
Abstract
A series of blend fibers based on a certain weight polypropylene (PP) and poly(butyl methacrylate-co-hydroxyethyl methacrylate) (PBMA-co-HEMA) were prepared via melt spinning in a corotating twin screw extruder. The morphology and structure of the blend fiber was studied by stereoscopic microscopy, polarized optical microscopy, wide-angle x-ray diffraction and field emission scanning electronic microscopy. Thermal properties were carried out by means of differential scanning calorimetry and melt flow index. The results demonstrated that the crystalline structure is proven to be greatly destroyed owing to the entanglement and cross-linking of molecular chains caused by hydrogen bond network of the copolymer in the blend. Oil absorbency decreases with increasing mass fraction of PP in the blend fiber. Moreover, the enthalpy of crystallization and supercooling temperature decrease as the content of PBMA-co-HEMA component increases.
Introduction
The oil spill occurs quite frequently with influences on artificial factor, natural disaster and accident. 1 There are some oil spill remediation products available including but not limiting to dispersants, absorbents, bioremediation agents and other miscellaneous products like surface cleaners, gelling agents, demulsifiers, solidifiers, and so on. 2,3 Oil-absorptive materials of acrylate polymers are expected as a novel functionalized material, having a bright future in the industrial application. In the recent years, extensive research efforts have been devoted to the development of polymorphic oil-absorptive materials such as pellets or films and fibrous materials to adapt various situations. Oil-absorptive fiber has an inimitable advantage of further processing in textile or nonwoven industry, such that it is facile to absorption and treatment. 4,5 However, the fibration desire to produce improved products development is frustrated by acrylate copolymers’ poor processability. As generally known, preparation of polymer blend is one of the most effective ways for upgrading its spinability. New materials may exhibit a favorable combination of properties, depending on the selection of blend components. 6 Moreover, polypropylene (PP) fiber-based oil-absorptive products have been found to be mostly used to oil spill cleanup. 7 PP shows relatively low modulus, low density, excellent thermal and mechanical properties and good processability as well as its low price. Alkyl acrylate copolymers have hydrophobicity, lipophilicity and gel-type structure consisting of a three-dimensional elastic network, making them excellent candidates for swelling and entrapping oil molecules. 8–10
The advantage of both the comprehensive PP and alkyl acrylate copolymer is used for developing novel oil absorptive blend fiber. In our research, a series of blend fibers based on certain weight of PP and poly(butyl methacrylate-co-hydroxyethyl methacrylate) (PBMA-co-HEMA) have been prepared via melt spinning. As most polymers are immiscible, their blends form multiphase systems with various morphologies and synergistic properties. In this study, as-prepared blend fibers are extensively explored by the morphology (phase structure and crystalline behavior) and thermal behaviors. These studies have been performed by means of wide-angle x-ray diffraction (WAXD); differential scanning calorimetry (DSC); field emission scanning electronic microscopy (FESEM), polarized optical microscopy (POM) and optical microscopy; melt flow index (MFI). In addition, oil absorbency and gel fraction measurement are investigated in this work. The miscibility of PP/PBMA-co-HEMA blends in the ambient and melt will also be discussed. The blend fibers exhibit higher oil absorbency than PP fiber. Such blend fibers are expected to have potential applications in oily wastewater cleanup.
Experimental
Materials
PP (PP H-T03) was supplied by Sinopec Corp., China); butyl methacrylate (BMA, M w = 142.20, d = 0.896 g/cm3, distilled before use) was purchased from Tianjin Fuchen Chemical Reagents Co. Ltd., Tianjin, China; hydroxyethyl methacrylate (HEMA) was purchased from Tianjin Chemical Research Institute (Tianjin, China) and used as-received; benzoyl peroxide (BPO; recrystallized) was purchased from Shanghai Chemical Reagent Co. Ltd., Guangdong, China; poly(vinyl alcohol) (PVA; purified by washing with deionized water) was purchased from Tianjin Chemical Reagent Co. Ltd., Guangdong, China.
Synthesis of PBMA-co-HEMA copolymers
PVA of 24 g was added as the dispersant agent for the suspension polymerization, dissolved in a 10-L polymerizer containing 4.8 L deionized water before copolymerization. A mixture of 1.53 L BMA, 67.3 mL HEMA and 7.2 g BPO was stirred to obtain a solution, and this homogeneous solution was transferred into the polymerizer. The experiment was carried out under a nitrogen atmosphere using standard suspension polymerization techniques. The mixed solution was stirred at 85°C to react for 5 h, and then another 1 h at 95°C. Finally, the product was washed and dried to obtain a white copolymer.
Preparation of oil-absorptive fiber
Both PP and PBMA-co-HEMA were dried in a vacuum oven at 60°C for 48 h before melt blending to remove moisture, completely. PP and PBMA-co-HEMA blends with different compositions were prepared using a SHJ-20 corotating twin-screw extruder (Nanjing Jieya Corporation, China) at 200°C. The composition ratios by weight of PP and PBMA-co-HEMA were 10/0, 9/1, 7/3, 5/5, 3/7, 1/9 and 0/10, respectively. Note that pure PBMA-co-HEMA exhibited no spinnability with poor melting fluidity. Primary strip blends were pelletized by granulating process of uniform mixture, while the constituent polymers were mixed to achieve intimate mixing by a grinder into the powder. The melt spinning was carried out using a twin-screw extruder equipped with a spinneret having a diameter of 0.5 mm and 36 holes (L/D 30) followed by air-cooling. The barrel temperatures were set to 195, 195, 195, 200, 205 and 205°C in the direction of hopper-to-nozzle for the first zone, the second zone, the third zone, the fourth zone, the fifth zone and the sixth zone, respectively. In addition, the melt temperature at the outlet and spinneret temperature were 200°C and 180–190°C. The rotating speed of the screws was maintained at 30 r/min and the melt pump rotation speed was about 80–90 r/min. These processing conditions resulted in a final diameter of the fibers between 0.6 and 0.7 mm. Subsequently, the fiber was processed by winding, cutting off, washing by distilled water and drying at room temperature, and eventually, the fiber was preserved for the test and analysis.
Oil absorbency and gel fraction measurement
Oil absorbency of the fiber was evaluated by ASTM (F726-81); briefly, 0.1 g fiber was put into a stainless steel mesh (4 × 4 × 2 cm3) and the mesh was immersed in toluene. The fiber and the mesh were together picked up from toluene or crude oil solution (crude oil diluted with toluene, 10% oil), drained for 20 s, tapped with filter paper to remove excess oil from the bottom of the mesh, and then weighted. The oil absorbency (Q) was calculated by the following equation (equation (1))
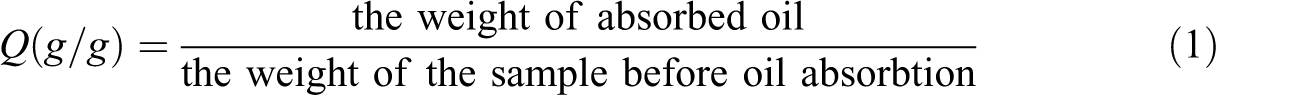
A weighed quantity of the fiber was put in Soxhlet extractor and continuously extracted for 8 h, using butanone as solvent because of its low boiling point and ability to dissolve methacrylate. After extracting, the samples were dried in vacuum at 60°C until a constant weight. The gel fraction (G) was calculated according to the following equation (equation (2))
11
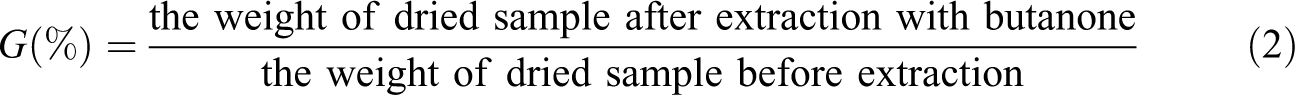
MFI measurement
MFI of polyethylene was obtained from the supplier Sinopec corp., (MFI 3.8 g/10 min, 230°C, 2.16 kg load, according to manufacturer). The blends were mixed using SHJ-20 corotating twin-screw extruder at 200°C. MFI was investigated using a XNR-400A MFI instrument (Chengde Testing Machine Co., Ltd, China). According to ASTM D1238-04 standard test method for melt flow rates of thermoplastics by extrusion plastometer, the data on the composition of the PP/PBMA-co-HEMA blends by mass were displayed in Table 1. The fluidity reflected by MFI measurement can be considered as a guide for processing condition.
MFI parameters of the PP/PBMA-co-HEMA blends.
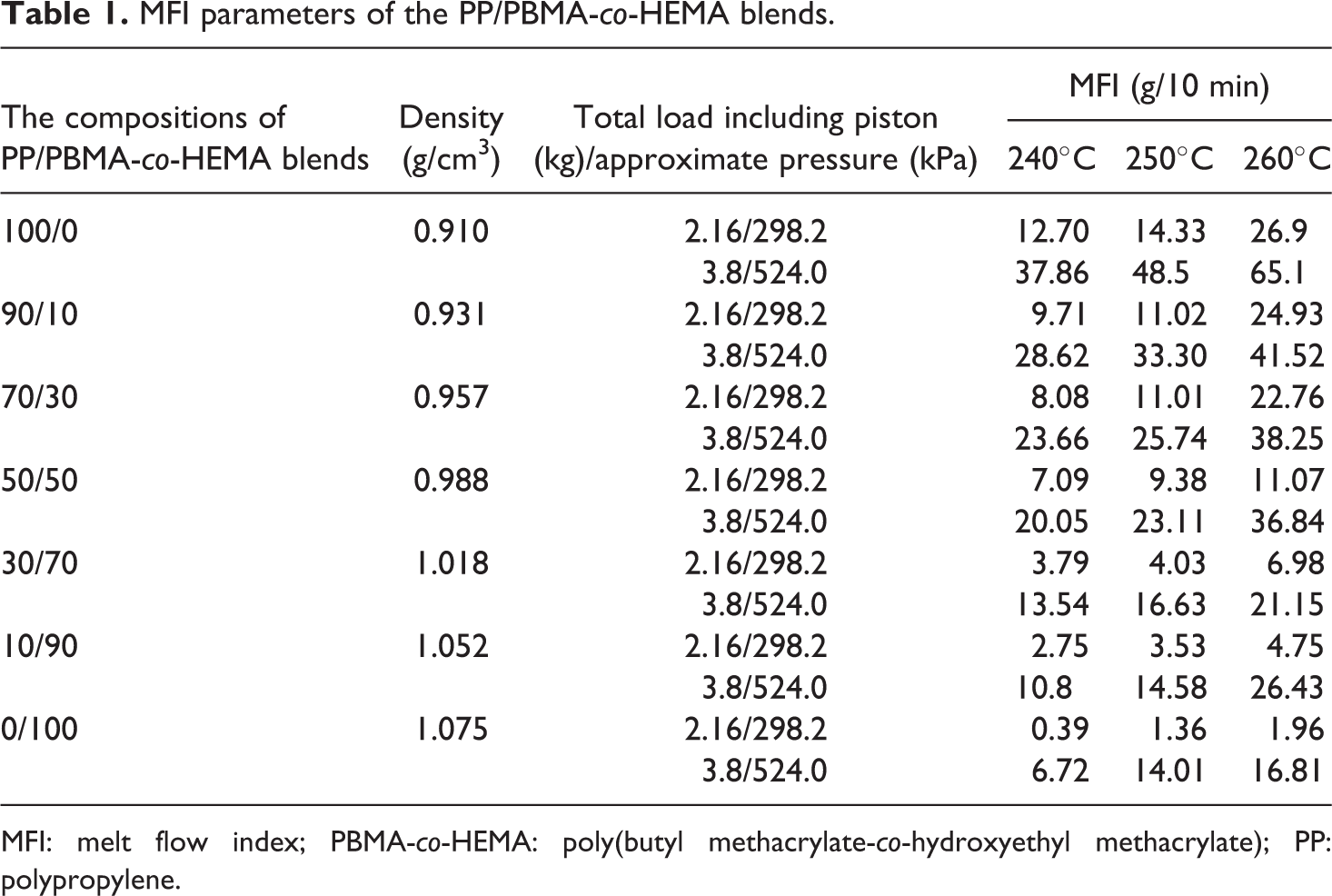
MFI: melt flow index; PBMA-co-HEMA: poly(butyl methacrylate-co-hydroxyethyl methacrylate); PP: polypropylene.
Morphology analysis
Dynamic absorption of the oil-absorptive fiber in toluene was observed by the photograph from a stereoscopic microscope (XTS30, TaiKe, Beijing). Crystallization of PP was observed with a POM. The morphology of the blend fiber was observed using an Olympus BX51 POM, with a Linkam-THMSE-600 automatic thermal control hot-stage (the controlling temperature precision of 0.1°C, Olympus Optical, Japan). The samples were heated to T max (185°C) annealing for 2 min and then cooled to crystallization temperature for crystallizing processing. The morphology change during the whole process was recorded for further analysis. All optical micrographs presented in this article were taken under crossed polarizers.
The morphologies of the fiber were monitored by a field emission scanning electron microscopy (FESEM, Hitachi S-4800, Japan). The cross-section and longitudinal surface of the fiber were coated with gold by an electrodeposition method to impart electrical conduction before recording the FESEM micrographs.
WAXD study
Crystalline structures of the fibers were analyzed by wide angle x-ray diffractometer (Bruker AXS Cooperation/D8 DICOVER with GADDS, German) with 4°r/min scanning speed. The fiber was laid on the fiber sample holder. Diffractometer with copper cathode was operated using Ni-filtered Cu-Kα radiation (λ = 1.5406 Å) that was generated at a voltage of 40 kV and a current of 40 mA and on a rotation from 10° to 60° at a 2θ scale.
DSC analysis on melting and crystallization
The DSC experiments were carried out using a DSC instrument (Netzsch DSC 200 F3, Germany). The sample of round 5 mg of the fiber was sealed in an aluminum pan and placed in the DSC cell. All the measurements were performed under nitrogen atmosphere at a heating rate of 10°C/min, and the temperature range was from ambient to 210°C, held isothermally for 2 min, and then cooled to ambient and heated to 210°C again.
The crystallization temperature (T c), the melting temperatures (T m) and enthalpies of fusion (▵H m) of the blends were observed and calculated from the maximum and the area under the endothermic peak, respectively.
Results and discussion
Oil absorbency
Oil absorbency of the sample fiber is a very important index in application. It has been reported that the excellent oil absorbency of the materials depends on the bulkiness and length of the alkyl substituents 12 and especially the porosity of the microstructure that can be controlled by cross-linking. It has been verified that a plenty of hydrogen bonding, interlocking and entanglement were constructed by the hydroxyls among the macromolecular chain in BMA and HEMA copolymer in our previous articles. 4,13
Crude oil with high viscosity contains innumerous individual components with varying proportions. Toluene is a prominent representative of light oil fractions. Because of its low viscosity and density, toluene is the most applicable solvent that is used to dissolve crude oil. Hence, an attempt was made to study the absorption of toluene and 10% crude oil in Figure 1.
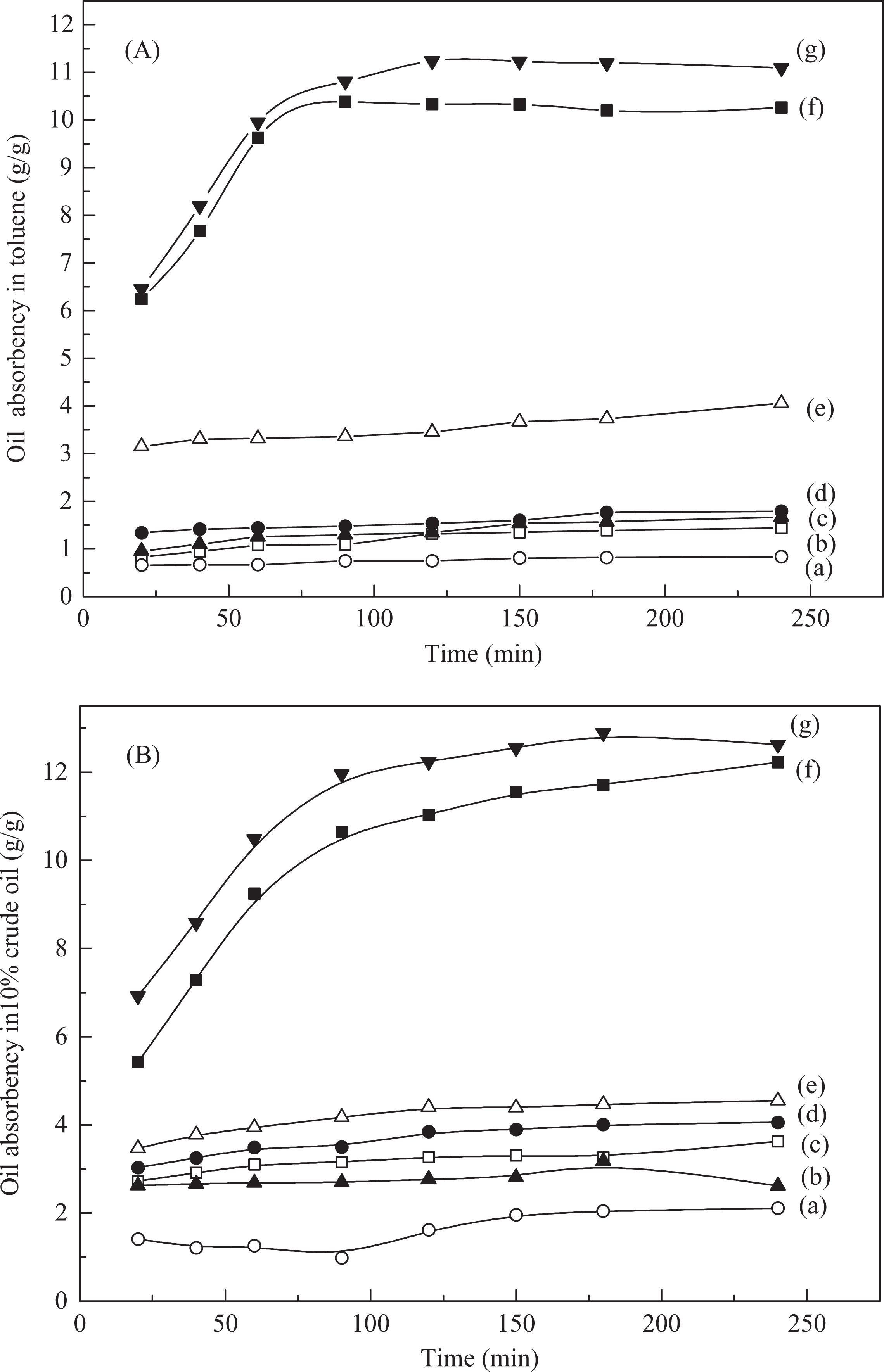
Oil absorbency of as-prepared samples in toluene (A) and in 10% crude oil (B) to the immersion time at different blend ratios (PP/PHEMA-co-PBMA: (a) 10/0, (b) 9/1, (c) 7/3, (d) 5/5, (e) 3/7, (f) 1/9, (g) 0/10). PP: polypropylene; PHEMA-co-PBMA: poly(hydroxyethyl methacrylate)-co-poly(butyl methacrylate).
These figures show that the oil absorbency increases with increasing immersion time and ultimately attains its equilibrium. On the other hand, Figure 1(a) shows oil absorbency of the blend fiber with increasing mass fraction of PP decreases with time in toluene. The same trend is seen in Figure 1(b) for blended fibers immersed in 10% crude oil. High PP content may be negative to hydrogen bonding, interlocking and entanglement and obstructs in the formation of polymer network structure and decreases the swelling further. However, matrix dispersed phase structure in the blends is full of small cavities of the polymeric network, which can provide larger absorption surfaces and give higher swollen rate. Gel fraction as a parameter commonly represents the solubility of polymer materials in a certain solvent. Here, as the PP amount was beyond 10 wt%, insoluble fraction significantly increased as shown in Figure 2. The results indicate that the PP, as a skeleton column, restrains the swelling and resolution of PBMA-co-HEMA in extract processing. The high PP content is negative to oil absorption, but have an adequate physicomechanical property for spinability.
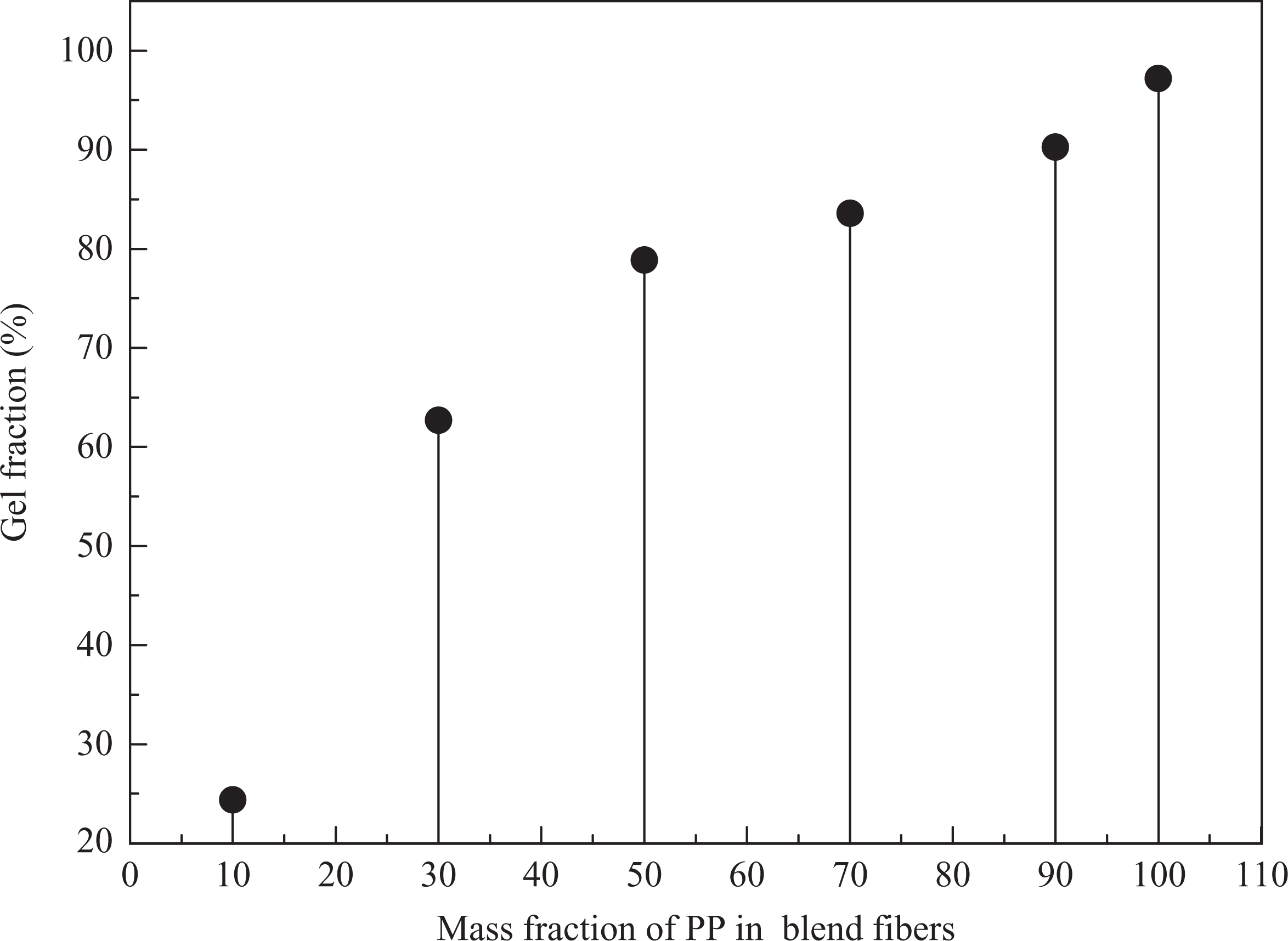
Relationship between gel fraction and the contents of PP in blend fibers. PP: polypropylene.
Optical microscopy results
There is a significant change in cross-section of the fiber in toluene treated by absorption, as depicted in Figure 3. Optical cross-section of the as-prepared fiber has a uniform and flat surface. After being immersion in toluene, the appearance displays a coarse pane. The raised star points represent the blend fiber skeleton. The component is probably presumed to be PP fibrils, as it is verified in Figure 4(b).

Cross-section changes of the fiber in toluene: PP fiber: ×40 (a), ×100 (b); blend fiber: ×40 (c); ×100 (d). PP: polypropylene.

Cross-sectional FESEM images of PP fiber (a) and PP/PBMA-co-HEMA fiber (b); FESEM micrographs for surface morphologies of PP fiber (c) and PP/PBMA-co-HEMA fiber (d). PBMA-co-HEMA: poly(butyl methacrylate-co-hydroxyethyl methacrylate); PP: polypropylene; FESEM: field emission scanning electronic microscopic.
Figure 4(a) and (b) shows the morphology of the fracture surface of pure PP and binary (noncompatibilized) blend fiber. Cross-section of PP fiber (Figure 4(a)) appears plain with smooth edges; FESEM analysis from cross-section micrographs (Figure 4(b)) reveals two-phase morphology with the polydispersity of the blend fiber. Poor adhesion between the phases can be observed in Figure 4(b). It is noteworthy that the smaller dispersed PP particles in the blends matrix as a result of high interfacial tension and coalescence at fiber cross-section have been presented by the high-magnification SEM image shown in Figure 4(b). Once a part of PP is removed from the surface, some particles are exposed.
In addition, it is interesting to note that the naked PP microrods provide the skeletal structure of fiber composite. Furthermore, PP, also known as an excellent engineering material, possesses strong mechanical property; evidently, the increasing PP incorporation exhibits higher performance. It is also verified that the well-regulated crystalline PP is of good mechanical properties with respect to Figure 5. Both of the polymers are immiscible, and therefore, there is a kind of interfacial microcavities and rugged topography for organic liquid adsorption and storage. Furthermore, PP is dispersed in the core of the copolymer matrix as fibrillate representing a potentially new approach and valuable reference to attain a sea-island fiber microstructure. In terms of surface micrographs (as shown in right inset of Figure 4(c) and (d)), the blend fiber exhibits a coarse and lumpy appearance.

Typical polarized optical micrographs of the fiber: PP and PP/PBMA-co-HEMA (wt%, 5/5) with crossed polars. PBMA-co-HEMA: poly(butyl methacrylate-co-hydroxyethyl methacrylate); PP: polypropylene.
Concerning crystalline morphologies, a continuous–discontinuous type crystalline morphology is shown in Figure 5. Pure PP fiber indicates a large bright prolate shape, heated to T max annealing for 2 min and then quenched to crystallization temperature (as depicted in the right inset of Figure 5); it seems that a uniform spherulite growth and particle size distribution are obtained. The blends with an interlocked morphology bring in scintillate droplets when melting. The featureless crystallizing agglomeration shows obscured phase morphology from the blend fibers. On the whole, the increase in the copolymer results in the variation of spherulite growth and the decrease in the crystalline perfection.
WAXD analysis
PP has a complex crystal system, which exhibits several crystalline forms: monoclinic or α-form, trigonal or β-form and triclinic or γ-form, even little information on secondary crystallization, depending on specific crystallization conditions. At low nucleation barrier, as a kinetic result of crystallization, the formation of α-crystal is facilitated. 14,15 As shown in Figure 6, the characteristic reflections of the α-form of PP can be found at scattering angles 2θ of 14.0° (100) plane, 17.08° (040) plane, 18.6° (130) plane and 21.2° (111), implying that there is good orientation of sharp peak shape at 13.9°. For merely PHEMA-co-BMA copolymer, it should be noted that the reflections of crystal at 11° and 15° are very weak, a board diffraction peak appears in the vicinity of 18°. Unobvious crystallization is subjected to the entanglement of long alkyl substituent and hydrogen bonding. However, the majority in the amorphous state is of benefit to oil molecules invasion. The macromolecular chains are relatively easier to stretch in the loose network structure at the occurrence of swelling, while imperfect orientation and crystallization lead to poor mechanical properties of fibers. In addition, judging by the figures, we can draw a conclusion that the blends of PBMA-co-HEMA copolymer have frequently been related to the obvious fact of decreasing crystallization even for the quietly limited copolymer content.
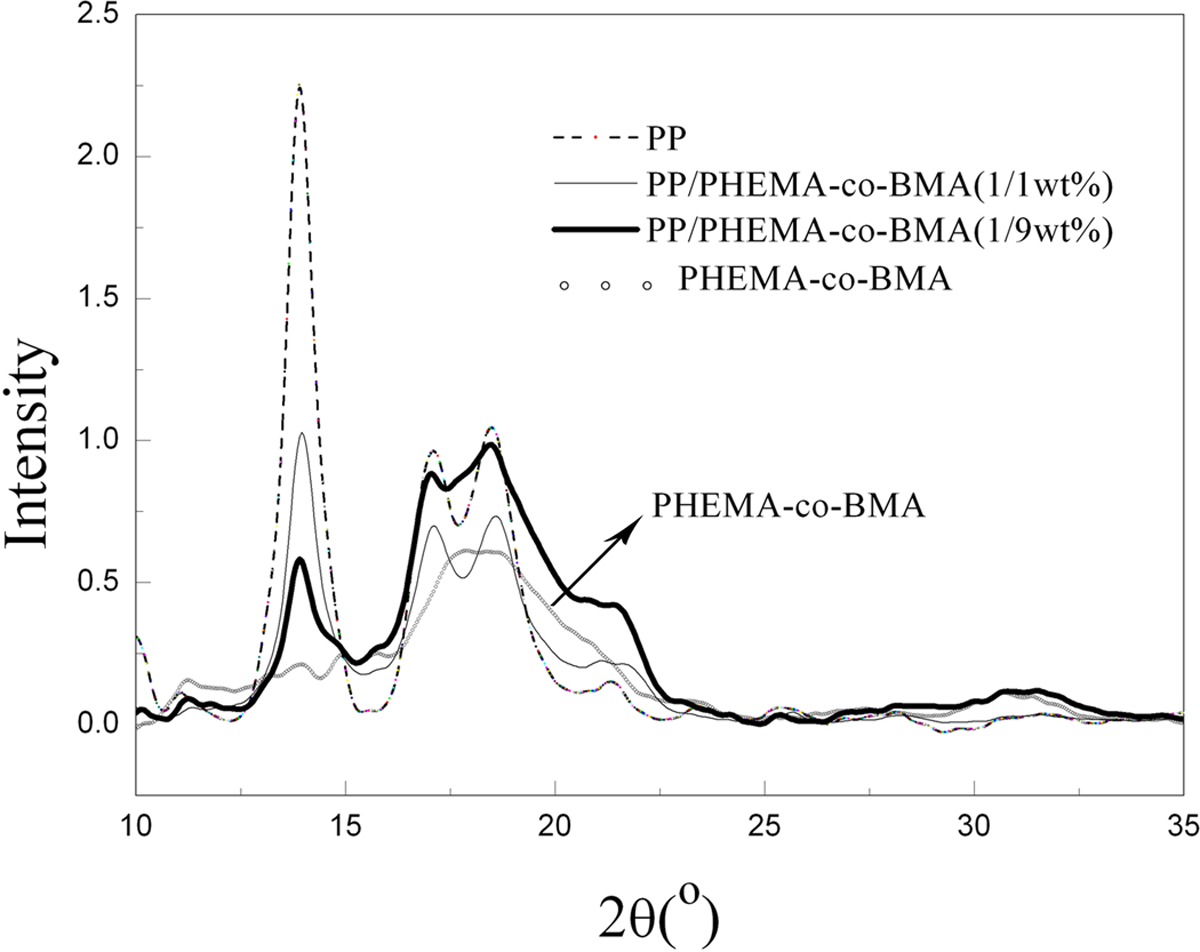
Wide-angle x-ray diffraction patterns.
DSC results
DSC measurement is used to estimate the mass fraction crystallinities of PP and their corresponding phases in blends. Some information can be obtained about the crystalline structure changes occurring in the blends. 16 Melting thermograms of the blends display only one melting peak due to amorphous structure of physical cross-linked PBMA-co-HEMA. In order to analyze the effect of the copolymer on the PP crystallization, the melting curves, as recorded from the blends versus the copolymer percentage, are graphically represented in Figure 7. The relative parameters are listed in Table 2. There is a decrease in the degree of crystallinity for PP with the increasing PBMA-co-HEMA, which can be revealed by the decline of ▵H c (enthalpy of crystallization). The melting peak of the blend fiber occurs at varying temperatures, although it always melts in the range 160–165°C as depicted in Figure 7(b). The increasing T c displayed in Table 2 demonstrates that the addition of PBMA-co-HEMA promotes the initial crystallization,
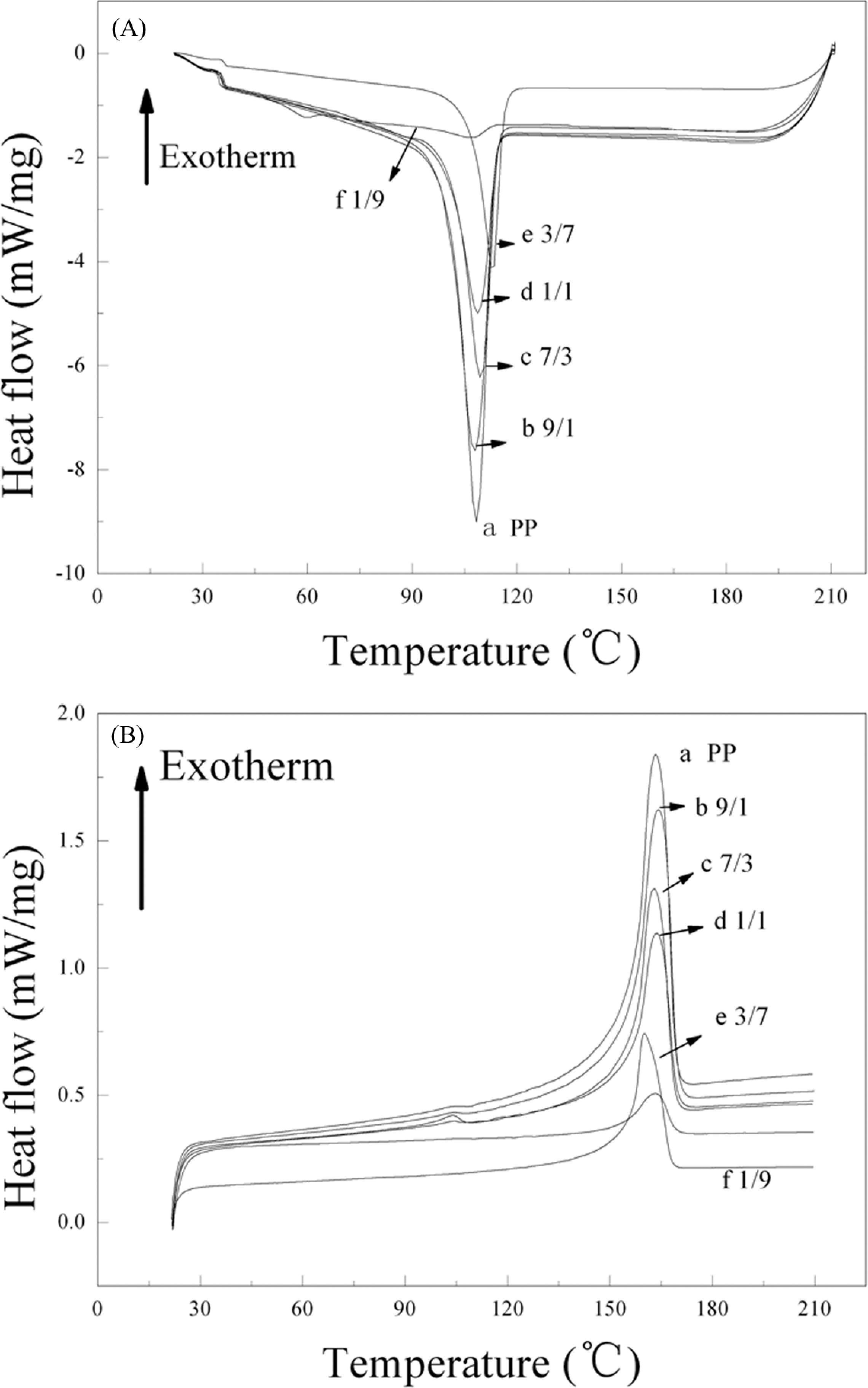
DSC curves of (A) cooling scans and (B) the secondary heating scans (PP/PBMA-co-HEMA: (a) 10/0, (b) 9/1, (c) 7/3, (d) 5/5, (e) 3/7, (f) 1/9). PBMA-co-HEMA: poly(butyl methacrylate-co-hydroxyethyl methacrylate); PP: polypropylene; DSC: differential scanning calorimetry.
Melting parameters of the blend fibers.
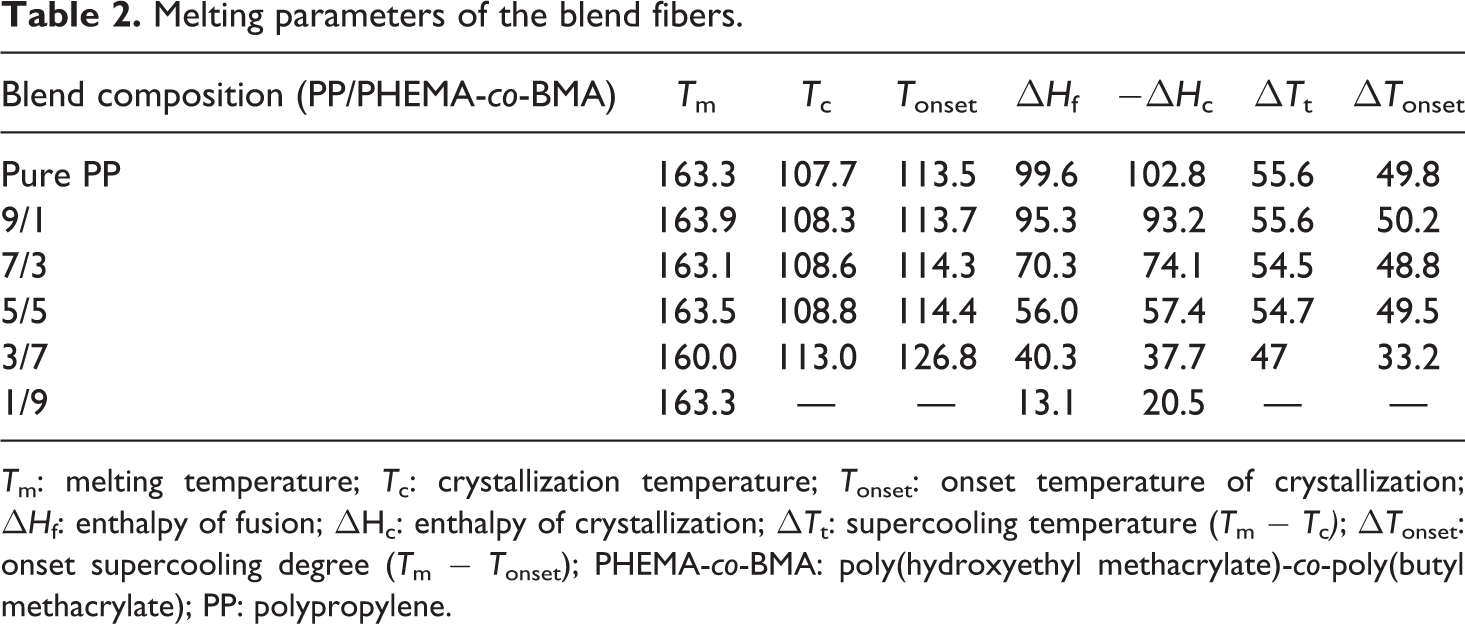
T m: melting temperature; T c: crystallization temperature; T onset: onset temperature of crystallization; ▵H f: enthalpy of fusion; ▵Hc: enthalpy of crystallization; ▵T t: supercooling temperature (T m − T c ); ▵T onset: onset supercooling degree (T m − T onset); PHEMA-co-BMA: poly(hydroxyethyl methacrylate)-co-poly(butyl methacrylate); PP: polypropylene.
Moreover, ▵T t in the cooling experiment decreases as the content of the copolymer in blends increases. The decline of ▵T t is observed in this study in comparison with the plain PP, which can be attributed to a nucleating effect of PBMA-co-HEMA on the PP crystallization. Meanwhile, the nucleating effect of the copolymer causes an increase in initial crystallizing rate. A similar crystallization mechanism has been previously presented in other literatures. 17,18 The decline of ▵H c indicates that some molecular chains or segments are not preferentially recrystallized during cooling process, that is, the process of crystalline is hindered due to long-alkyl substituents entanglement interaction in polymers. Therefore, it can be concluded that PBMA-co-HEMA plays two roles in the crystallization of the blend: it acts as a heterogeneous nucleating agent to facilitate crystallization in the initial phase and as a physical hindrance to retard further crystallization.
Conclusion
In this study, PP and PBMA-co-HEMA blend fibers are prepared and investigated by melt spinning and various testing. The results demonstrate that the fiber swelling in toluene is subjected to fetter from PP skeletal structure, the polymers with higher PP content have lower oil absorbency. A systemic study on morphology suggests two-phase morphology composite structures in the blends. The addition of the copolymer restrains excellent melt fluidity of PP. Moreover, crystalline structures of individual polymers are obviously affected by blending. The limited copolymer content has a marked impact on the PP crystallization, which was analyzed using WAXD and DSC.
Footnotes
Funding
This work was supported by the National Natural Science Foundation of China (Project number: 50673077).