
Abstract
The treatment of artificial marble wastes (AMWs) is a difficult aspect of the artificial marble industry and has enormous impacts on the environment. Herein, a series of polybutylene terephthalate (PBT)/AMWs composites were prepared by in situ polymerization. The effects of AMWs content (1–10 wt%) on the thermal stability, crystallization rate, and impact strength of the PBT/AMWs composites were investigated in detail. The results showed that the composites containing 10 wt% of AMWs exhibited the best performance. Due to the high thermal resistance and uniform distribution of the AMWs particles, the heat deflection temperature reached 67.40°C with optimal thermal stability. Additionally, under heterogeneous nucleation, the crystallization rate was also the highest, and impact strength reached 17.29 kJ/m2 due to the partial core-shell structure and hardened matrix effects of the AMWs particles.
Keywords
Introduction
The marble industry has developed rapidly in recent years due to the continuous development of mining resources. However, during marble mining and processing, as much as 60% of the total product generated is solid waste material, including waste, waste powder, and waste mud.1-3 These solid waste materials occupy large volumes in landfills, causing serious environmental pollution and threats to public health.4-7 Artificial marble wastes (AMWs), which are by-products of artificial marble sawing or polishing, are one of the most common types. 8 Thus, converting AMWs into valuable resources remains a significant but imperative challenge.
Over the past few years, considerable research has been conducted on the recycling of AMWs. Ahmed et al. 9 prepared a hybrid composite material consisting of marble waste, shell ash, and natural rubber, and the results showed improved mechanical properties such as flexural strength, compression strength, hardness, and wear resistance. Kar et al. 10 studied how the addition of marble waste materials into polyethylene terephthalate (PET) composites improved the hardness, bending strength, and conductivity of the composites. Meanwhile, Lu et al. 11 produced high-strength ceramic containers through surface modification and low-temperature sintering of the AMWs. Awad et al. 12 and Pappu et al. 13 prepared a novel composite material by mixing marble dust particles with low density polyethylene, and the flexural strength and thermal stability of the composite material were effectively improved. In addition, Nayak et al. 14 added AMWs as secondary fillers to a glass-resin composite system, which effectively reduced the wear rate of the composite material while also reducing cost. Zhang et al. 15 prepared polyvinyl alcohol (PVA)/AMWs composites via melt processing. The results showed that the melt-processing window of 76.9°C for PVA significantly increased when combined with 10 wt% of AMWs, which was due to the interfacial hydrogen bonding interactions between the AMWs and the PVA molecules.
Polybutylene terephthalate (PBT) is an important thermoplastic resin that is widely used in applications such as automobile and mechanical parts manufacturing due to its excellent mechanical properties, corrosion resistance, and dimensional stability.16-19 However, neat PBT has disadvantages such as low heat deflection temperature and weak impact strength, which limit its applications.20-22 Therefore, it is commonly combined with reinforcing materials to meet the requirements for high-performance materials.23-28
In this work, PBT/AMWs composite materials were successfully prepared by in situ polymerization, and the effects of AMWs content on the thermal stability, crystallization rate, and impact strength of the PBT/AMWs composites were investigated. The purpose of this study was to utilize the AMWs as a resource and provide a new perspective for the applications of PBT composites in order to efficiently recover AMWs for economic and environmental benefits.
Experimental
Materials
AMWs were collected from Guangxi Lisheng Stone Industry Co., Ltd, Hezhou, China. Dimethyl terephthalate (DMT) (Aladdin, 99% purity) and 1,4-butanediol (BDO) (Aladdin, 98% purity) were used to synthesize PBT. Tetrabutyl titanate (Aladdin, ≥99% purity) was used as the catalyst for trans-esterification.
Material preparation
The AMWs used in this study were treated using 1000 mesh wet sieving and then dried at 80°C for 10 h. The trans-esterification of DMT (97.09 g, 0.5 mol) with BDO (99.13 g, 1.1 mol) was carried out in the presence of tetrabutyl titanate as the catalyst in a 250 mL three-neck flask. The system was equipped with a mechanical stirrer, a vacuum pump, and a condensing device. The DMT, BDO, and catalyst mixture were continuously stirred throughout the reaction at 190°C. When the methanol evaporation rate reached 93% of the theoretical value, 1 wt% (1.21 g) of AMWs was added to the reaction and stirred continuously for 10 min. Then, to start polycondensation, the mixture was vacuumed at 250°C, and a composite containing 1 wt% of AMWs was obtained. Neat PBT was synthesized using the same steps as previously stated but without the addition of AMWs. According to the weight percentage of AMWs, the composites were denoted by PBT-0 (0 wt% of AMWs), PBT-1 (1 wt% of AMWs), PBT-3 (3 wt% of AMWs), PBT-5 (5 wt% of AMWs), PBT-7 (7 wt% of AMWs), and PBT-10 (10 wt% of AMWs). Afterward, the composites were crushed and melted into three-dimensional strip samples (80 mm × 10 mm × 4 mm), using an SZS-20 micro-injection machine (Wuhan Ruiming Experimental Instrument Co., Ltd, Wuhan, China) at a melting temperature of 260°C, a mold temperature of 60°C, and a holding time of 40 s.
Characterizations
Particle size analysis
The particle size analysis of the AMWs was conducted using a BT-2600 laser particle size analyzer (Dandong Baite Instrument Co., Ltd, Dandong, China), with water as the dispersant. The shading index was set to 10%–15%, and the rotation speed was 1600 r/min.
Gel permeation chromatography measurements
The molecular weights and polydispersity indices of the PBT/AMWs composites were tested at 35°C by gel permeation chromatography (GPC) using a Waters GPC device (Agilent, USA) equipped with a Watersmodel 1515 autosampler, a 2414 refractive index detector, and an Agilent PLgel 5-μm MIXED-C chromatographic column (made in GB). Hexafluoroisopropanol was used as the eluent, and the flow rate was 1 mL·min−1. Additionally, polymethyl methacrylate (PMMA) was employed as the standard.
Wide-angle X-ray diffraction (XRD) measurements
X-ray diffraction (XRD) measurements were performed on an Ultima IV instrument (Rigaku, Japan), using Cu-Kα radiation (operated at 40 kV and 40 mA) between 5° and 80° at 10°/min.
Thermal gravimetric analysis
Thermal gravimetric analysis (TGA) measurements were obtained using a TGA4000 instrument (PerkinElmer, USA). Each sample was approximately 9–10 mg, and the analysis was carried out in a nitrogen atmosphere, ranging from 30°C to 850°C, and with a heating rate of 10°C/min.
Heat deflection temperature tests
Heat deflection temperature (HDT) tests were conducted using an HDT/V-1103 instrument (Chengde Jinjian Testing Instrument Co., Ltd, Chengde, China) according to the GB/T 1634–2004 standard, and each sample was injected with a micro-injection machine and tested a minimum of five times.
Differential scanning calorimetry analysis
Differential scanning calorimetry (DSC) analysis was carried out on a DSC25 instrument (TA, USA). The non-isothermal crystallization and isothermal crystallization measurements were conducted in a nitrogen atmosphere; each sample was approximately 9–10 mg and sealed in an aluminum pan. Each sample was first heated from 30°C to 260°C at a heating rate of 20°C/min and was held at this temperature for 2 min to eliminate the effect of the thermal and processing history. For non-isothermal crystallization, each sample was cooled down from 260°C to 0°C for six cycles at various cooling rates of 1.25°C/min, 2.5°C/min, 5°C/min, 10°C/min, 20°C/min, and 40°C/min, respectively. After each cooling, heating to 260°C at 10°C/min was performed. For isothermal crystallization, each sample was rapidly cooled from 260°C to different crystallization temperature of 204°C, 206°C, 208°C, 210°C, 212°C, and 214°C, respectively. And then, each sample was kept at the corresponding temperature until the crystallization finished. Finally, heating to 260°C at 5°C/min was carried out.
Impact strength tests
The impact strength tests were performed using a CEAST 9050 impact testing machine (INSTRON, USA) according to the ISO 180 standard with unnotched samples. Each sample was tested a minimum of five times.
Flexural strength tests
The flexural strength tests were conducted employing an INSTRON 3367 universal material testing machine (INSTRON, USA) according to the ISO 178: 2001 standard. Each sample was tested a minimum of five times.
Tensile strength tests
The tensile strength tests were executed using an INSTRON 3367 universal material testing machine (INSTRON, USA) according to the ISO 527-1: 1993 standard. Each sample was tested a minimum of five times.
Scanning electron microscopy (SEM)
The surface morphology of the AMWs powder was observed using a scanning electron microscope (SEM) (S4800, Hitachi, Japan) operated at an accelerating voltage of 5 kV. Surface morphologies of the PBT/AMWs composites were also observed using an SEM (JSM-7610F, JEOL, Japan) operated at an accelerating voltage of 1.6 kV. The AMWs powder and composite sample surfaces were sputter coated with a light layer of gold.
Results and discussion
Surface characteristics of AMWs
Figure 1(a) shows the size distributions of the AMWs particles. The average particle size was 5.358 μm with a narrow particle distribution. The SEM image of the AMWs powder is shown in Figure 1(b)b, indicating that AMWs have the advantages of tiny particle sizing and appropriate specific surface areas, both of which are beneficial for the dispersion of AMWs in the PBT matrix.15 The TGA curves for the AMWs and neat CaCO3 are shown in Figure 1(c), clearly showing there are three steps in the TGA curve for the AMWs between 220°C and 600°C compared with neat CaCO3. This can be interpreted as the unsaturated resin coating on the surface of AMWs, which occurred due to decomposition. (a) Particle size distribution and (b) SEM image of the AMWs powder. (c) TGA curves for the AMWs and neat CaCO3.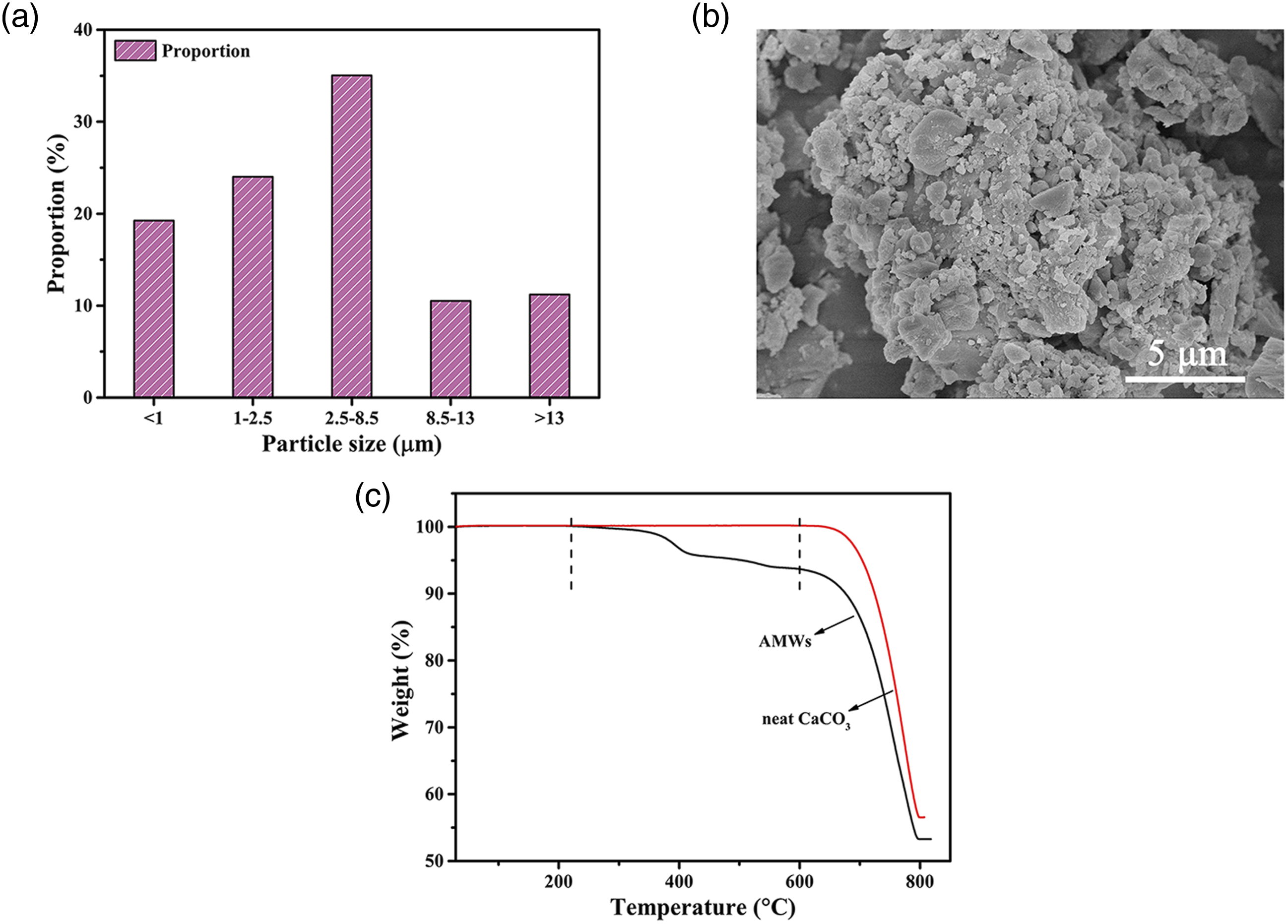
Gel permeation chromatography measurements
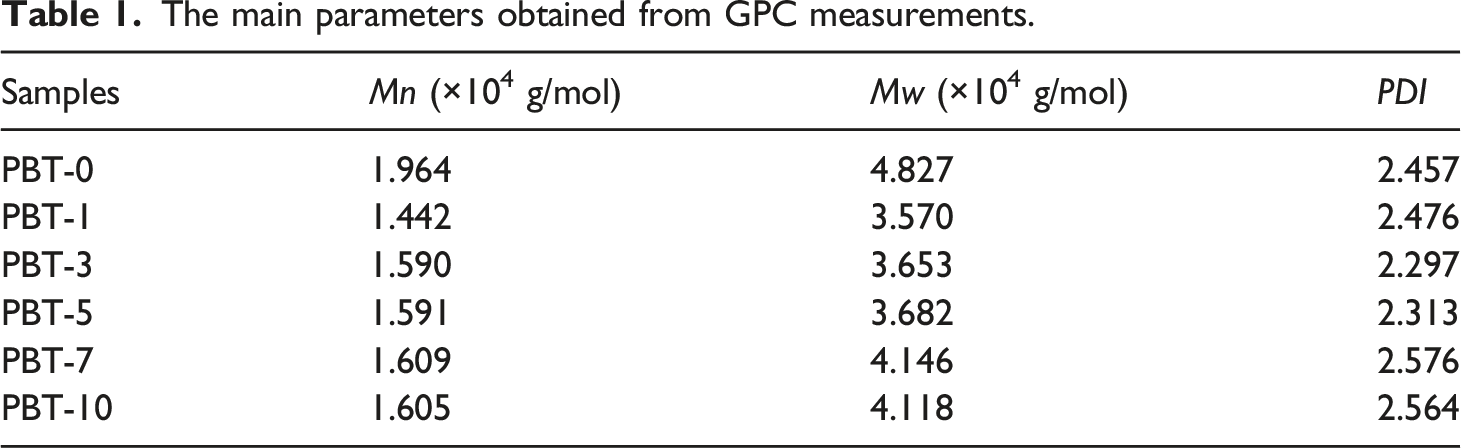
The main parameters obtained from GPC measurements.
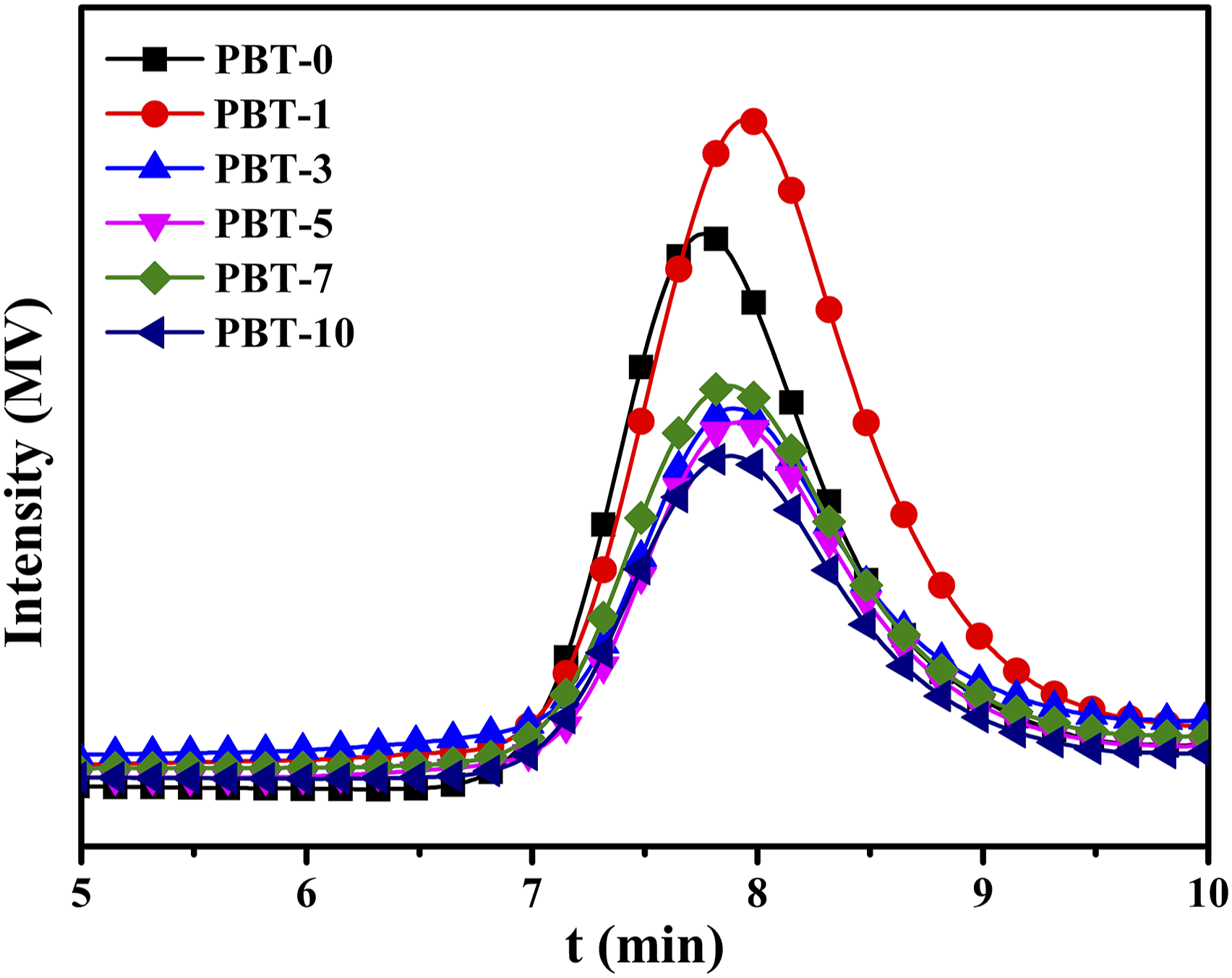
GPC outflow curves of the PBT/AMWs composites.
XRD analysis
XRD was carried out to assess the crystalline structure and crystallite sizes of the PBT/AMWs composites. Crystalline Bragg peaks were evident for all the XRD patterns, as shown in Figure 3(a). The new peaks for the composites were similar to the expected AMWs characteristic peaks, indicating that the composition did not change. (a) XRD patterns of the AMWs and PBT/AMWs composites. (b) Crystallite sizes of the PBT/AMWs For Peer Review composites.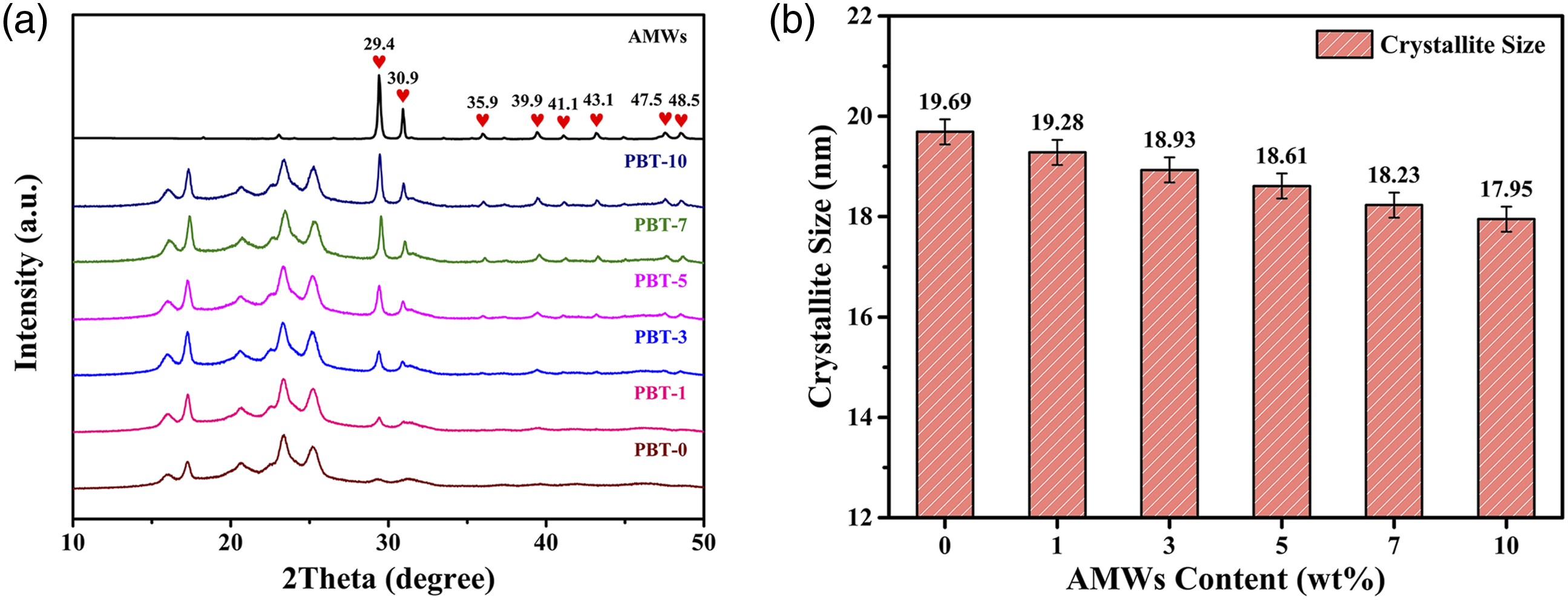
The effects of adding AMWs on crystallite size were quantitatively evaluated through the Debye—Scherrer equation,
29
as follows
Thermal stability analysis
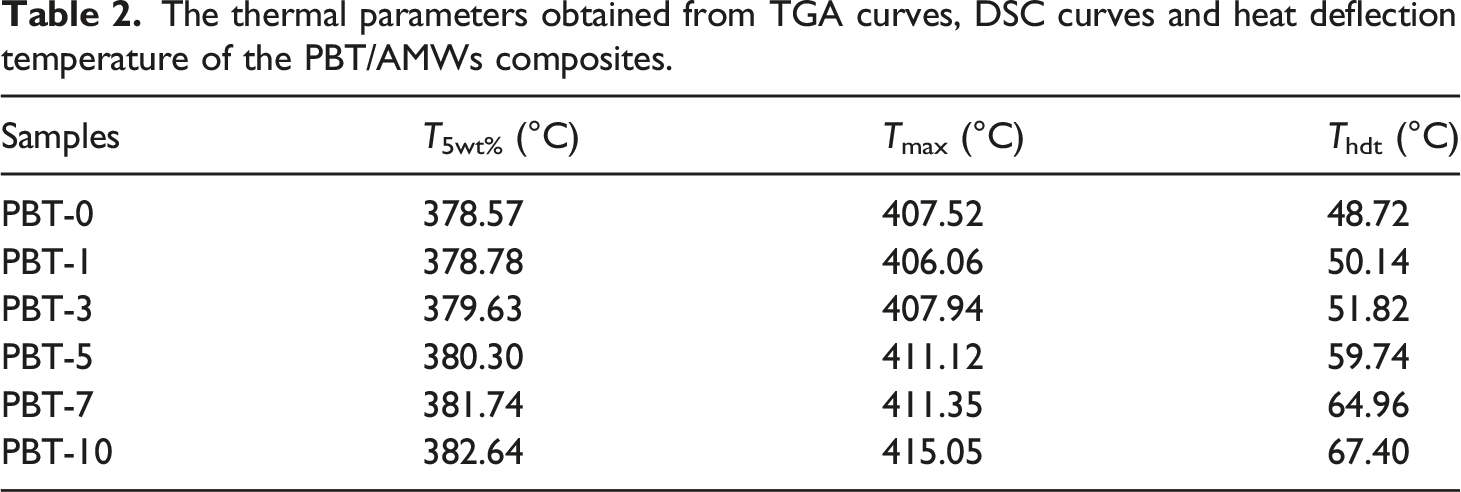
The TGA curves, Derivative Thermogravimetry (DTG) curves, and heat deflection temperature (Thdt) for the PBT/AMWs composites are shown in Figure 4, and the thermal parameters are listed in Table 2. The T5wt% and Tmax represent the initial decomposition temperature and maximum weight loss temperature, respectively. Both exhibited an overall upward trend as the AMWs content increased, which indicated that AMWs were conducive to improving thermal stability.31,32 This may have been due to the high thermal resistance of the AMWs and the strong interactions between the PBT matrix and AMWs filler, resulting in the restricted movement of the polymer chain.
33
In addition, the addition of AMWs can also promote the entanglement of polymer molecular chains. Therefore, as the AMWs content increased, the higher the energy required for molecular chain fracture, the higher the corresponding temperature.
34
Furthermore, the heat deflection temperature of the composites increased from 48.72°C to 67.40°C, which may have been due to the uniform distribution of AMWs particles in the matrix.
35
(a) TGA curves, (b) DTG curves, and (c) heat deflection temperature for the PBT/AMWs composites. The thermal parameters obtained from TGA curves, DSC curves and heat deflection temperature of the PBT/AMWs composites.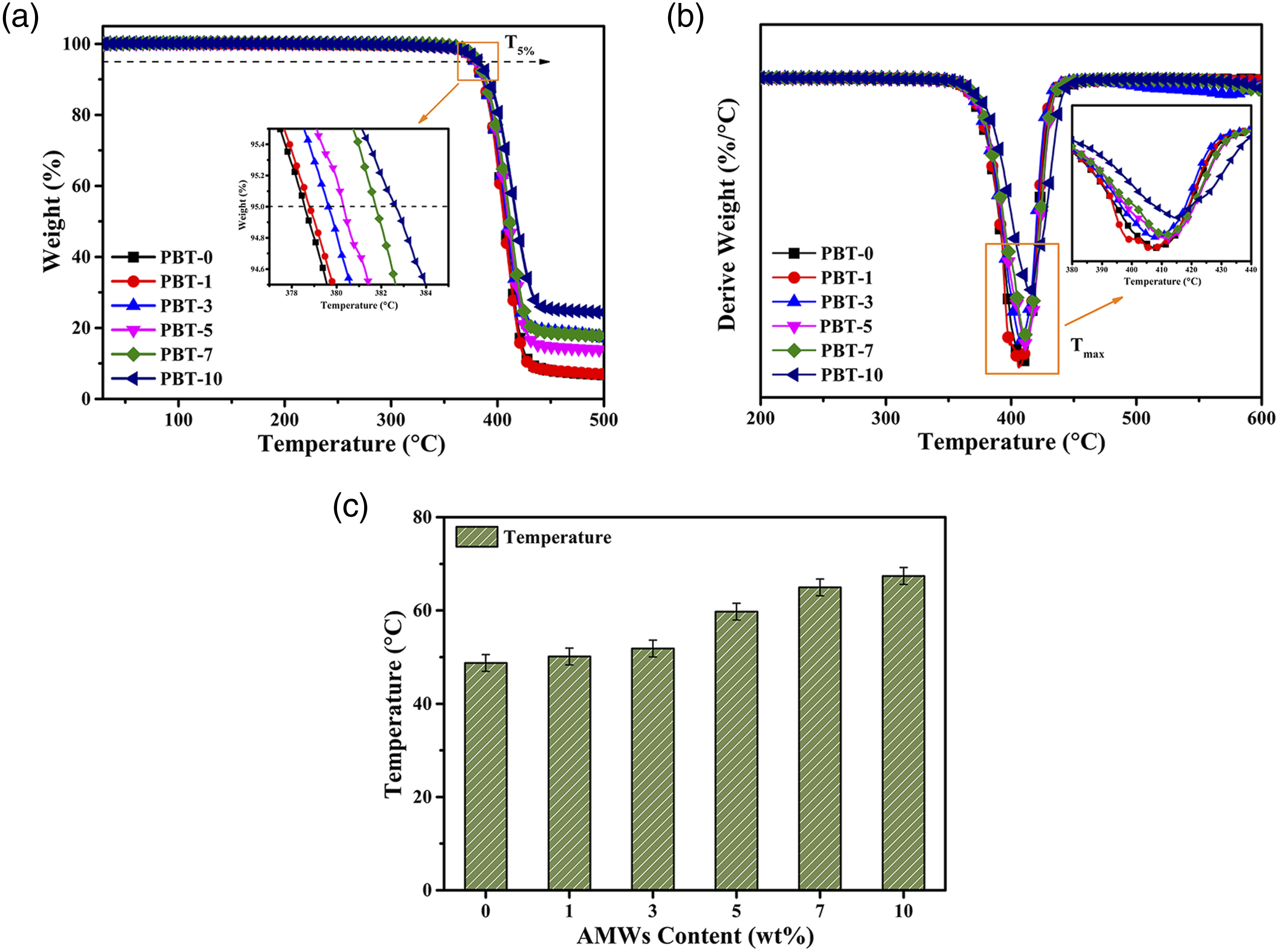
Crystallization behavior analysis
Figure 5 shows the changes in the crystallization curves as the AMWs content increased at different cooling rates. It was apparent that the crystallization onset temperature Tonset c and the crystallization peak temperature Tpeak c increased with the addition of AMWs when the cooling rate was maintained, which may have been due to the heterogeneous nucleation of the AMWs particles in the PBT matrix.36–38 In addition, when the AMWs content was constant, the crystallization curves moved to the low-temperature region when the cooling rate increased. This was attributed to the molecular chains not having enough time to arrange into a crystalline phase during rapid cooling,
39
thus chain segment activity was poor and the crystallization was delayed, which lowered the crystallization temperature. Non-isothermal DSC curves for the PBT/AMWs composites at cooling rates of (a) 1.25°C/min, (b) 2.5°C/min, (c) 5°C/min, (d) 10°C/min, (e) 20°C/min, and (f) 40°C/min, respectively.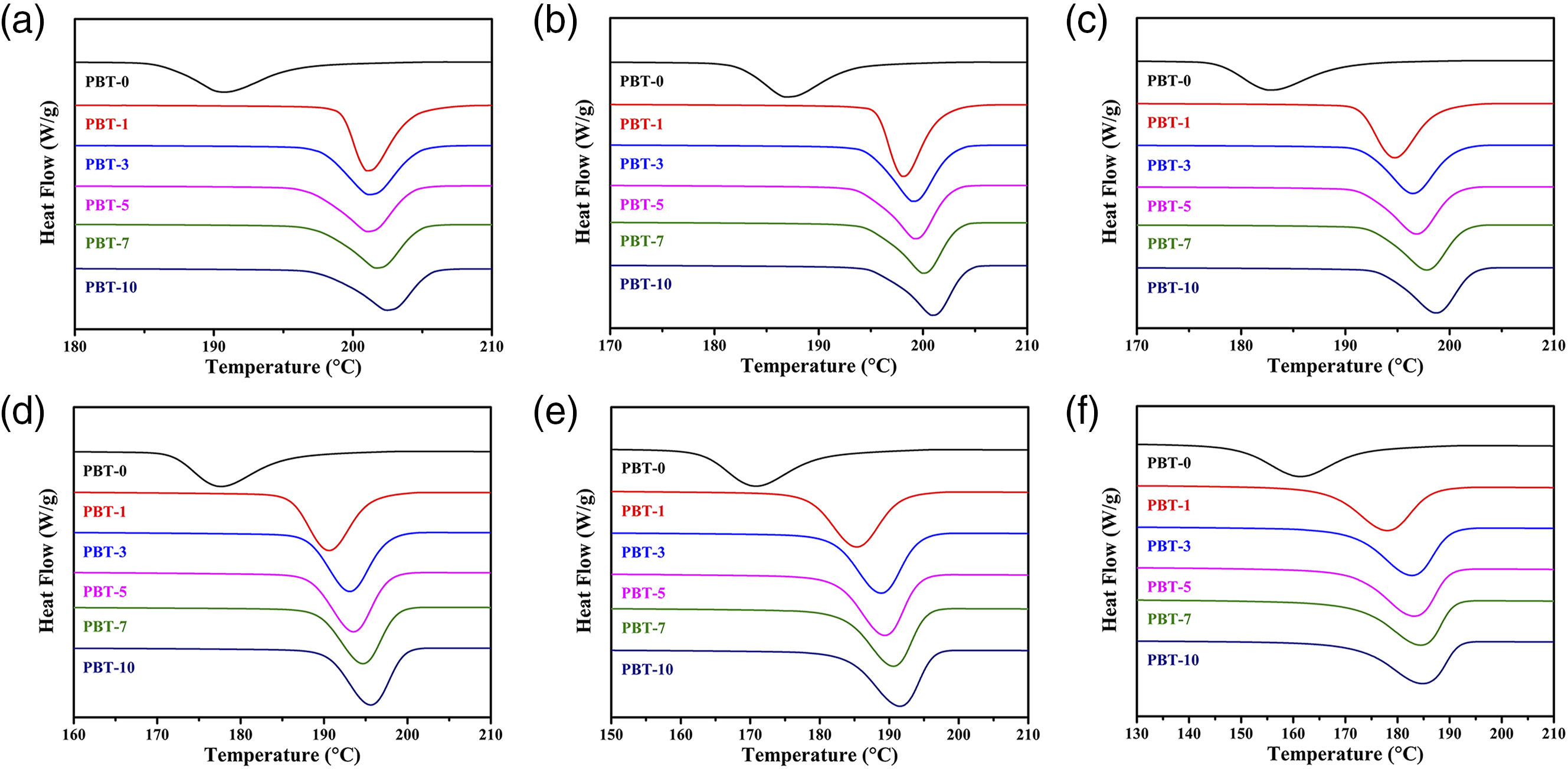
Figure 6 shows the comparative differences in crystallization peak temperatures for the PBT/AMWs composites with different content. At a cooling rate of 1.25°C/min, when only 1 wt% of AMWs was added, the Tpeak c increased by about 10°C compared with neat PBT and continued to increase with increased AMWs content. Similar results were obtained for other cooling rates, and the faster the cooling rate, the more significant the difference. The reason for this phenomenon was due to the addition of the AMWs material, which gradually improved the heterogeneous nucleation of PBT. Crystallization peak temperature Tpeak c of the PBT/AMWs composites at different cooling rates.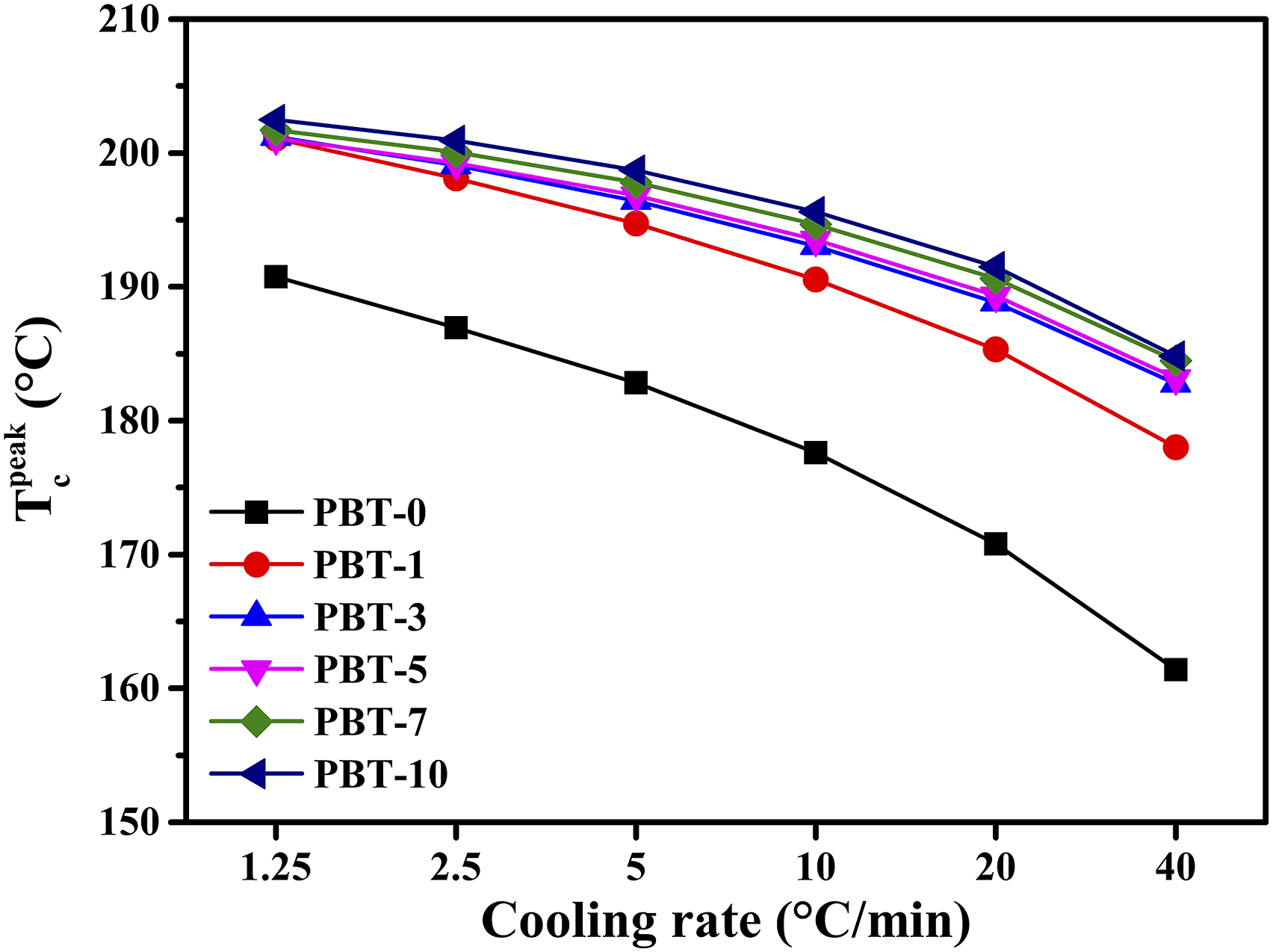
To investigate the isothermal crystallization behavior of the PBT/AMWs composites, crystallization kinetics were assessed, which are quantified by relative crystallinity Xt as a function of time t at different temperatures and obtained by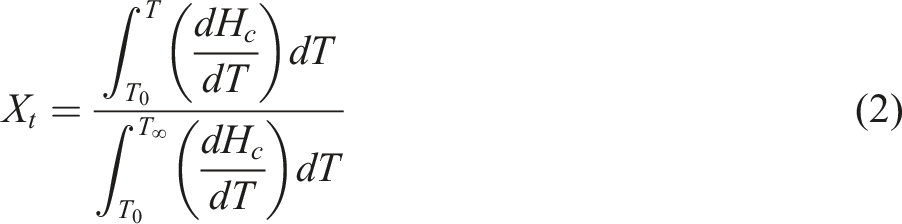

As shown in Figure 7, as the temperature increased, the neat PBT required more time for crystallization, indicating that the crystallization rate decreased gradually. This was attributed to the fact that the crystallization rate was controlled by the nucleation rate at a high temperature, and under this condition, polymer chain mobility improved and the crystalline nucleus was unstable.
40
Thus, as shown in Figure 7(b)–(f), the crystallization rate of the PBT/AMWs composites increased gradually compared with neat PBT. This phenomenon was more apparent at higher temperatures, suggesting that sufficient nucleation sites were made available by the AMWs, which accelerated the crystallization process. Relative crystallinity Xt as a function of time t for the PBT/AMWs composites with the AMWs content of (a) 0, (b) 1 wt%, (c) 3 wt%, (d) 5 wt%, (e) 7 wt%, and (f) 10 wt%, respectively.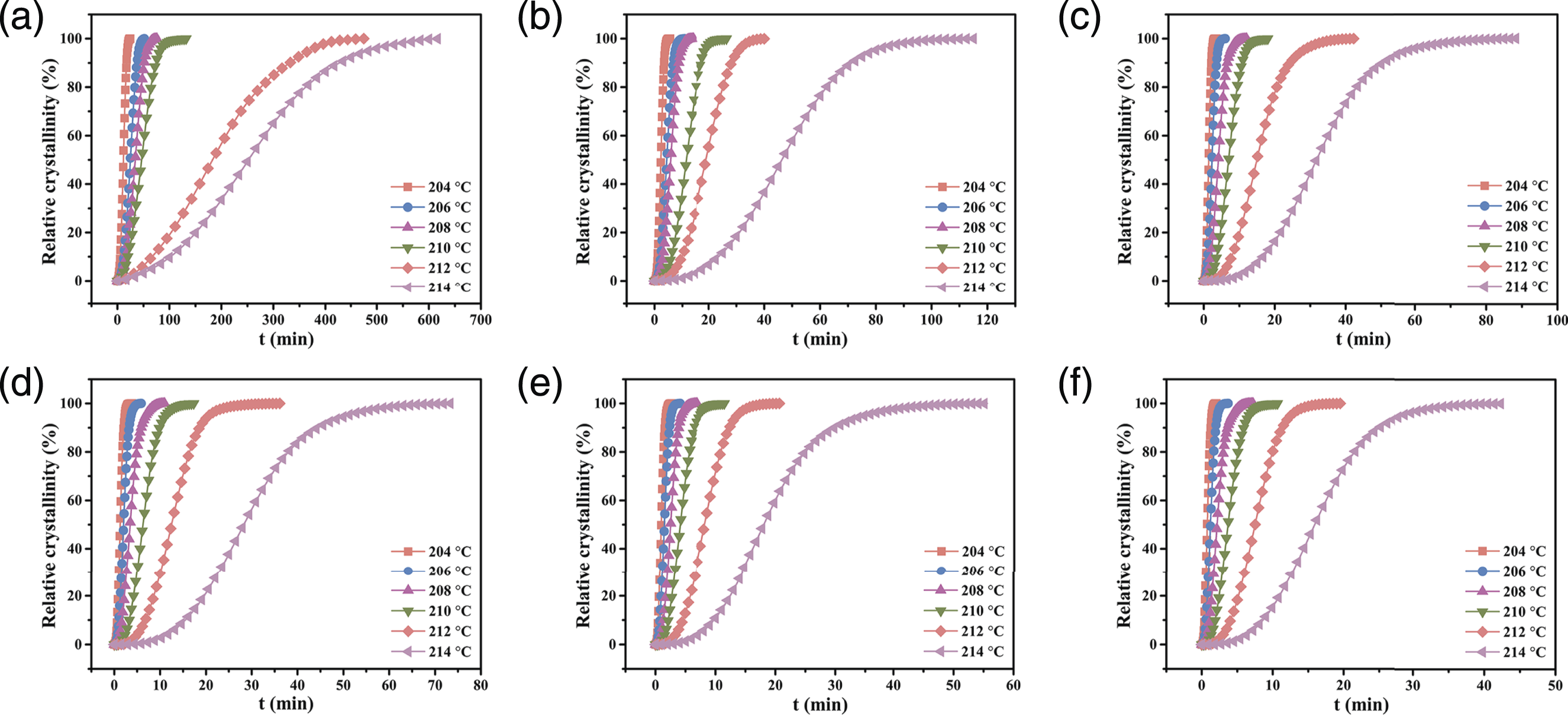
The Avrami model describes the relationship between relative crystallinity and crystallization time, and can be used to analyze the isothermal crystallization kinetics of most semicrystalline polymers and is expressed41,42 as follows Avrami plots of log [-ln(1-Xt)] versus log t for the PBT/AMWs composites with the AMWs content of (a) 0, (b) 1%, (c) 3%, (d) 5%, (e) 7%, and (f) 10%, respectively. The isothermal crystallization kinetic parameters obtained from the Avrami plots of the PBT/AMWs composites.
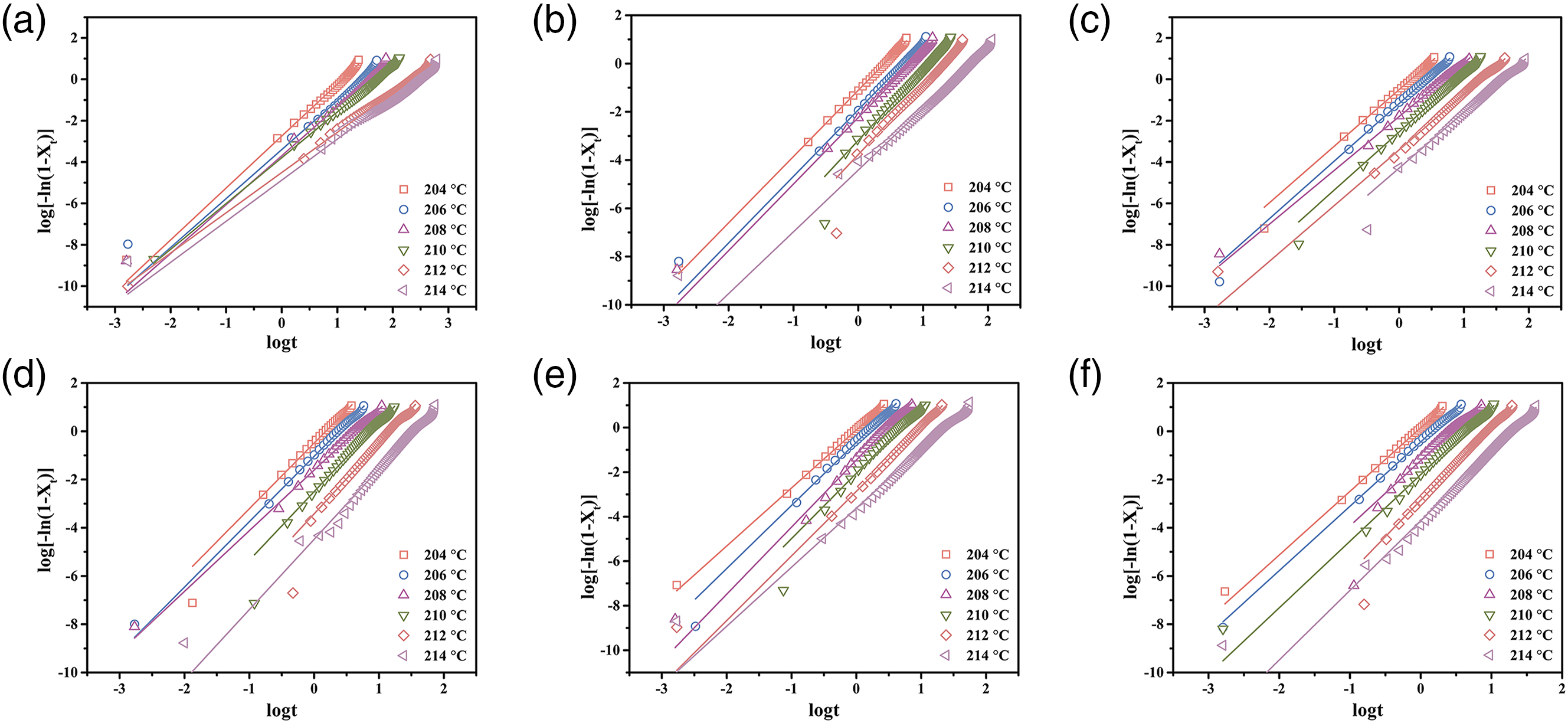
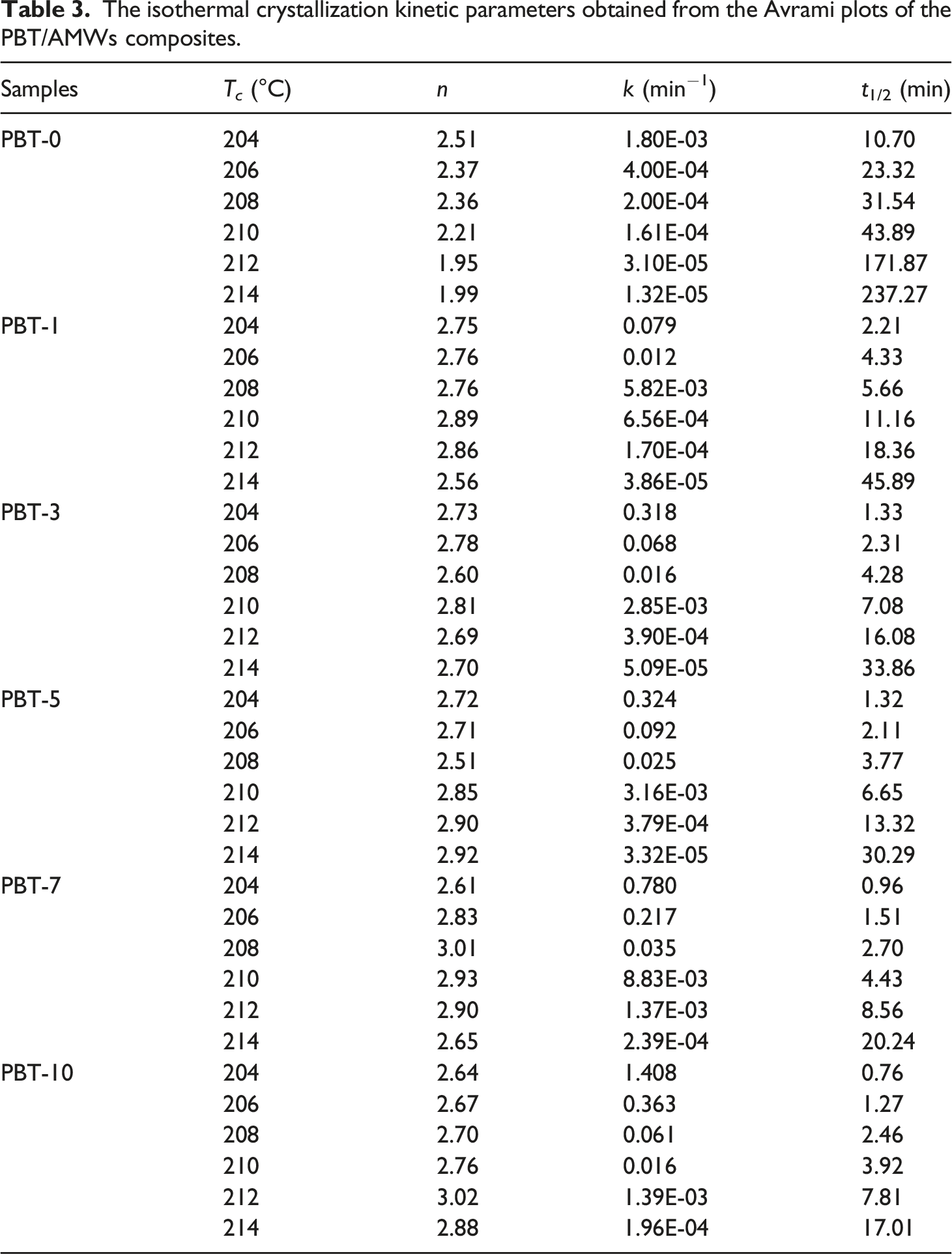
To more thoroughly study the effects of AMWs content on crystallization rate during the isothermal crystallization process, an important parameter t1/2 was considered, which can be estimated
43
as follows
As shown in Figure 9, with an increase in crystallization temperature Tc, the t1/2 value for the PBT/AMWs composites increased significantly, and the corresponding crystallization rate decreased. This indicated that the isothermal crystallization of the PBT/AMWs composites was controlled by nucleation. Therefore, it was evident that the crystallization time decreased rapidly at different crystallization temperatures after the AMWs were added, which agreed with the phenomenon observed in Figure 7. t1/2 of the PBT/AMWs composites at different crystallization temperatures.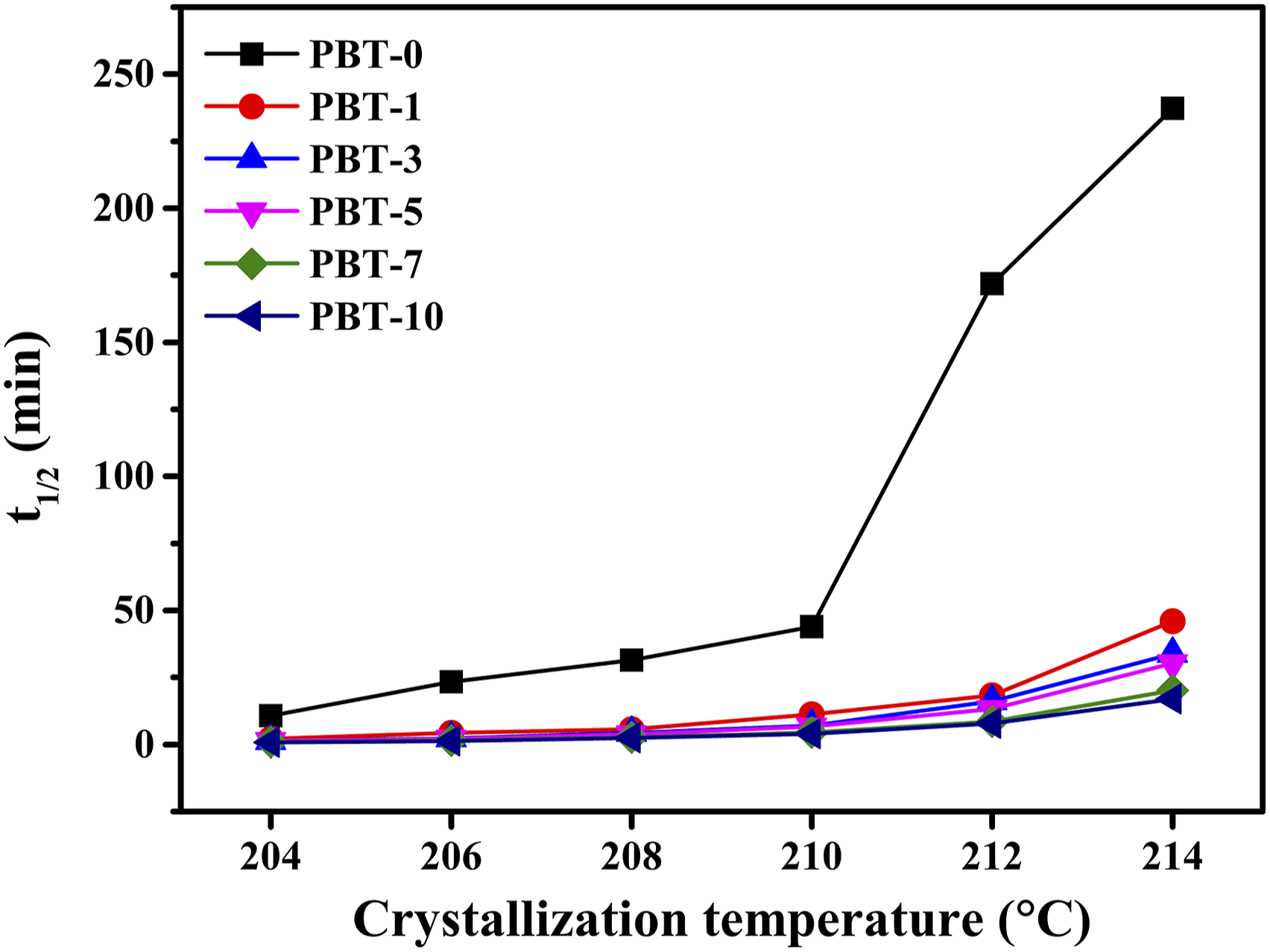
Mechanical properties analysis
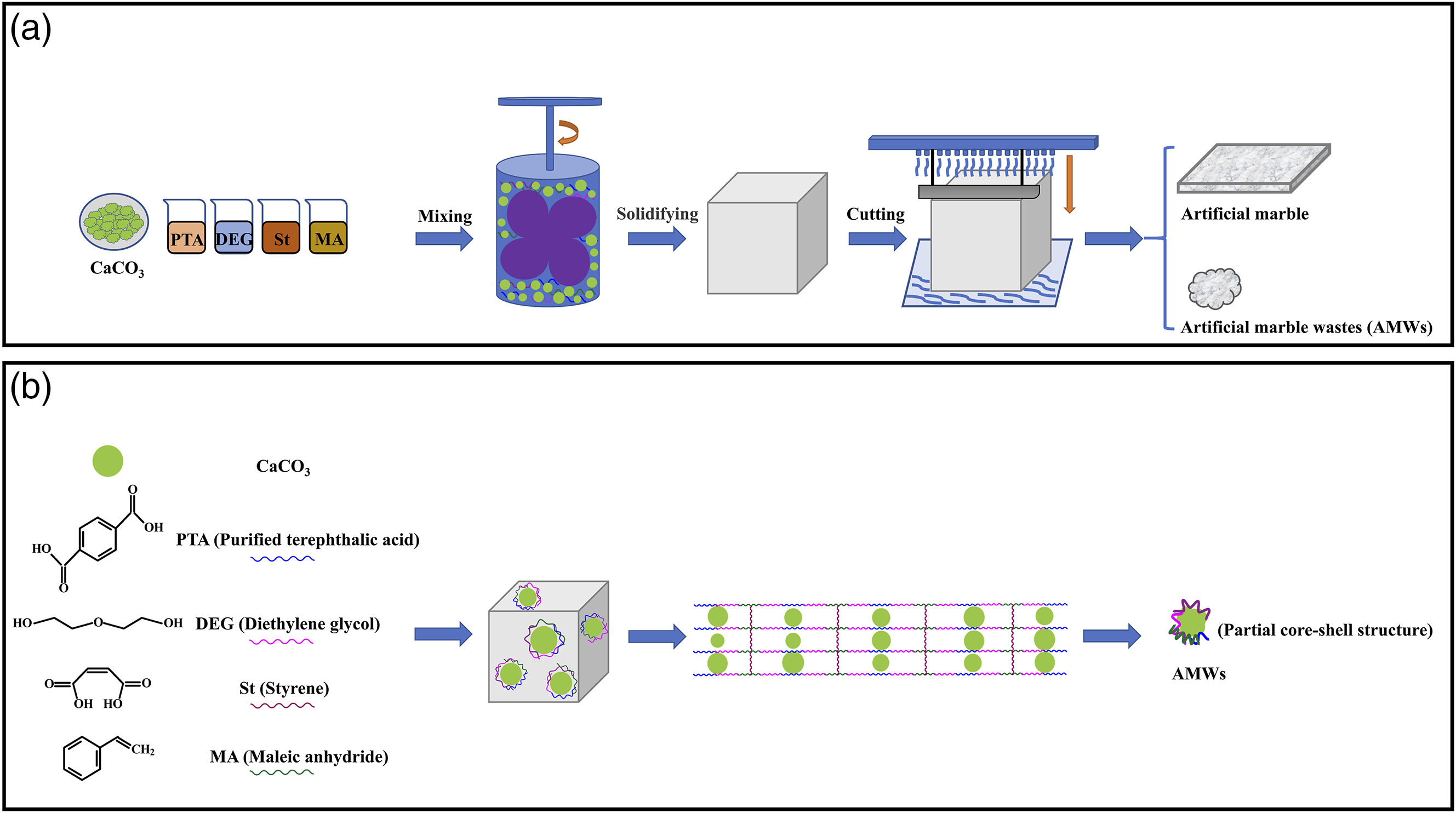
After composite preparation, the samples were crushed and fabricated into a specific size (80 mm × 10 mm × 4 mm) using a micro-injection machine for impact strength tests, as shown in Figure 10(a)–(c). By gradually adding the AMWs, the impact strength of PBT significantly improved from 5.79 to 17.29 kJ/m2, as shown in Figure 10(d). This was attributed to the AMWs particles, which had a partial core-shell structure during the formation process, as shown in Figure 11. The AMWs used in this experiment were treated through 1000 mesh wet sieving, the particle size was far smaller than the initial heavy calcium powder, which used in the synthesis of artificial marble, suggesting that the initial heavy calcium powder was cut open again in the cutting process, and the new section produced was not covered by unsaturated polyesters. This structure produced a toughening effect,45–47 which absorbed the impact energy and hindered the further propagation of microcracks. In addition, the hardness of the AMWs particles was higher than that of the PBT matrix, and the hardened matrix effect may have also contributed to the improvement in impact strength.
48
(a) Injection molding simulation diagram, (b) actual diagram, and (c) impact simulation diagram of a PBT/AMWs composite sample. (d) impact strength of the PBT/AMWs composites. (a) Formation of artificial marble and AMWs. (b) microstructure model of AMWs with partial core-shell structure.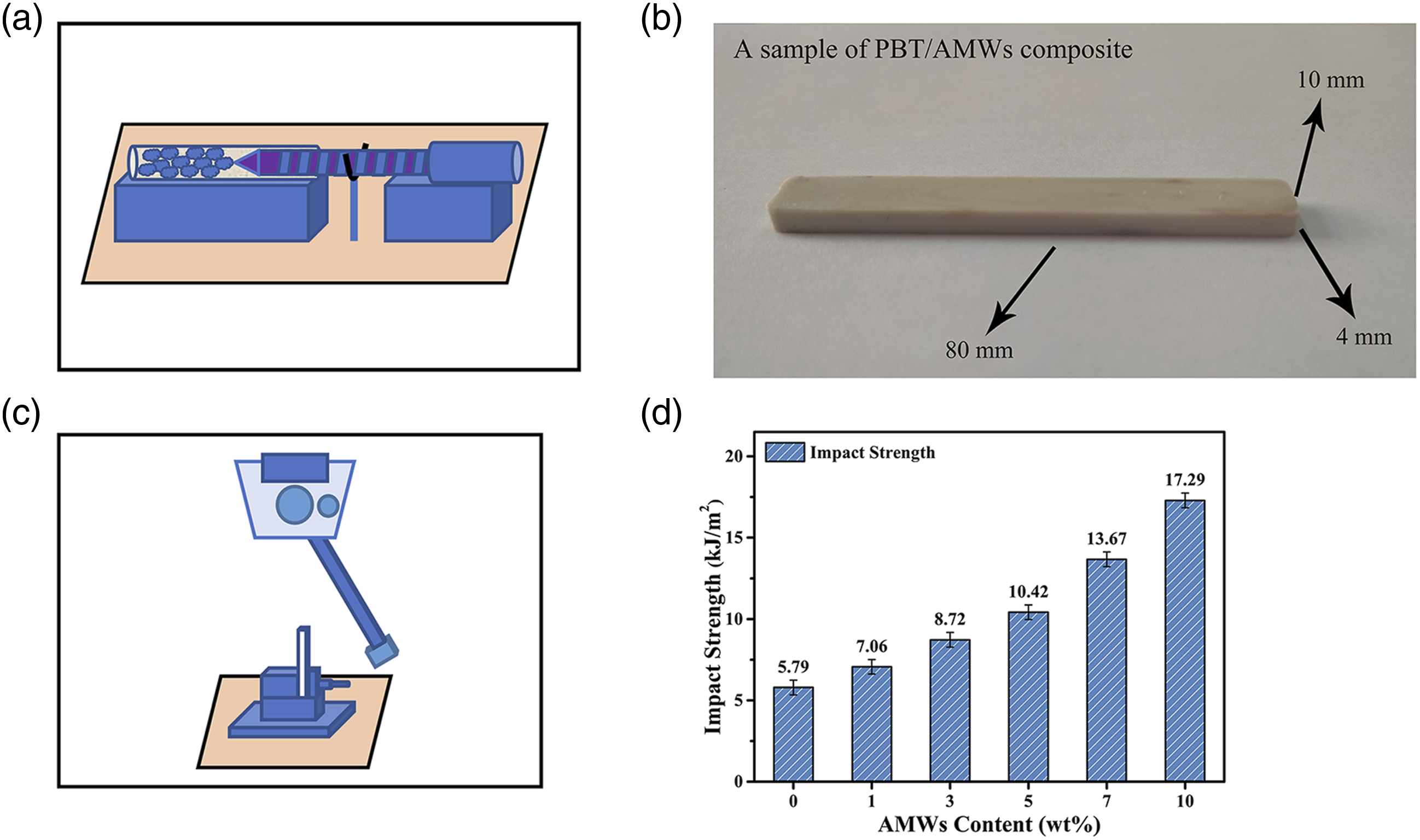
As shown in Figure 12, the flexural strength and tensile strength of PBT/AMWs composites were also investigated in this work. The flexural strength and tensile strength increased with the increase of AMWs content, and reached the maximum at 3 wt%. It was due to that the AMWs filled PBT composites had higher stiffness and hampered the free mobility of PBT chains.49 This was also related to the appropriate dispersion of AMWs in PBT matrix and the proper stress transfer between the PBT matrix and AMWs.50 When the AMWs content exceeded 3 wt%, the decrease of flexural strength and tensile strength may be caused by the increment in stress concentration. The flexural strength and tensile strength of PBT/AMWs composites.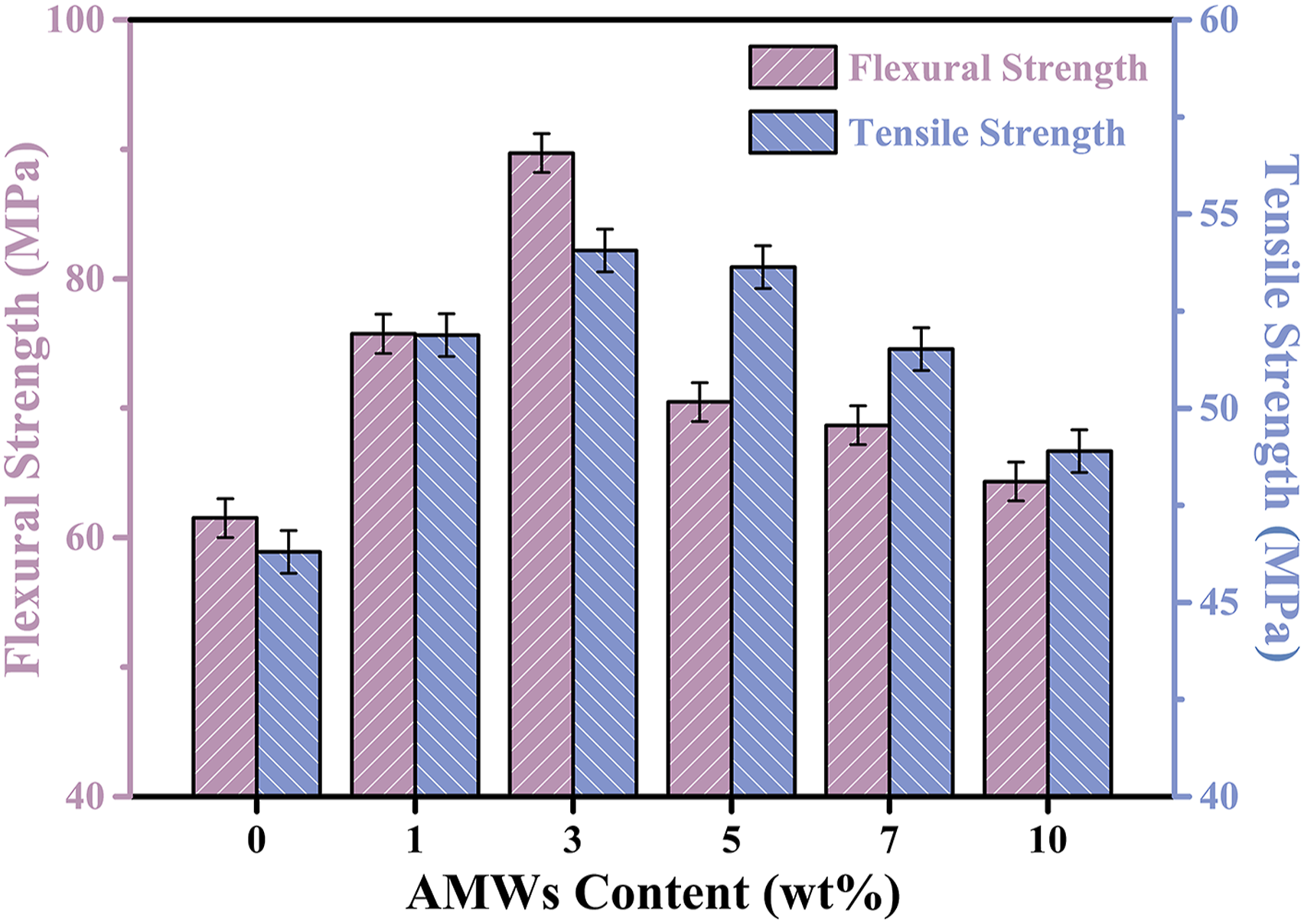
SEM analysis
SEM results indicated many fluvial-like steps on the impact fracture surface, which were not on the same plane when AMWs content increased, indicating enhanced interface interactions (Figure 13(a)–(f)). Starting with an AMWs content of 5 wt%, a wire drawing phenomenon appeared at marks ①, ②, and ③. Afterward, marks ③ and ④ become locally enlarged (Figure 13 (g) and (h)), and the wire drawing effect was evident and the distribution of the AMWs particles was uniform. This also indirectly indicated that the AMWs with partial core-shell structures toughened the PBT matrix and improved the impact strength. As a result, this agrees well with the impact strength test results. SEM images of impact fracture surface of the PBT/AMWs composites with the AMWs content of (a) 0, (b) 1 wt%, (c) 3 wt%, (d) 5 wt%, (e) 7 wt%, and (f) 10 wt%, respectively. (g) and (h) locally enlarged images of two places in graph (e) and (f), respectively.
Conclusion
In this study, PBT/AMWs composites were prepared by in situ polymerization, and the addition of AMWs into PBT enhanced the properties of the composite material, that is, thermal stability, crystallization rate, and impact strength. The results were attributed to two main reasons. Firstly, the AMWs particles possessed partial core-shell structures and were distributed uniformly throughout the matrix. Secondly, heterogeneous nucleation occurred at the interface between the AMWs particles and PBT. It is, thus, likely that the development of PBT/AMWs composites will provide opportunities for expanded PBT applications and the effective recycling of AMWs into high-value and high-performance composites.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of Guangxi Province (2018GXNSFAA281218 and 2019GXNSFAA245017), the Innovation-Driven Development of Hezhou (Hekechuang PT0710004), the Undergraduate Teaching Reform Project for Higher Education of Guangxi Province (2019JGZ150), and the Science and Technology Major Project of Guangxi Province (AA18242006 and AA18242008).