
Abstract
Phenyl molybdate-modified phenolic fibers (PMoPFs) were prepared by melt spinning from the corresponding resin which was polymerized from phenol, formaldehyde, and phenyl molybdate, followed by solution curing and then heat curing processes. The molecular structures, including characteristic groups and molecular weight, were examined by nuclear magnetic resonance, Fourier transform infrared spectroscopy, and gel permeation chromatography. The influences of the curing processes were demonstrated by mechanical properties, scanning electron microscopy, and thermogravimetric analysis characterization. PMoPF with 8 wt% molybdic acid and cured in an oven possessed a tensile strength as high as 187 MPa, initial decomposition temperature of 300°C, and a char yield under nitrogen atmosphere at 800°C as high as 66.0%, with the molybdate (MoO4 2−) groups being introduced into the phenolic main chain.
Introduction
Phenolic fibers have been widely applied in flame resistance fabrics, friction composites, and as precursors of carbon and activated carbon fibers for almost a half century, 1,2 because of their high thermal stability properties, good bonding performance, and high char yield. 3 -5 For many extreme conditions, such as in the aerospace and nuclear industries, the high heat and ablative resistance properties of phenolic fibers are needed. Boron-containing phenolic fibers and resin with high bond energy and the long bond distance of B–O were prepared to improve flame resistance and heat resistance of fibers 6 -8 but resulted in decline of mechanical performance and spinnability because of the uncontrollable cross-linked network formed by the tri-functionality of B–O during the synthesis and spinning. Molybdenum containing phenolic resin has shown a high heat resistance in fiber-reinforced composites 9,10 and grinding applicability as the matrix resin binder for diamond grinding tools, 11 where the molybdate was added into the flask after the addition reaction between phenol and formalin. But the low reactivity of molybdate and hydroxymethyl phenol had an adverse effect on the thermal and mechanical performance induced by the resulting inhomogeneity of the macrostructure. The modified resins with the unreacted molybdate particles showed poor spinnability. As a result, molybdenum-modified novolac fibers remain unused in industry.
In this article, we describe phenyl molybdate (PMo)-modified phenolic resins (PMoPRs) synthesized and melt-spun at 125°C. The PMo-modified phenolic fibers (PMoPFs) were cured in a combined solution of formaldehyde and hydrochloric acid and then heat-cured up to 240°C under nitrogen (N2) atmosphere. A number of characterization techniques, including Fourier transform infrared (FTIR) spectroscopy, nuclear magnetic resonance (NMR), scanning electron microscopy (SEM), thermogravimetric analysis (TGA), and tensile tests, indicated that the molybdenum-modified novolac fibers possessed a homogenous micro- and macrostructure, high char yield, and excellent mechanical properties.
Experiment
Materials
All chemicals were of analytical grade and used without further purification. Phenol, hydrochloric acid, and zinc acetate were supplied by Tianjin Fengchuan Chemical Reagent Co. Ltd (China). Formaldehyde (37 wt%) and sulfuric acid (98 wt%) were supplied by Xilong Chemical Co. Ltd (China). Molybdic acid (MA) was supplied by Tianjin Guangfu Fine Chemical Research Institute (China).
Preparation of PMo
PMo was prepared by the reaction of phenol and MA in the presence of hydrochloric acid. 12 Phenol (1 mol), MA (0.5 mol), and hydrochloric acid (1/30 mol) were added to a three-necked flask equipped with a reflux condenser, stirrer, and thermometer and then the solution was heated, stirred, and maintained at 60°C for 2 h, and the reaction mixture was then distilled for 2 h under vacuum to remove the hydrochloric acid. The reaction is shown in Figure 1. The resultant molten PMo was filtered to remove the particles of the unreacted MA.

Reaction scheme for the preparation of PMo.
Preparation of PMoPF
Phenol (100 g), formaldehyde (71.8 g), and zinc acetate (2.0 g) were added into a three-necked flask with various amounts of PMo, equipped with a reflux condenser, stirrer, and thermometer. The mixture was heated and maintained at 94°C for 4 h, and then sulfuric acid (0.3 mL) was added, followed by further heating under reflux for 50 min. The reaction mixture was distilled under vacuum to remove water and unreacted phenol at an elevated temperature, up to 115°C, for 3°h. The reaction is shown in Figure 2. A series of novolac resins (PMoPR) was obtained by changing the concentrations of the PMo (0, 2, 4, 6, 8, 10, or 12 g), all relative to the 100 g of phenol.
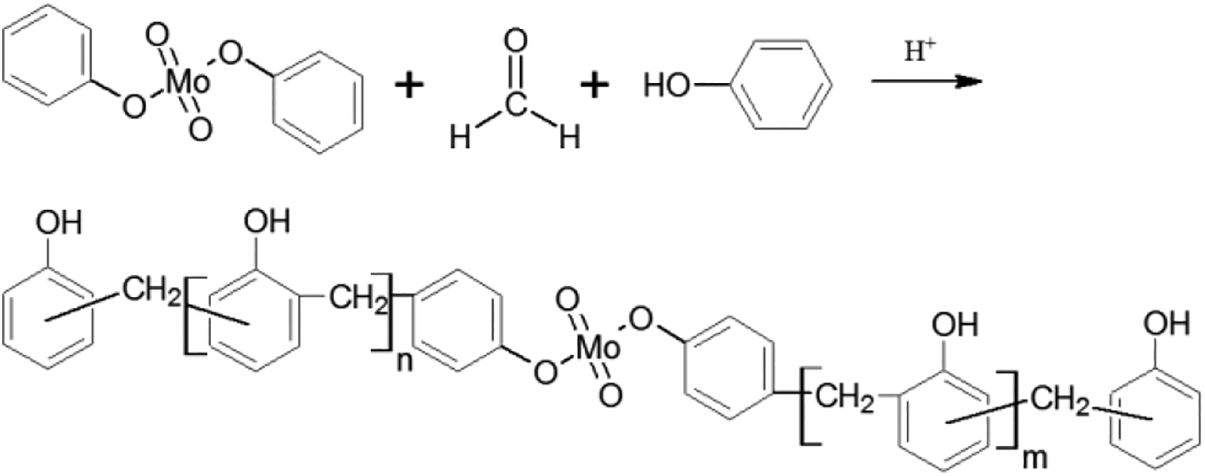
Synthesis route for the PMoPR.
PMoPFs were prepared by melt-spinning using the molten PMoPR around 125°C with a winder speed of 430 m min−1. The as-spun fibers were cured in a combined solution of hydrochloric acid (12.5 wt%) and formaldehyde (18.0 wt%) in a bath equipped with a thermometer and stirrer. The solution was heated from room temperature to 95°C at a heating rate of 15.4°C h−1, and then maintained at 95°C for 2 h. The fibers were removed from the curing bath, washed and dried, and then heated in a vacuum oven at a rate of 2.5°C min−1 to 240°C and then held for 2 h under N2 atmosphere. Finally, the cooled samples were washed with water and dried at room temperature. The cross-linking reactions occurring in the solution stage and heat curing are shown in Figure 3.
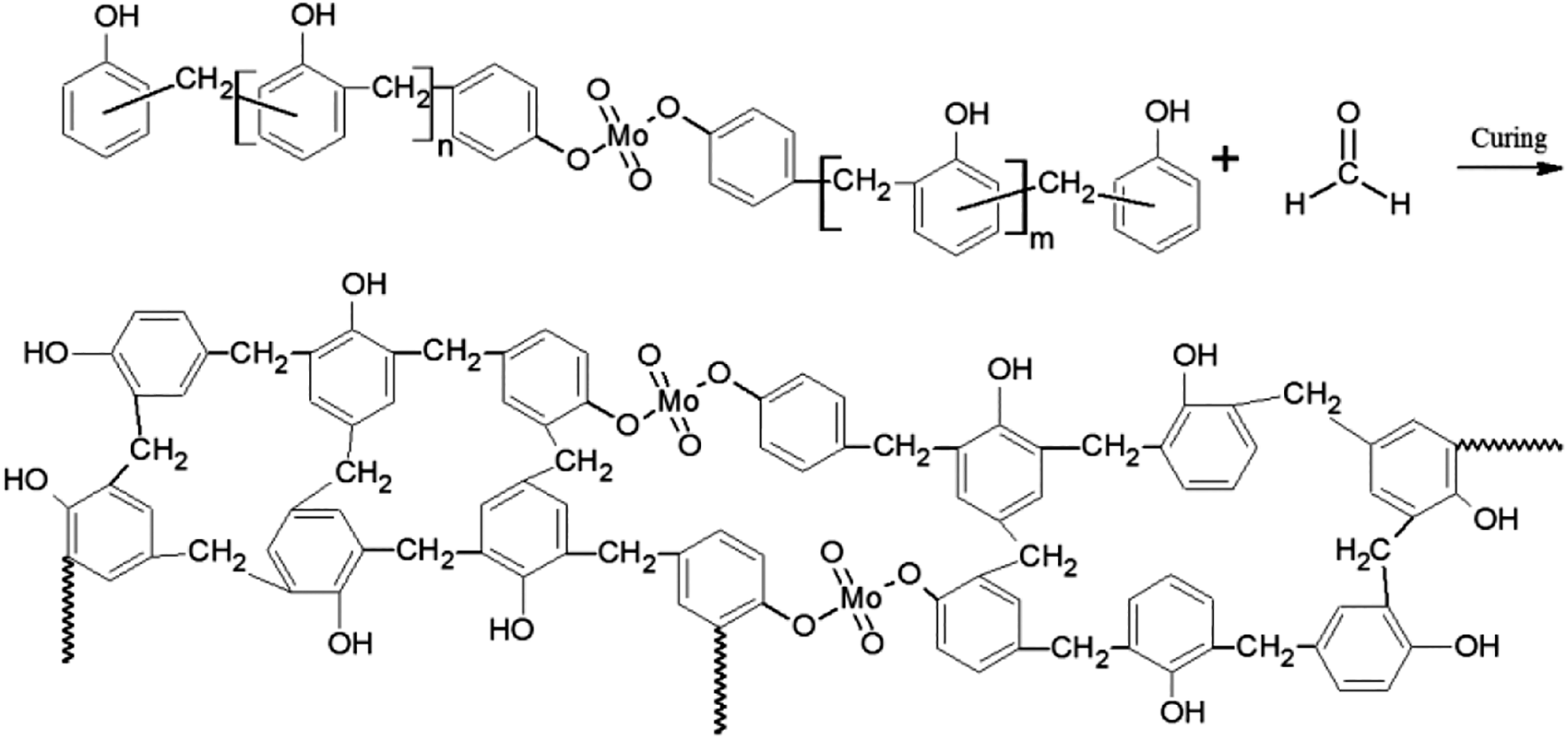
A possible cross-linking reaction for the preparation of PMoPFs while in the solution and/or during the heat curing.
The obtained resins contained PMo of 0, 2, 4, 6, 8, 10, and 12 g per 100 g phenol, which were denoted as PMoPR-0, PMoPR-2, PMoPR-4, PMoPR-6, PMoPR-8, PMoPR-10, and PMoPR-12, respectively. The as-spun fibers (PMoPF-n-0) cured in solution or by heat curing are denoted by PMoPF-n − 1 and PMoPF-n − 2, respectively, where n represents the amount of PMo added.
Characterization
The chemical structure of the PMoPR was analyzed with carbon nuclear magnetic resonance (13C NMR) spectroscopy (Avance 400 MHz, Bruker Corporation Co. Ltd, MA, USA) at 293 K in deuterated chloroform (CDCl3) solvent with TMS (tetramethylsilane) as an internal standard.
FTIR were recorded on a Nicolet Magan-750 spectrometer (Nicolet Instrument Corp., WI, USA) using the potassium bromide (KBr) disk technique to investigate the functional groups of the resins and fibers. The fibers were pulverized and mixed with KBr before being pressed into a disk.
GPC was applied to determine the molecular weight and the molecular weight distributions of the PMoPR. The measurement was performed on a GPC-717/1515/2414 (Waters Corp., MA, USA), using tetrahydrofuran as the carrier solvent and polystyrene as the reference polymer.
The fibers were fractured in liquid nitrogen to get their cross sections. The fiber surfaces and cross sections were coated with 5–10 nm of gold and then observed with a JEOL JSM-6360LV scanning electron microscope (Japan Electronics Optics Laboratory Co, Ltd, Japan).
The tensile performance of the PMoPFs was measured by an XQ-1A fiber tensile tester (Shanghai New Fiber Instrument Co. Ltd, China). The gauge length was 20 mm, and a cross-head speed of 10 mm min−1 was used. The fiber diameters were observed using optical microscopy (LWT300LPT, Xian Cewei Optoelectronic Technology Co. Ltd, China).
TGA up to 800°C, with a NETZSCH STA 409 PC/PG TGA system (NETZSCH Group, Germany) under an inert atmosphere of N2 was employed to investigate the high-temperature behavior of the PMoPFs. The heating rate was 10°C min−1.
Results and discussion
13C NMR analysis
Figure 4 shows the 13C NMR spectra of both PMoPR (PMoPR-8) and the phenolic resin without PMo (PMoPR-0). The peaks of ortho–ortho, ortho–para, and para–para methylene bridges appeared at 31ppm, 36 ppm and 41 ppm, respectively. 13 The chemical shifts of the ortho–ortho and ortho–para bridges are in good agreement with previously reported studies with the very weak peak at 41 ppm indicative of the low proportion of para–para links in the phenolic resins. We attribute this to the catalyst of zinc acetate benefiting the preparation of ortho phenolic resin. 14 The unstable dimethylene ether bridges, at around 72 ppm, disappeared in the modified resins. The disappearance of unstable groups helped increase the thermal stability of the PMoPR. The aromatic regions at 113–118 and 118–122 ppm are assigned to the resonances of the methine carbons in the ortho-position at the end of the chain and of the methine carbons at the free para positions. The peaks at 122–132 ppm and 132–137 ppm are assigned to the resonances of the meta carbons and the substituted ortho carbons, and substituted para carbons, respectively. Finally, the broadened resonances at 152–158 ppm could be assigned to phenoxy carbons, which were complicated by the molybdate groups. The shift of the resonances of the phenoxy carbons to higher chemical shifts (158 ppm) are attributed to the electrophilic effect of molybdate groups reducing the shielding of the phenoxy carbons. 15 The broadened chemical shifts (152–158 ppm) of the phenoxy carbons illustrate that the molybdate groups entered the phenolic molecular chain.
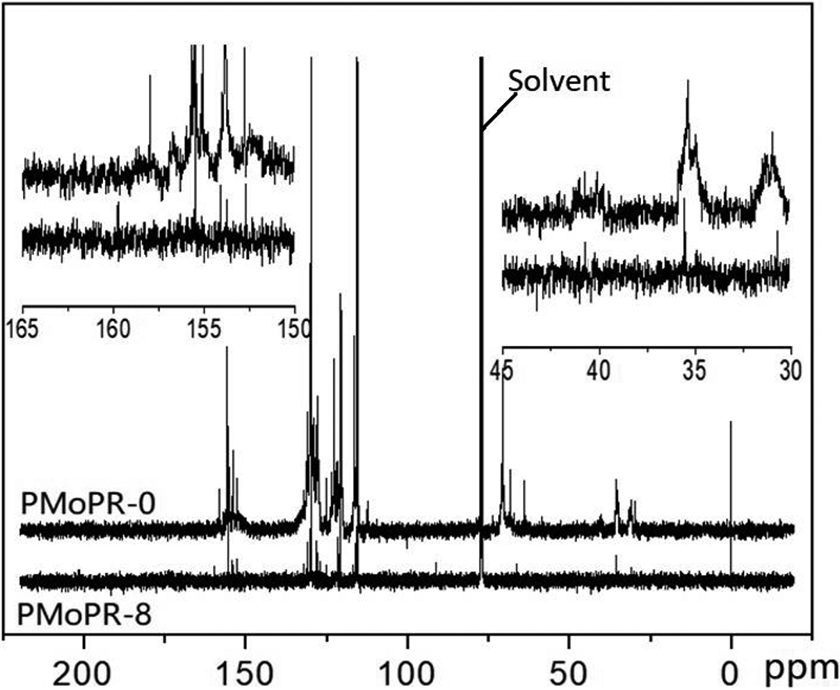
13C NMR spectra of resins from modified phenolic resins of PMoPR-0 and PMoPR-8 in CDCl3, the peak at 75 ppm was assigned to the resonances of solvent (CDCl3).
FTIR and GPC analyses
The FTIR spectra of the heat-cured phenolic fibers with various PMo concentrations are shown in Figure 5. The fibers exhibited the characteristic stretching vibrations of aromatic C–H at 3041 cm−1 and C=O at 1634 cm−1. The C=C stretching vibrations of the phenolic ring is at 1600 cm−1. The peak at 1457 cm−1 corresponds to the C–H2 scissoring vibrations. 16 The phenolic hydroxyl group (PhO–H) stretching vibrations is at 1230 cm−1. 17 The peak at 947 cm−1 is associated with the bending vibrations of the Mo–O bonds of PMoPF. 18 -21
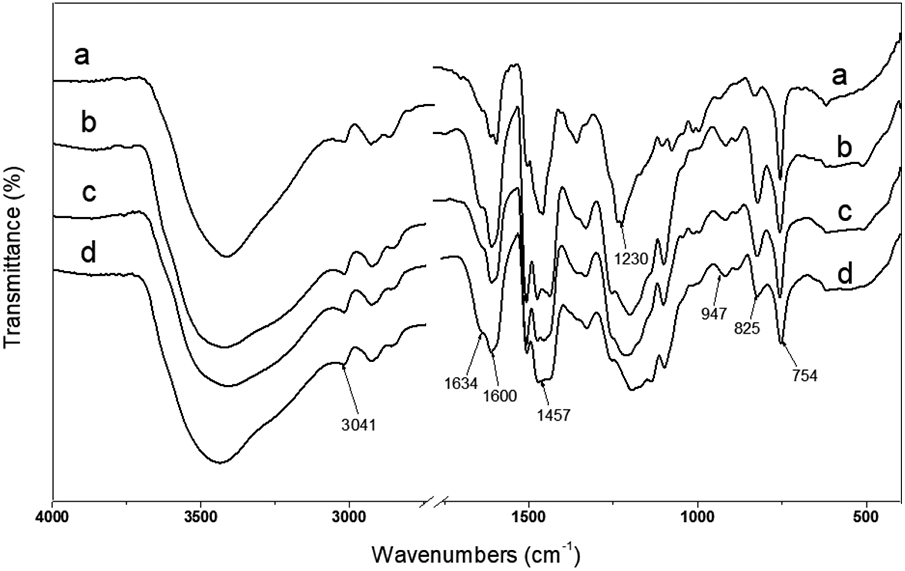
FTIR spectra of the heat-cured fibers ((a) PMoPF-0-2, (b) PMoPF-4-2, (c) PMoPF-8-2, and (d) PMoPF-12-2) with various PMo contents.
According to the Beer–Lambert law, the C=C groups stretching vibrations of the phenolic ring (1600 cm−1) can be adopted as an internal reference peak because of the stabilization of the phenolic ring during the synthesis and curing. 14,17,22 A new absorption peak appeared at 947 cm−1 in Figure 5(b) to (d) after MA modification. The relative absorptions of the peak at 947 cm−1 to the peak at 1600 cm−1 were 0.21 for PMoPF-4-2 and 0.26 for PMoPF-8-2, 0.28 for PMoPF-12-2, respectively. Relative to the internal reference peak of C=C stretching, the peak intensity of the Mo–O bending vibrations increased with the increasing concentration of PMo added during the polymerization, which is attributed to an increase of the Mo–O bonds in PMoPFs due to more reactions between phenol and PMo. As a result, the addition of PMo was expected to enhance the thermal stability because the methylene bridges were replaced by the higher bond energy Mo–O groups, which is 601.7 kJ mol−1, much higher than that of the C=C bond (345 kJ mol−1). 23 The higher thermal stability is demonstrated by the TGA curves.
The differences in transformation of the various groups during curing in solution and the oven are shown by the relevant FTIR spectra of PMoPF-8 (Figure 6). The reactions in the curing process can again be reflected by the relative absorption of the functional groups to that of the aromatic C=C groups (1600 cm−1). In the FTIR spectra of PMoPF-8-1, the intensities of the peaks at 1040 cm−1, 1457 cm−1, and 3428 cm−1 of the sample cured in solution, relative to the internal reference peak were 0.56, 1.67, and 2.35 respectively. These values were all higher than the values of the sample without curing (0.24, 1.48, and 1.24, respectively). These relative intensity increases of the O–H, CH2 bridges, and C–O–C groups indicate the formations of hydroxymethyl groups between phenol and formaldehyde, methylene bridges, and ether linkage reactions between hydroxymethyl groups, respectively. It is evident that a cross-linking structure began to appear between the molecular chains, including methylene bridges and hydroxymethyl groups between phenolic rings, when cured in solution. The ratios of the CH2–O–CH2, CH2 bridges, and O–H groups after heat curing (0.51, 1.59, and 2.02, respectively) were all slightly reduced compared with the values for the solution cured fibers. It suggests that some of the hydroxymethyl groups were consumed by the cross-linking and the unstable ether linkages broke up into methylene bridges.

(a) FTIR spectra of PMoPF-8 as-spun fibers, (b) the solution-cured fibers, and (c) the heat-cured fibers.
The peaks at 825 cm−1 and 754 cm−1 are attributed to the characteristic absorption peaks of the para and ortho hydrogen substitutions on the phenolic ring, respectively. 9,14,24 Therefore, the O/P value (substituted ortho and para carbon ratio) can be determined by the relative intensities of the absorptions at 825 cm−1 and 754 cm−1 (Iortho/Ipara) of the phenolic fibers. As presented in Table 1, the O/P ratios of the fibers changed with the curing processes and the content of PMo. When the element molybdenum enters into the backbone of the phenolic chains, the large O=M=O group between the phenoxy groups possibly restrict the ortho hydrogen addition reaction between phenol and formaldehyde and decreases the number of ortho CH2 bridges. As a result, the O/P value dropped from the high O/P value of phenolic as-spun fibers without MA modification when the content of PMo was increased for the as-spun fibers (Table 1). During the solution and heat curing processes, the O/P values fluctuated. The para hydrogen of the phenolic rings has a lower activation energy than that of ortho hydrogen. This is helpful for the addition reaction of formaldehyde and para hydrogen, and the condensation reaction of hydroxymethyl group and the hydrogen. The reactions will decrease the O/P value with the consuming of the para hydrogen. For the heat-cured fibers, the solution cured fibers were further heated to 240°C under an inert atmosphere of N2. Under high temperature the unreacted cross-linkage points continued to react, including the hydroxymethyl groups and the ortho-hydrogen with high steric effect. The heat curing reaction slightly raised the O/P values and thus the cross-linkages of the PMoPFs. The cross-linking networks are favorable to the mechanical properties.
Molecular weight, molecular weight distributions (measured by GPC) for the resins, and O/P values (calculated from the FTIR spectra) for the fibers with increasing PMo content.
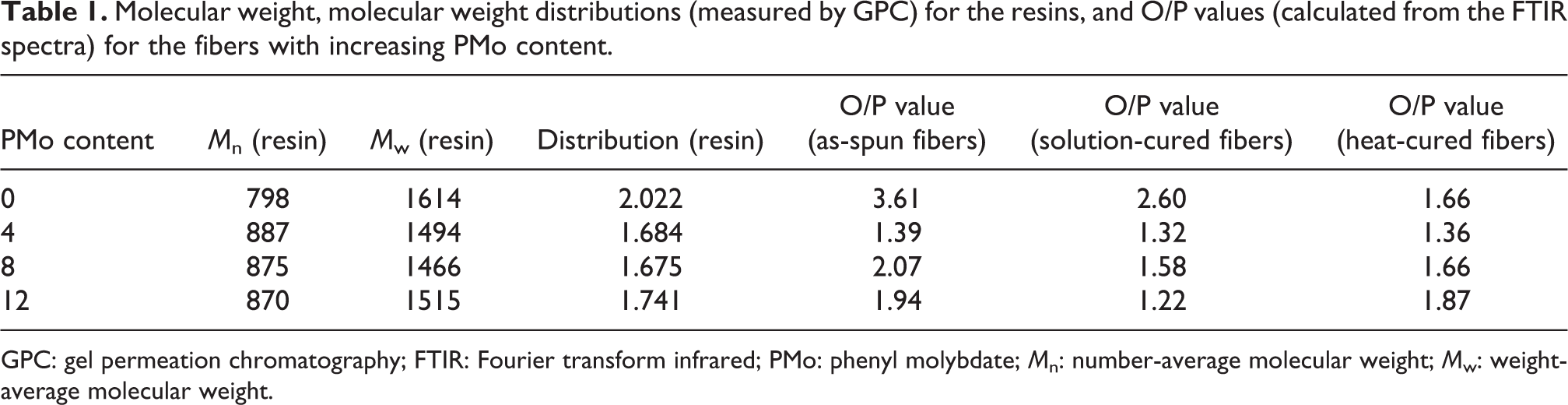
GPC: gel permeation chromatography; FTIR: Fourier transform infrared; PMo: phenyl molybdate; Mn: number-average molecular weight; Mw: weight-average molecular weight.
To analyze the influence of the element of molybdenum on the molecular structure, the number- and weight-average molecular weights of the phenolic resin were determined using GPC. As presented in Table 1, the number-average molecular weights were slightly increased from 798 for phenolic to 870–887 for PMo-modified phenolic. A possible reason is that the molybdenum enters into the backbone of the phenolic resin and leads to an increase of the number-average molecular weight of the modified phenolic resins. It showed that the PMo entering into the macromolecules was beneficial to decreasing the molecular weight distribution. The narrow distributions were helpful for the process of melt-spinning.
SEM analysis
The effect of the difference in the reaction during solution curing and heat curing can be displayed by the morphology of the fibers as shown in the SEM micrographs. The cross-sectional and surface structures of the as-spun filaments and cured fibers observed by SEM are shown in Figure 7. Compared with the as-spun fiber with a smooth exterior surface and circular cross section (Figure 7(a)), some of the solution-cured PMoPF-8-1 fibers appeared rough with some grooves along the axial direction (Figure 7(b)). The characteristics of the smooth surface and circular cross section were obtained by the melt spinning. We suggest the axial grooves were caused by the diffusion of both formaldehyde and water occurring easily between the oriented molecular chain axes in the solution curing process, where the formaldehyde diffuses into fiber and the water diffuses out during the condensation reactions.

Surface and cross-sectional SEM micrographs of the as-spin (a) PMoPF-8-0, (b, c) PMoPF-8-1, and (d) PMoPF-8-2.
The fracture cross sections of the as-spun, solution cured, and heat-cured fibers are shown in Figure 7. The round cross section of the as-spun fiber deformed into an irregular ellipse during the curing processes. This is attributed to the skin–core structure formed by the double diffusions of formaldehyde and water in the solution and curing shrinkage in the oven. The sustained contraction core and the harder skin caused the cross-sectional shape of the heat-cured fiber to transform into elliptical shape. The rough cross sections of the solution-cured and heat-cured fibers were compared with the smooth as-spun fiber fracture cross sections. The toughness improvement of the fiber was due to the properly increasing cross-linkage during each curing process.
Mechanical properties
The changes of mechanical properties with the content of PMo are shown in Figure 8. The tensile strength first increased slowly to the peak value of 187 MPa for PMoPF-10-2 with the rise of the PMo concentration and then decreased to 131 MPa for PMoPF-12-2. The tensile strength of the modified fibers increased with the introduction of molybdenum. However, the strength began to decrease with the excess Mo–O groups could not enter the phenolic molecular main chain. The value of elongation reached a peak at PMoPF-8-2 and then decreased slowly, due to the longer bond length of Mo–O entering into the main chain of phenolic fibers.

Mechanical properties of the phenolic fibers with the various contents of PMo.
TG analysis
The thermal resistance of the as-spun fibers, solution-cured fibers, and heat-cured fibers indicated the transformation of the molecular chains during the curing, as shown in Figure 9. The thermal stability improved significantly due to the solution curing and heat curing. The rapid mass loss of the as-spun fibers within the temperature range of 110–240°C can be attributed to the loss of weak end groups and residual small molecules in the as-spun fibers. The solution-cured fibers showed slight mass loss starting at 50°C, which is due to escape of formaldehyde from the interior of the fibers. The cross-linked networks formed during the solution curing were increased further during the heat curing, resulting in further improvement in the thermal stability.
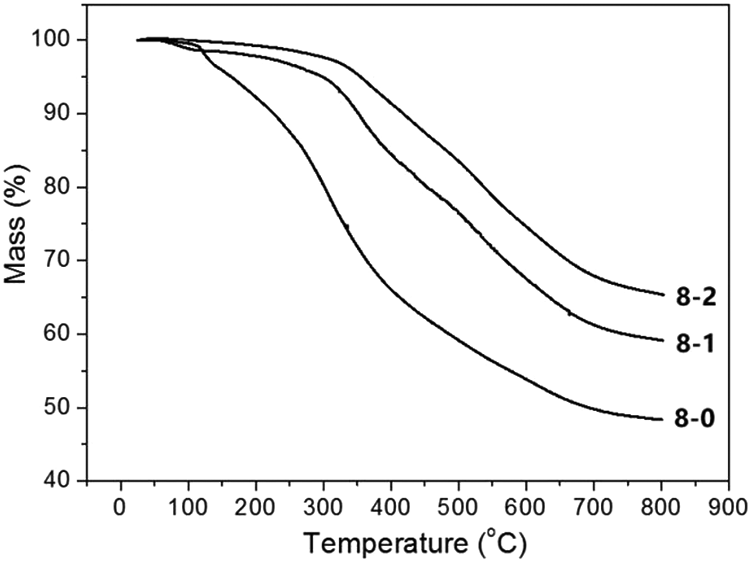
TGA curves of PMoPF-8-0 (8-0), PMoPF-8-1 (8-1), and PMoPF-8-2 (8-2) in N2 atmosphere.
As shown in Figure 9, the rapid weight loss of the as-spun fibers appeared at 307°C, which was due to the rapid decomposition of the linear phenolic molecular chains at high temperature. Compared with the heat-cured fibers, the solution-cured fibers without the additional heat curing process at 240°C, displayed similar thermal decomposition processes in the TGA curves as those of the heat-cured fibers above at 240°C, but showed a more rapid weight loss and had a lower char yield. It demonstrates further that the process of heat curing enhances the heat stability not only under 240°C but also above at 240°C. We suggest that a high degree of cross-linkage was caused among the hydroxymethyl groups and the aromatic C–H groups in the heat curing. The elimination of the weak inner groups (methylene ether) of phenolic molecular and the fastening of the free molecules of formaldehyde (residual formaldehyde from the solution curing and the decomposition of methylene ether) to the polymer backbone, and the formation of biphenyl structures occurred during the heat curing. The resulting high degree of cross-linkage and stable structure together enhanced the thermal stability of the heat cured fibers.
The effect of the different concentrations of molybdenum in the heat-cured fibers is shown in Figure 10. The thermal stabilities of the fibers increased with the incorporation of high bond energy Mo–O groups. The heat-cured fibers without the element molybdenum began to lose mass at 250°C and exhibited two main mass losses around 450°C and 540°C (Figure 10). The first mass loss was due to the condensation reaction between the phenolic hydroxyls and methylene bridges, which yields diphenyl ether linkages and aliphatic methyls. 25 Around 540°C, carbonyl groups were formed by the oxidation of the methylene bridges, and polyaromatic compounds were formed from the removed phenolic hydroxyl groups and hydrogen. 26 The char yield at 800°C increased regularly from 49.0% for PMoPF-0-2 to 59.1% for PMoPF-4-2, 65.4% for PMoPF-8-2, and to 66.0% for PMoPF-12-2, respectively. It is reasonable to conclude that the thermal stability increased continuously with the rise of concentration of PMo.

TGA curves of PMoPF-0-2 (0-2), PMoPF-4-2 (4-2), PMoPF-8-2 (8-2), and PMoPF-12-2 (12-2) in N2 atmosphere.
Conclusions
Novolac-type phenyl molybdate-modified phenolic resins were prepared with those of fibers obtained by melt spinning and subsequent solution and heat curing processes. Stabilization groups were introduced into the cross-linking network of the phenolic fibers with the addition of MA and the heat curing process. Tensile strength as high as 187 MPa for PMoPF-8-2 with conversion from brittle as-spun filaments to tenacious ones was obtained and was suggested to be due to the more uniform and higher cross-linkage obtained by the two curing processes. Furthermore, an initial degradation temperature as high as 300°C and a 65.4% char yield at 800°C of PMoPF-8-2 proved that the introduction of Mo–O groups increased the thermal stability. To apply in the flame-resistant fields, the combustion mechanism with PMo embedded in the phenolic fibers will be investigated in future work.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Natural Science Foundation of China (51803245, 51973246), the Science and Technology Foundation of Henan Province (182102210129), the Program for Science and Technology Innovation Talents in Universities of Henan Province (19HASTIT024), Young Backbone Teacher Training Program of Universities in Henan Province (2019GGJS145), and the Interdisciplinary Direction Team in Zhongyuan University of Technology, China.