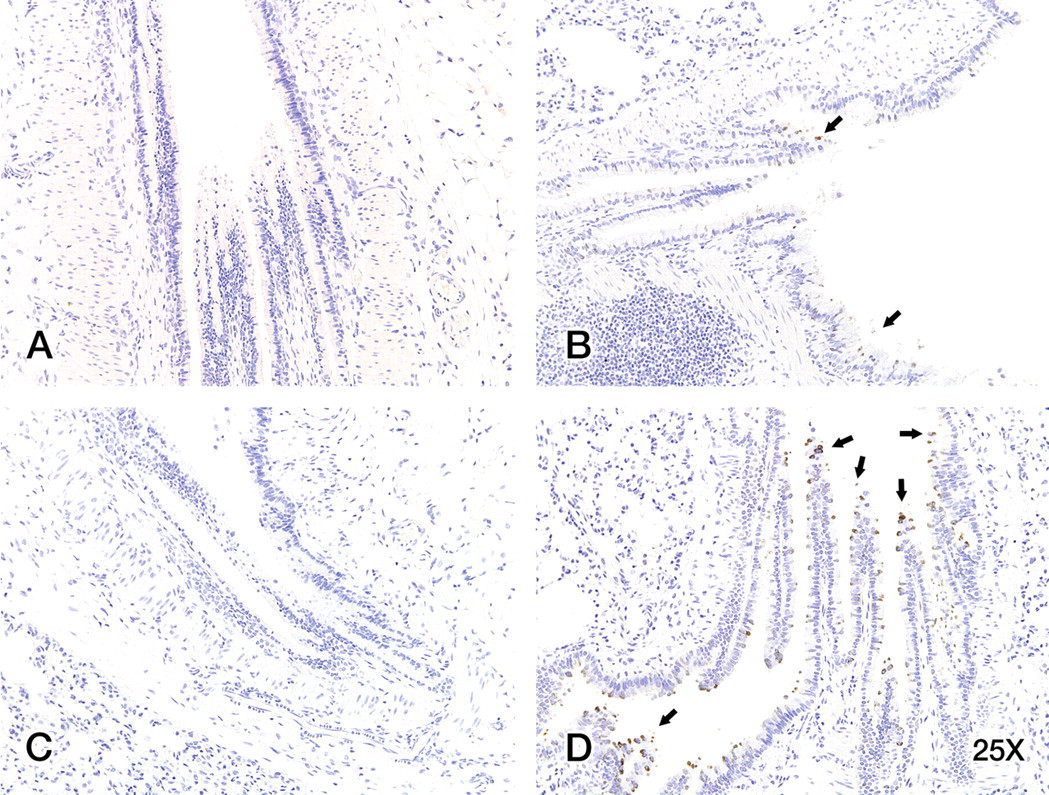
A decrease in JNK staining was observed in bronchiolar epithelial cells and in macrophages of smoke-exposed tissues compared with controls. Staining intensity for JNK increased in epithelial cells deeper within the lung parenchyma. Unexposed female lung tissues showed positive JNK staining in bronchiolar epithelial cells and in alveolar macrophages (Figure 3A). There was a decrease in positive staining for JNK in bronchiolar epithelial cells and alveolar macrophages with increasing smoke concentrations (Figures 3B and C). Unexposed male lung tissues showed positive JNK staining in bronchiolar epithelial cells and in alveolar macrophages (Figure 3D). A decrease in JNK staining was seen with increasing smoke concentrations (Figures 3E and F).
Staining intensity for c-Jun NH2-terminal kinases (JNK) increased in epithelial cells deeper within the lung parenchyma for all animals (bronchiolar intensity > bronchial intensity). There was a slight decrease in positive staining for JNK in alveolar macrophages in smoke-exposed tissues. Control female (A) and male (D) lung displayed JNK staining in bronchiolar epithelial cells. JNK staining decreased slightly in the 200-mg TPM/m3–exposed female (B) and male (E) lung. A more pronounced decrease in JNK staining was observed in the 400-mg TPM/m3–exposed lungs in females (C) and males (F), with the most obvious decreases in lung tissues from the 400-mg TPM/m3–exposed females. Magnification = 20×.
COX-2 showed only slight increases in staining in male and female rat lung at the 400-mg TPM/m3 dose. The increased COX-2 staining was scattered, displaying no homogenous staining pattern throughout the tissue. Control female (Figure 4A) and male tissues (Figure 4C) lack COX-2 staining, but 400-mg TPM/m3–exposed female (Figure 4B) and male (Figure 4D) lung tissue displayed significant COX-2 staining in some bronchial epithelial cells.
When control, 75-, 200-, and 400-mg TPM/m3–exposed lung tissues were stained for cyclooxygenase-2 (COX-2), only the 400-mg TPM/m3–exposed tissues displayed significant positive staining. Unexposed male (A) and female (C) lung did not stain for COX-2, while 400-mg TPM/m3–exposed lung tissues displayed positive COX-2 staining in some epithelial cells in the male (arrows; B) and female (arrows; D). Magnification = 25×.
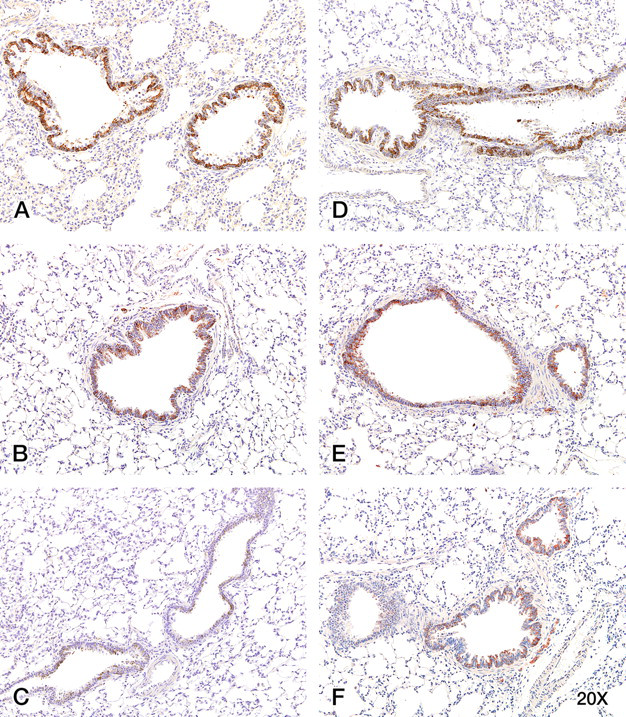
MMP-12 staining was obvious in alveolar macrophages and bronchiolar epithelia in unexposed female and male lung tissue (Figure 5A, D ). A larger percentage of alveolar macrophages stained positive for MMP-12 in exposed animals (Figure 5B, C, E, F), especially in 400-mg TPM/m3–exposed female lung tissue (Figure 5C). Bronchiolar epithelial cells also displayed increased positive staining for MMP-12 in 400-mg TPM/m3–exposed female lung tissue (Figure 5C) compared with female unexposed tissue.
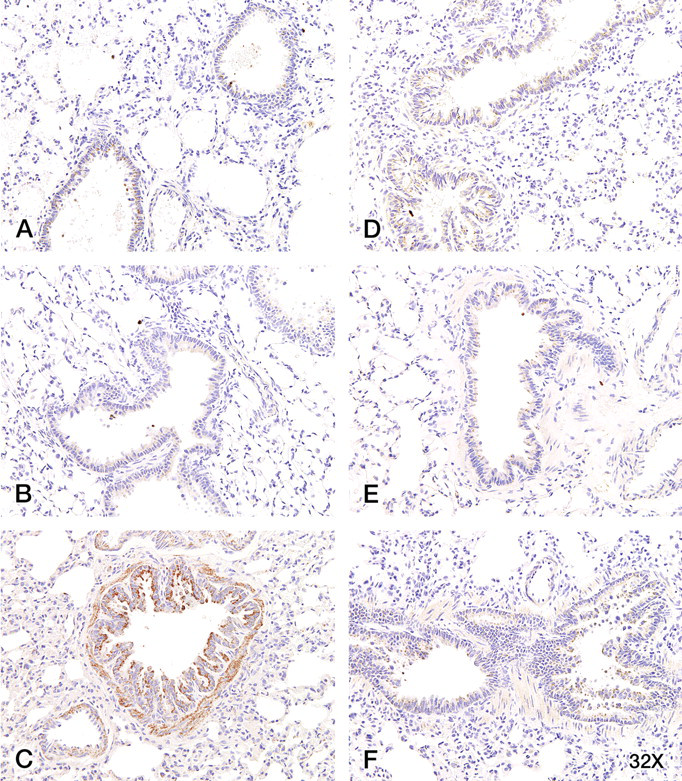
Matrix metalloproteinase (MMP-12) staining was obvious in alveolar macrophages and in bronchiolar epithelia, with a larger percentage of alveolar macrophages staining positively for MMP-12 in exposed animals. Control female (A) and male (D) lung displayed MMP-12 staining of alveolar macrophages and bronchiolar epithelia. An increased faint stain for MMP-12 in more alveolar macrophages was observed in the 200-mg TPM/m3–exposed female (B) and male (E) lung and the 400-mg TPM/m3–exposed female (C) and male lung (F). The most pronounced increase in MMP-12 staining occurred in bronchiolar epithelia in the 400-mg TPM/m3–exposed female lung (C), which also displayed MMP-12 staining of smooth muscle cells. Magnification = 32×.
Discussion
Fischer 344 rats have been widely used in long-term toxicity studies. In Fischer 344 rats exposed to cigarette smoke, dose-related reductions in body weights were noted after 13 weeks (Gaworski et al. 1998; Heck et al. 2002). In a short-term study, F344 rats displayed significantly suppressed body weight gains compared with controls at and after day 7 (Ohtsuka et al. 2000). Likewise, we show that doses of 200 and 400 mg TPM/m3 resulted in decreased body weights and body weight gains. Male rats exposed to 400 mg TPM/m3 exhibited decreased body weights. Similarly, male rats exposed to 200 and 400 mg TPM/m3 displayed a decrease in body weight gains, while female rats displayed a decrease in body weight gains at all smoke concentrations. Our data showed increased heart/aorta weights in males at 75 and 200 mg TPM/m3, similar to increases observed in response to smoke treatment when Fisher 344 rats were exposed to 350 mg TPM/m3 for 1 hour/day, 5 days/week for 13 weeks (Heck et al. 2002). Results from our study also showed decreased lung weight in male and female rats at 75 and 200 mg TPM/m3. Our data also showed increased salivation at the 200- and 400-mg TPM/m3 doses, similar to another study showing that F344 rats exhibited salivation after every cigarette smoke exposure (Ohtsuka et al. 2000). Our results showed a slight increase in alveolar histiocytosis (relative increase in the number of alveolar macrophages) in all smoke-exposed females and 200- and 400-mg TPM/m3–exposed males. This is of interest because 200 mg TPM/m3 has been equated to a heavy human smoking pattern (Finch et al. 1995), and alveolar macrophages remove debris and play important roles in lung inflammation (Hodge-Bell et al. 2007). Similar to our results, when male Sprague-Dawley rats were exposed to whole-body smoke, starting at day 5, macrophage numbers in the lungs increased (Stevenson et al. 2007).
We sought to evaluate proteins that are commonly altered in response to smoke based on the literature on smoke-exposed rodent and cell models in order to evaluate the utility of these proteins as biomarkers. The proteins that showed an obvious change in this short-term study based on immunohistochemistry were PKC-α, MMP-12, JNK, and COX-2 (Figure 6 ). Not only do these proteins play roles in inflammation, immune function, and disease, but most are in the activator protein–1 (AP-1) pathway. AP-1 is a major target of JNK and PKC-α, while MMP-12 is activated by AP-1 (Figure 6). AP-1 has been suggested to play an important role in tobacco smoke–induced pathogenesis. When male Wistar rats were exposed to 30 mg/m3 or 80 mg/m3 tobacco smoke for 6 hours/day, 3 days/week for 14 weeks, tobacco smoke dramatically induced cell proliferation and squamous metaplasia in a dose-dependent manner, and these effects paralleled activation of AP-1 (Zhong et al. 2005).
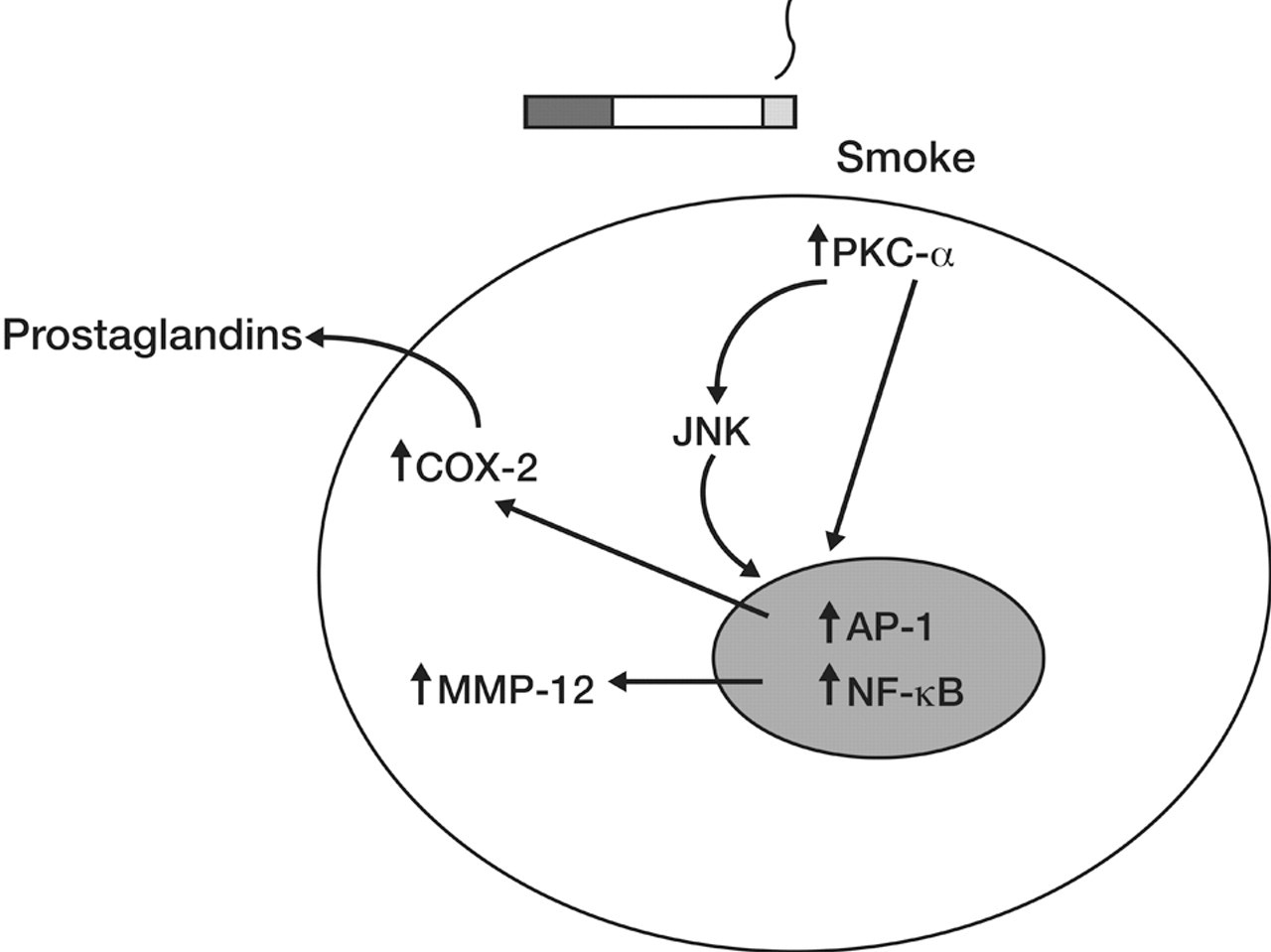
Schematic illustration of the effects of smoke on selected cellular proteins. Cigarette smoke treatment of lung cells causes activation of protein kinase C (PKC)-α, which activates the transcription factors activator protein-1(AP-1) and nuclear factor-kappa B, either directly or through c-Jun NH2-terminal kinases. Activation of PKC-α can also induce activation of cyclooxygenase-2, which stimulates the release of prostaglandins. AP-1 activation activates matrix metalloproteinase 12. These proteins are involved in inflammatory, immune, and stress pathways in response to toxins.
PKC may be a central player in inflammation and cigarette smoke–induced lung protein changes. PKC is either required for activation or is implicated to be an integral mediator in a pathway of other proteins that tested positive in our study. PKC mediates MMP-12 protein secretion (Raza et al. 2000). PKC-α appears to be required for COX-2 expression (Chen et al. 2000). PKC-α and -ϵ act cooperatively to activate JNK in non–small lung cancer cells, and activated JNK is decreased when cells are exposed to PKC inhibitors (Lang et al. 2004).
PKC is often activated in disease including cancer, atherosclerosis, diabetes, and respiratory diseases, including non–small cell lung cancer, asthma, and COPD (Carter 2000; Dempsey, Cool, and Littler 2007). PKCs are important in many cellular lung responses, including permeability, contraction, migration, hypertrophy, proliferation, apoptosis, differentiation, motility, and inflammation (Dempsey, Cool, and Littler 2007; Nakashima 2002). Our data showed that cigarette smoke–exposed rat lung tissues displayed an increase in activated PKC-α staining and an increase in the number of activated PKC-α–stained alveolar macrophages compared with controls. Cigarette smoke exposure activates PKC-α in human bronchial BEAS-2B cells (Carter and Hamm 2009) and in tracheal epithelial cells from tracheal ring explants from Sprague Dawley rats (Vander Top, Wyatt, and Gentry-Nielsen 2005). Alterations in PKC occur in lung cancers (Bae et al. 2007) and PKC-α is highly expressed in patients with lung adenocarcinoma (Lahn et al. 2004).
JNK is a mitogen-activated protein kinase and has essential roles in cellular processes including differentiation, apoptosis, and chemotaxis (Huang, Jacobson, and Schaller 2004; Karin and Gallagher 2005). JNK is activated primarily by cytokines and exposure to environmental stress (Weston and Davis 2002). JNKs phosphorylate and activate members of the AP-1 transcription factor family and regulate cellular survival and proliferation in response to cytokines and growth factors, noxious stimuli, and oncogenic transformation (Manning and Davis 2003). JNK plays a pro-oncogenic role possibly due to its ability to promote proliferation (Weston and Davis 2007). However, other studies have linked JNK to tumor suppression possibly due to the proapoptotic effects on JNK activation (Kennedy and Davis 2003). This concept may apply to our study because our data showed that JNK staining decreased in tissues from smoke-exposed rats, perhaps permitting proliferation of damaged cells.
Inflammation has a specific purpose in protecting the organism from damage. Acute inflammation often plays a protective role, but chronic inflammation may lead to disease. One protein activated by inflammation is COX-2. COX-2 is inducible by phorbol esters, cytokines, growth factors, reactive oxygen species, and chemical carcinogens (Jones et al. 1993; Williams and DuBois 1996). We showed that COX-2 was increased by smoke only at the high 400-mg TPM/m3 dose. Pathological overexpression of COX-2 is found in early carcinogenesis (Martey et al. 2004) and is often overexpressed in premalignant and malignant tissues (Liao and Milas 2004). By promoting chronic inflammation, COX-2 may cause a predisposition to malignancy. The associated inflammation induced by COX-2 is central to the pathogenesis of COPD. COX-2 is induced when human lung fibroblasts are exposed to cigarette smoke extract, thus creating a proinflammatory environment (Martey et al. 2004). This chronic inflammatory state may act as one of the first steps in epithelial transformation.
MMP-12 is a potent proinflammatory and oncogenic molecule (Qu et al. 2009). Upregulation of MMP-12 plays a critical role in the emphysema to lung cancer transition that is facilitated by inflammation (Qu et al. 2009). MMP-12 expression correlates with early cancer-related deaths in non–small cell lung cancer, especially those associated with cigarette smoke exposure (Hofmann et al. 2005). Blocking MMP-12 is a potential avenue for treating diseases including COPD (Nenan et al. 2005). Cigarette smoke treatment of alveolar macrophages, smooth muscle cells, and epithelia induces MMP-12 production (Gueders et al. 2006; Lavigne and Eppihimer 2005). MMP-12 is a key molecule in the recruitment of inflammatory cells and the release of TNF-α. MMP-12 preferentially degrades elastin and is implicated in COPD development (Cawston et al. 2001; Joos et al. 2002). Macrophages may be one target of MMP-12, with TNF-α release from macrophages initiating an inflammatory cascade (Churg et al. 2003). When AJ mice were nose-only exposed to cigarette smoke for three weeks and gene microarray analysis performed on lung tissues, the most upregulated gene in the smoke group was MMP-12 (Meng et al. 2006). Knock-out mice lacking expression of mouse MMP-12 have a markedly diminished capacity to degrade extracellular matrix, and these mice do not develop emphysema in response to long-term cigarette smoke exposure (Shipley et al. 1996; Hautamaki et al. 1997). Whole-body cigarette smoke exposure increased MMP-12 gene expression in male Sprague-Dawley rats, with increased expression starting at three days and continuing for the thirty-four weeks of the study (Stevenson et al. 2007). Our data support the idea that MMP-12 is increased in response to cigarette smoke exposure because macrophages from one week smoke-exposed rats display elevated MMP-12, especially in the 400-mg TPM/m3–exposed females. In these animals, MMP-12 is likely participating in inflammatory pathways and playing a role in cellular matrix degradation.
Unlike previous short-term studies that used gene expression profiling (Stevenson et al. 2007), we used immunohistochemistry to evaluate protein localization in the lung. Our data show increased staining of PKC-α, COX-2, and MMP-12, especially at the 400-mg TPM/m3 dose. Increases in hyperplasia, alveolar histiocytosis, PKC-α, COX-2, and MMP-12 are known to participate in COPD and cancer and thus may serve as early biomarkers of disease. These proteins play key roles in inflammation. Inflammation is a common thread among lung diseases such as asthma, COPD, and acute and chronic forms of lung injury and fibrosis (Dempsey, Cool, and Littler 2007). Targeting key effectors of inflammation may be an effective therapeutic intervention strategy in lung disease. As has been pointed out, “the challenge to toxicology is to develop confidence in the predictive power of early events by following their development over time as was previously done for pathology” (Witschi 2007). The development of short-term animal exposure studies, as an alternative to longer-term studies, with evaluation of targeted protein response should prove beneficial in further understanding mechanisms of smoke-induced disease as well as potentially serving as a method to screen potential toxins.
Footnotes
Acknowledgments
We thank the Illinois Institute of Technology Research Institute for performing the animal exposures and tissue collection. We thank the following people from Experimental Pathology Laboratories: Dr. Holly Kolenda-Roberts for performing the pathology analysis and Nancy Harris and Mary Parker for performing the histological and immunohistochemical staining. We appreciate the valuable scientific input of Dr. Robert R. Maronpot. We thank David Schickedantz for expert assistance in preparation of the figures.