
Abstract
Monodisperse core–shell poly(styrene) (poly(St))/poly(styrene-co-butyl methacrylate) spheres were fabricated from styrene (St) and butyl methacrylate (BMA) monomers by a two-step, soap-free emulsion polymerization process at the boiling point. The two-step process involves initial polymerization for a fixed period of time, followed by the addition of BMA monomer to generate the core–shell structure microsphere. Formation of the shell portion increased as the initial polymerization time period was decreased. Differential scanning calorimetric analysis showed that the core–shell microsphere exhibited glass transition temperatures (T gs), when the monomer conversion during the initial St polymerization step was higher than 40%. The T gs of the core and shell occurred at 107°C and 41.9–56.7°C, respectively. These core–shell structure spheres were used to fabricate a colloidal crystal film, the photonic band gap of which could be shifted from 455–631 nm by employing core–shell spheres of various sizes. These films having photonic band gaps in the visible region were obtained by self-assembly of the core–shell spheres at 30, 50, and 80°C. The pencil hardness of the films prepared using the core–shell spheres could be increased from 5B to HB by increasing the preparation temperature, whereas the hardness of the film prepared using simple poly(St) spheres was lower than 6B.
Introduction
Recently, research on photonic crystals or photonic bandgap (PBG) materials has received considerable attention due to the structure–specific periodic dielectric (or refractive index) property of these materials. Structural variation of PBG materials can be used to control the propagation of electromagnetic waves in certain frequency ranges 1 –4 and the display of the photonic band gaps. Certain PBG materials occur naturally as exemplified by some butterfly wings, 5 –8 arthropod cuticles, 9 and snake skin 10 ; these materials can also be synthetically generated such as in the artificial opal crystal. Artificial opal crystals have been extensively utilized in the applications including paints, bioassays, and photonic paper. 11 –18
PBG materials can be fabricated by a variety of methods; the simplicity and cost-efficiency of the methodology used to fabricate colloid crystals consisting of monodisperse spheres makes these PBGs particularly desirable for large-scale production. 19 Colloid crystals are generally fabricated from suspensions consisting of inorganic particles or organic polymer spheres, the components of which have been widely investigated. Polymer colloids of several hundred nanometers in diameter are generally utilized for the construction of materials having PBGs in the visible range. 20 Of the numerous methods used to synthesize polymer spheres, soap-free emulsion polymerization represents a particularly attractive approach due to the high monodispersity and diameter of the generated spheres. 21
The mechanical properties of opal films are also actively researched because of the potential utility of these films in optical devices. The current approaches for the fabrication of robust opal films can be roughly divided into three categories. 22 The first method, which focuses on improving the mechanical properties of the films, involves construction of composite opal films by incorporating the ordered particles into polymer materials, such as poly(dimethylsiloxane) (PDMS) 23 and polyacrylate. 24 In the second method, inverse opal films are constructed after the removal of a colloidal template incorporated into the deformable polymer, which is then responsive to slight compressive pressure. 25 –27 The third method involves the fabrication of functional core–shell spheres. Certain studies 28 –30 have indicated that hard-core/soft-shell structure spheres can be applied to fabricate reversible deformable opal films using the melt-flow method; other preparation methods have also been reported, 31 including vertical or horizontal deposition, which is used to construct elastic opal films by utilizing similar hard-core/soft-shell particles.
Generally, core–shell spheres are synthesized by means of layer-by-layer techniques that are governed by hydrophilicity/hydrophobicity or interface energies. The stepwise construction of multiple layers used in these methods is necessarily time-consuming, and requires complex procedures such as a swelling process, 28 construction of interlayers, 32 the use of phase transfer catalysts, 33 and so on. Recently, a simple approach for the synthesis of monodisperse spheres was published by Gu et al., 21 in which a soap-free emulsion polymerization technique employing the boiling liquid medium was used to synthesize submicrospheres. This method is markedly simplified relative to conventional methods and significantly shortens the reaction time. Based on this advantageous strategy, a novel approach is presented herein for the synthesis of hard-core/soft-shell structure spheres in a single two-step procedure using boiling liquid medium. This method eliminates the complex procedural requirements by utilizing core–shell spheres and efficiently shortens the reaction time (2 h). The core–shell spheres are further employed to construct opal films with improved film properties. The hard core functions to generate the periodic structure of the opal film, while the soft shell acts as a binder between the interfaces of spheres. Consequently, the distinct relationship between soft-shell thickness and thermal properties is important for the proper designing of robust films as illustrated herein. The results of this study are expected be provide a simple approach for the synthesis of core–shell spheres and can be employed to improve the film properties of opal films.
Experimental methods
Materials
Reagent-grade monomers styrene (St) and methacrylic acid (MAA) were purchased from SHOWA (Showa Chemical Co., Ltd., Tokyo, Japan). Butyl methacrylate (BMA) was obtained from Acros. All monomers were purified by vacuum distillation. Potassium persulfate (KPS), used as the initiator, was acquired from SHOWA.
Preparation of poly(St) core
Monodisperse poly(styrene) (poly(St)) cores were synthesized via soap-free emulsion polymerization of St at the boiling point. Initially, 85 g of deionized water was mixed with 10 g St and 490 µl MAA in a 250-ml three-necked flask equipped with a reflux condenser. The reaction mixture was mechanically stirred at 420 r/min, and the reaction temperature was then increased to achieve the boiling point. After 5 min, 5 g of deionized water containing 0.0876 g of dissolved KPS was added to the reaction mixture. The polymerization reaction was terminated after 2 h.
Preparation of poly(St)/Poly(St-co-BMA) spheres
Poly(St)/poly(styrene-co-butyl methacrylate) (poly(St-co-BMA)) spheres were fabricated by introducing the BMA monomer into the reactor, while the core polymerization was still ongoing. The typical synthesis procedure is as follows: 85 g of deionized water, 10 g St, and 490 µl of MAA were combined in a 250-ml three-necked flask. The reaction mixture was mechanically stirred at 420 r/min, and the reaction temperature was increased to achieve the boiling point. After 5 min, 5 g of deionized water containing 0.0876 g of dissolved KPS was added to the reaction mixture. The reaction mixture was polymerized for a specified period of time, after which an appropriate amount of BMA monomer was added into the latex mixture. The polymerization was stopped after 2 h.
Preparation of microsphere films
The deionized water was added into the latex to prepare sphere-suspension with fixed concentration. Then 3 µl of sphere-suspension was dropped onto a 2 × 2 cm2 slide glass and spread well. These above slide glasses were dried at specific temperatures (30, 50, and 80°C) for 6 h at room temperature in vacuum (−750 mm Hg) overnight.
Measurement
The particle diameter was estimated using field emission scanning electron microscopy (FE-SEM, HITACHI, model S80, Japan) observation for more than 100 particles to ensure accuracy. The monodispersity of the spheres was determined on the basis of the coefficient of variation (C v), which can be derived as follows




The glass transition temperatures (T gs) were determined by differential scanning calorimetry (DSC 7; PerkinElmer, Waltham, Massachusetts). The samples were kept at 200°C for 10 min and then they were cooled quickly to −20°C from the melt of the first scan. The T g values were obtained as the inflection points of the heat capacity jump at a scan rate of 10°C/min from −20 to 180°C during the second scan. All measurements were conducted in a nitrogen atmosphere.
Optical properties of the opal films were evaluated by measuring reflection spectra to determine wavelength of maximum reflected intensity (λ max), using an ultraviolet–visible spectrophotometer (UV–visible, JASCO, V-670) with an ARSN-733 absolute reflectance measurement accessory. The reflection spectra were obtained by fixing the incident angles at 6°. The reflected light with the spectral range of 350–800 nm was collected through an aperture.
Results and discussion
First step: Preparation of the monodisperse poly(St) core
Monodisperse poly(St)/poly(St-co-BMA) structure spheres were fabricated by two-step, soap-free emulsion polymerization at the boiling point. Polymerization of the monodisperse poly(St) core spheres was allowed to proceed for specified periods of time, after which the BMA monomer was added to prepare the core–shell structure microsphere. The relative composition of the poly(St) core portion and the poly(St-co-BMA) shell portion was significantly affected by the polymerization reaction time (first step). At longer first-step reaction times, the percentage of the poly(St) core constituent of the core–shell spheres increased. Moreover, addition of the monomer in the second step was not easily performed, when the initial polymerization step was allowed to proceed for longer times. The relationship between monomer conversion and reaction time of the first step is shown in Figure 1. Conversion during the first step increased with increasing polymerization time. The conversion reached 100% at a polymerization time of 50 min. Moreover, the theoretical conversion of poly(St) could be predicted for the addition of the BMA monomer at the various reaction times.
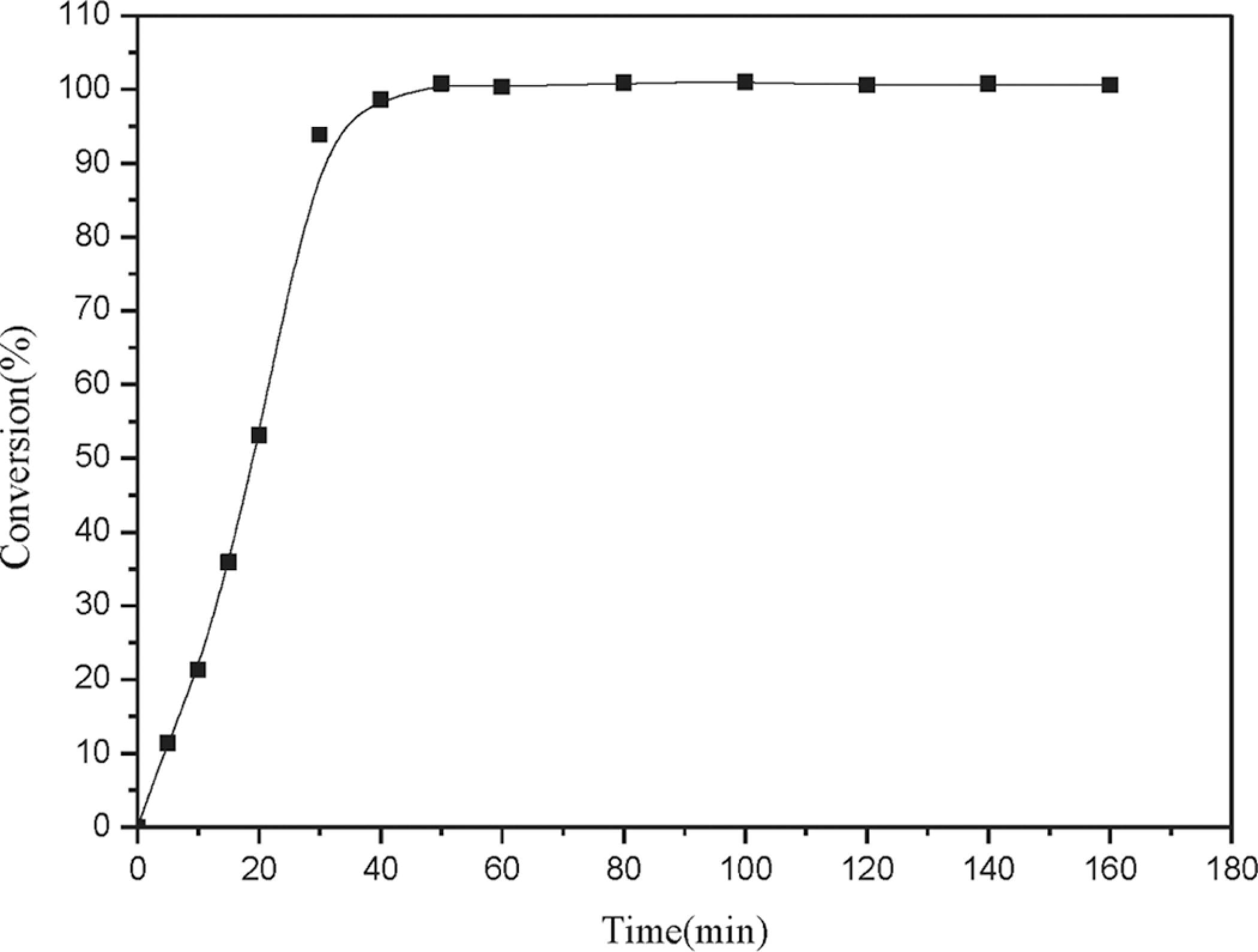
Poly(styrene) conversion as a function of reaction time.
The relationship between polymerization rate and poly(St) conversion is shown in Figure 2. Three distinct intervals of the polymerization process were indicated in the plot. During the first interval, at a monomer conversion below 10%, the polymerization rate increased dramatically; the increase in the polymerization rate was gradual in the second interval, in the monomer conversion range of 10–95%. The polymerization rate finally decreased during the third interval. Based on the literature, 34 the monomer dissolved in the aqueous phase undergoes waterborne free radical polymerization in the first interval to generate particle nuclei. The first interval is referred to as the particle nucleation stage. The number of particles and the degree of monodispersity can be determined in this interval. During the second interval, known as the particle growth stage, the process of free radical propagation takes place primarily in monomer-swollen particles, and these particles grow continuously by supplying monomer from monomer droplets. A steady polymerization rate results in a steady monomer concentration in the monomer-swollen particle. In the final interval, all of the monomer droplets disappear and the monomer concentration can no longer be maintained in the latex particle. Thus, the polymerization rate decreases toward the end of polymerization. In order to ensure a high degree of monodispersity, BMA monomer addition (second step) should be performed during the particle growth stage (interval II of the first step).
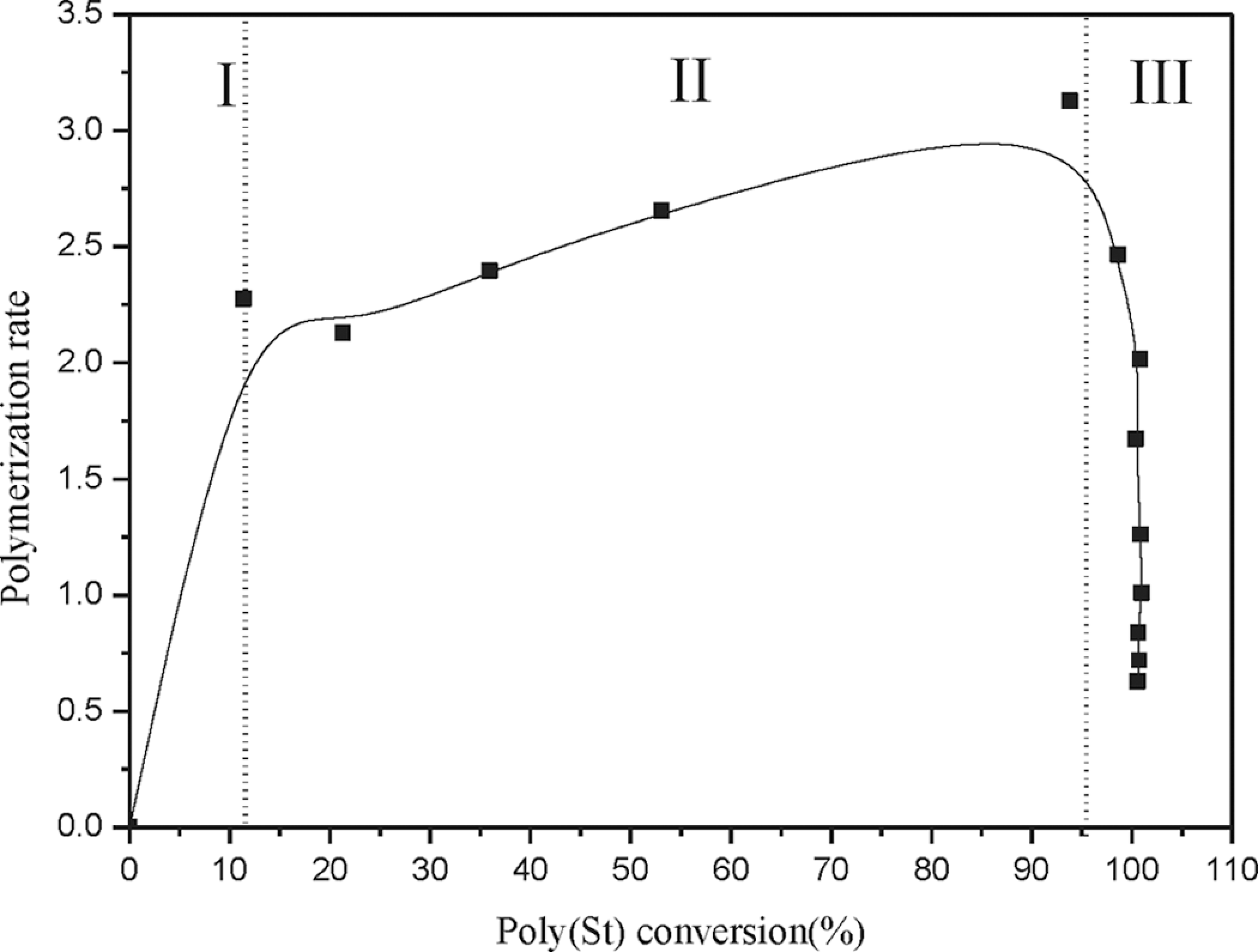
Poly(styrene) conversion as a function of polymerization rate.
The SEM images of the poly(St) particles at various degrees of monomer conversion are shown in Figure 3. As poly(St) conversion increased from 10–100%, the average particle size of the poly(St) spheres increased from 85 to 191 nm. The C v was below 8%, which indicates that the poly(St) spheres were all monodispersed.

Scanning electron microscopy images of poly(styrene) core at various degrees of monomer conversion: (a) 10%, (b) 35%, (c) 50%, and (d) 100%.
Preparation of core–shell microsphere at various degrees of first-step monomer conversion
It was demonstrated in the previous section that a high degree of monodispersity of the core–shell sphere could be obtained when the BMA monomer was added during the particle growth stage of the initial polymerization step, where the monomer conversion was in the range of 10–95%. To further define the reaction, the BMA monomer was added at intervals during the initial polymerization step corresponding to monomer conversions of 20, 40, 60, and 80%. The amount of BMA monomer was fixed at 10 g in this system, and the solid content of the reaction system was controlled at 20%. All of the preparation conditions and relative calculation data are summarized in Table 1. The SEM images of the core–shell spheres prepared at different degrees of poly(St) conversion are show in Figure 4. The particle sizes fell in the range of 234 ± 13 nm for all degrees of conversion of poly(St) in the core–shell spheres, consistent with the high monodispersity (C v < 8%). These observations indicated that a high degree of monodispersity of the core–shell sphere could be obtained when the second step of monomer addition was performed during the particle growth stage of the first step. DSC data, shown in Figure 5, were acquired to further investigate the structure of the core–shell spheres. Only one T g (T g = 52.4°C) was observed when the conversion of the core region, that is, poly(St) in the core–shell sphere, was 20%. However, two T gs were present when the poly(St) conversion increased from 40 to 80% in the core–shell spheres. Theoretically, the core–shell sphere is expected to have two T gs contributed by the poly(St) core and the poly(St-co-BMA) shell, respectively. In this case, poly(St-co-BMA) exhibited a lower T g at about 50°C and poly(St) had a higher T g at about 105°C. The ratio between poly(St) and poly(St-co-BMA) in the core–shell sphere was determined by the relative conversions of poly(St). A lower proportion of poly(St) resulted in no evident T g for poly(St) in the DSC plot. Increasing the conversion of poly(St) in the core–shell sphere (higher than 20%) produced a distinct second T g peak in the DSC profile, due to the increased proportion of poly(St) in the core–shell sphere. Moreover, increasing the poly(St) conversion in the first step resulted in a contrasting decrease in the concentration of St in the reaction system. Consequently, at higher poly(St) conversion in the first step, there is a lower concentration of St for copolymerization with the BMA monomer, which also resulted in the obvious decreasing tendency in T g (52.4–48.7°C) of poly(St-co-BMA) shown in Table 1. In addition, the T g of poly(St-co-BMA) was higher than that of poly(BMA) because the unreacted St was still present when the monomer was added in the second step.

Scanning electron microscopy images of poly(styrene)/poly(styrene-co-butyl methacrylate) submicrospheres prepared at various degrees of monomer conversion: (a) 20%, (b) 40%, (c) 60%, and (d) 80%.
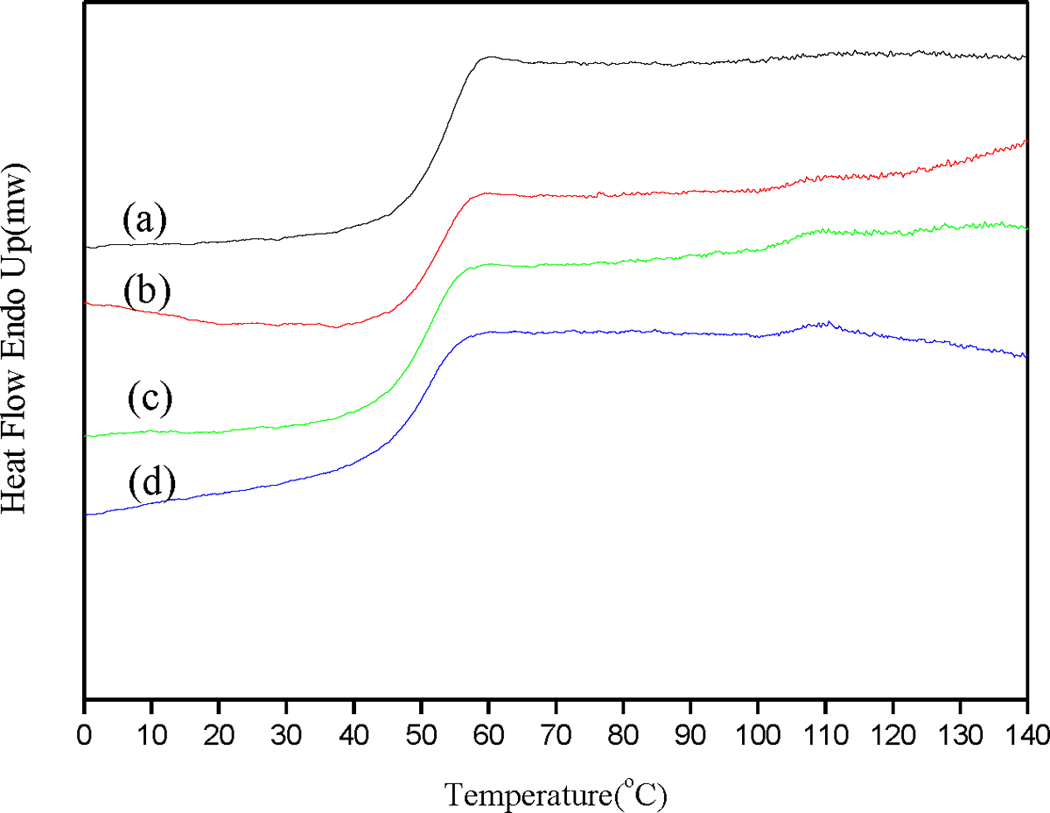
DSC curves of poly(St)/poly(St-co-BMA) submicrospheres prepared at various degrees of poly(St) conversion: (a) 20%, (b) 40%, (c) 60%, and (d) 80%. St: styrene; BMA: butyl methacrylate; DSC: differential scanning calorimetry.
Polymerization conditions, particle size, and T g of various core–shell spheres.
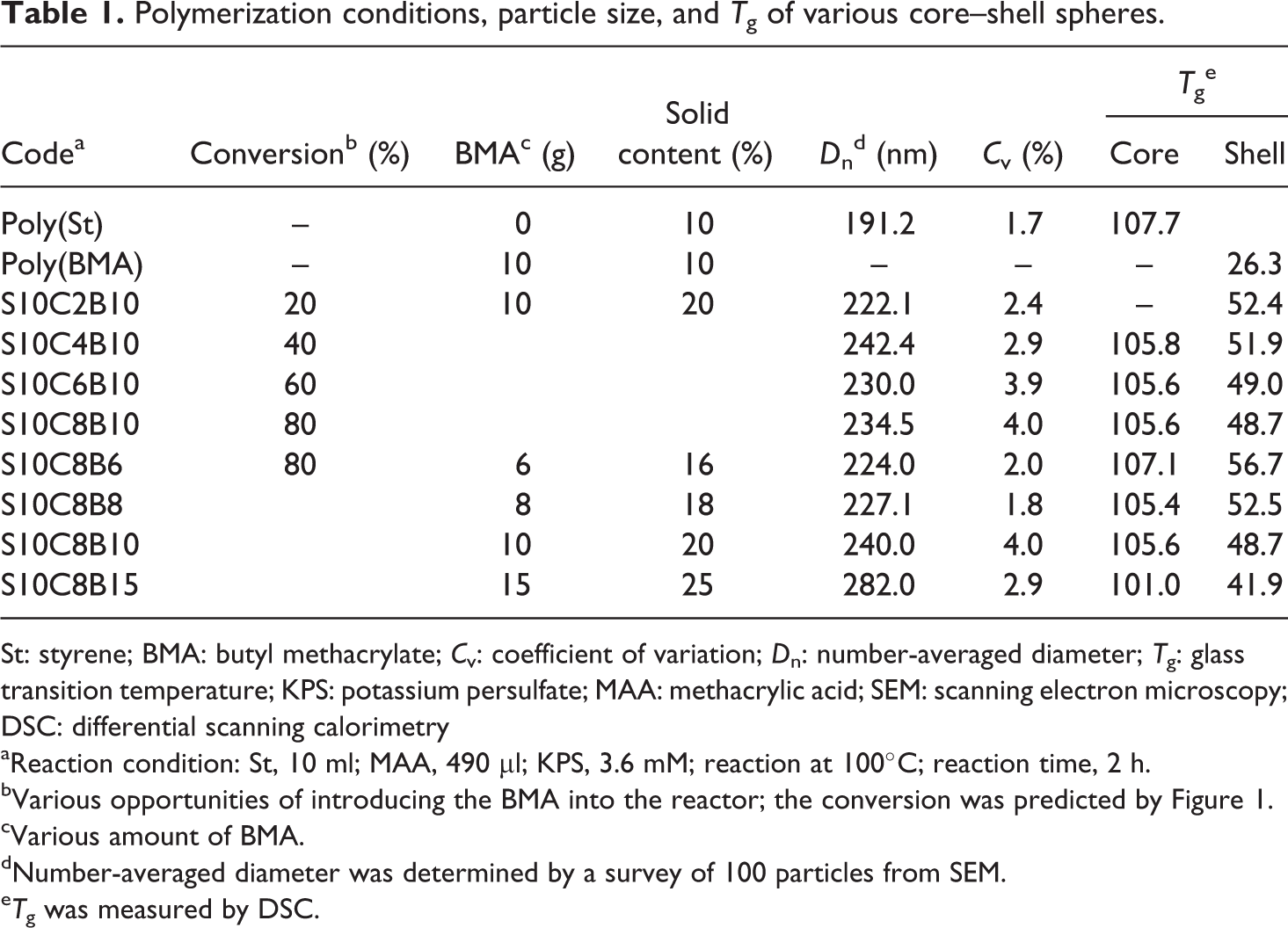
St: styrene; BMA: butyl methacrylate; C v: coefficient of variation; D n: number-averaged diameter; T g: glass transition temperature; KPS: potassium persulfate; MAA: methacrylic acid; SEM: scanning electron microscopy; DSC: differential scanning calorimetry
aReaction condition: St, 10 ml; MAA, 490 µl; KPS, 3.6 mM; reaction at 100°C; reaction time, 2 h.
bVarious opportunities of introducing the BMA into the reactor; the conversion was predicted by Figure 1.
cVarious amount of BMA.
dNumber-averaged diameter was determined by a survey of 100 particles from SEM.
e T g was measured by DSC.
Preparation of core–shell microsphere with various amounts of monomer addition in second step
The amount of monomer added in the second step was systematically varied from 6 to 15 g, while the poly(St) conversion was fixed at 80% to ensure the core–shell structure of spheres. It was observed that the particle size of the core–shell spheres and the thickness of the shell could be increased by increasing the monomer concentration. The preparation conditions and relative calculation data are also summarized in Table 1. SEM analysis (Figure 6) shows the significant observation that the particles had a uniformly spherical morphology irrespective of the solid content in the reaction system. By increasing the quantity of BMA monomer from 6 to 15 g, the solid content in reaction system increased from 16 to 25% with an attendant increase in the particle size of the core–shell spheres from 224 to 282 nm. This tendency was attributed to the increased thickness of poly(St-co-BMA) in the core–shell spheres. DSC analysis (Figure 7) shows the T g of the various core–shell structure spheres prepared by varying the monomer content. It was observed that the T g of poly(St) spheres without the core–shell structure was 107.7°C, and similar T gs (107–101°C) for poly(St) were obtained for the core–shell structure spheres prepared with various monomer contents. In addition, the T g of poly(St-co-BMA) in the core–shell structure spheres decreased (56.7–41.9°C) with increasing solid content of the reaction system (as shown in Table 1). Although the quantity of BMA monomer was varied in the system, the conditions of the initial polymerization step were controlled. Therefore, similar T gs of poly(St) were observed in the range of 101–107°C. In the second step, the final solid content was adjusted by adding different quantities of BMA monomer. With increasing BMA monomer addition, more BMA monomer was available for copolymerization with residual St monomer from the first step, resulting in a corresponding decrease in the T gs (56.1–41.9°C) of poly(St-co-BMA). In contrast, the proportion of poly(St) core in the core–shell sphere decreased with increasing amounts of BMA monomer. This lower proportion of poly(St) rationalizes the lack of observation of a T g corresponding to poly(St) in the DSC curves.

SEM images of poly(St)/poly(St-co-BMA) submicrospheres prepared with various amounts of BMA: (a) 0 ml, (b) 6 ml, (c) 8 ml, (d) 10 ml, and (e) 15 ml. St: styrene; BMA: butyl methacrylate; SEM: scanning electron microscopy.
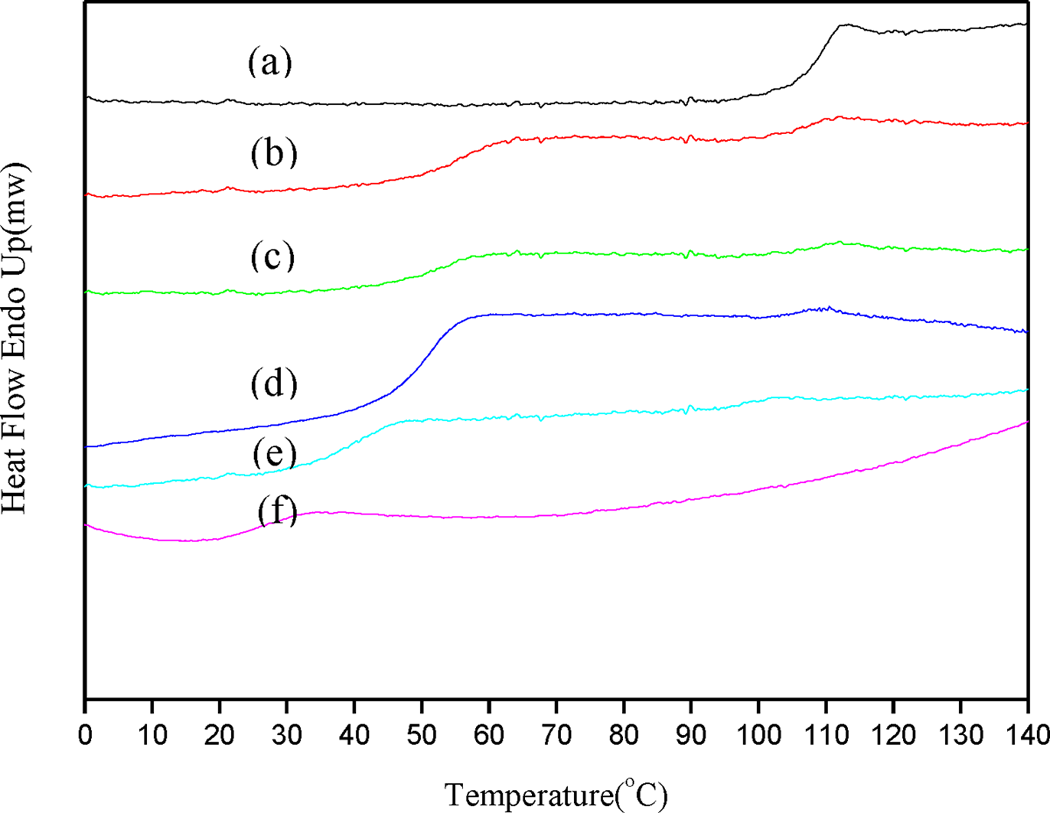
DSC curves of poly(St)/poly(St-co-BMA) submicrospheres prepared with various weights of BMA: (a) 0 g, (b) 6 g, (c) 8 g, (d) 10 g, (e) 15 g, and (f) poly(BMA). St: styrene; BMA: butyl methacrylate; DSC: differential scanning calorimetry.
Characteristics and fabrication of opal film
Latex from poly(St)/poly(St-co-BMA) spheres was used to prepare an opal film. The latex was spread on a slide glass substrate and self-assembled by capillary force during the drying time. The periodic structure comprised two different refractive index materials constituting the core–shell spheres and air, respectively. In this experiment, the poly(St-co-BMA) acts as an adhesive binder for the assembly of the core–shell spheres. The diffraction properties of the film could be altered by adjusting the proportion of poly(St-co-BMA) in the core–shell sphere. The reflection spectra of the opal films prepared with a series of poly(St)/poly(St-co-BMA) spheres at 30°C is shown in Figure 8. All of the films exhibited brilliant colors from indigo to red, resulting from the diffraction of light inside the periodic structure comprising the core–shell spheres. The sharpness of the reflection peaks indicated the high quality of the opal film; corresponding data for the photonic band gaps are presented in Table 2. The photonic band gaps were consistent with the apparent colors; a red shift was observed as the quantity of BMA monomer was increased. Comparison of the experimental and theoretical values calculated using Bragg’s law showed a deviation of less than 2%, indicative of seldom defects and disordered areas in the colloidal crystal.
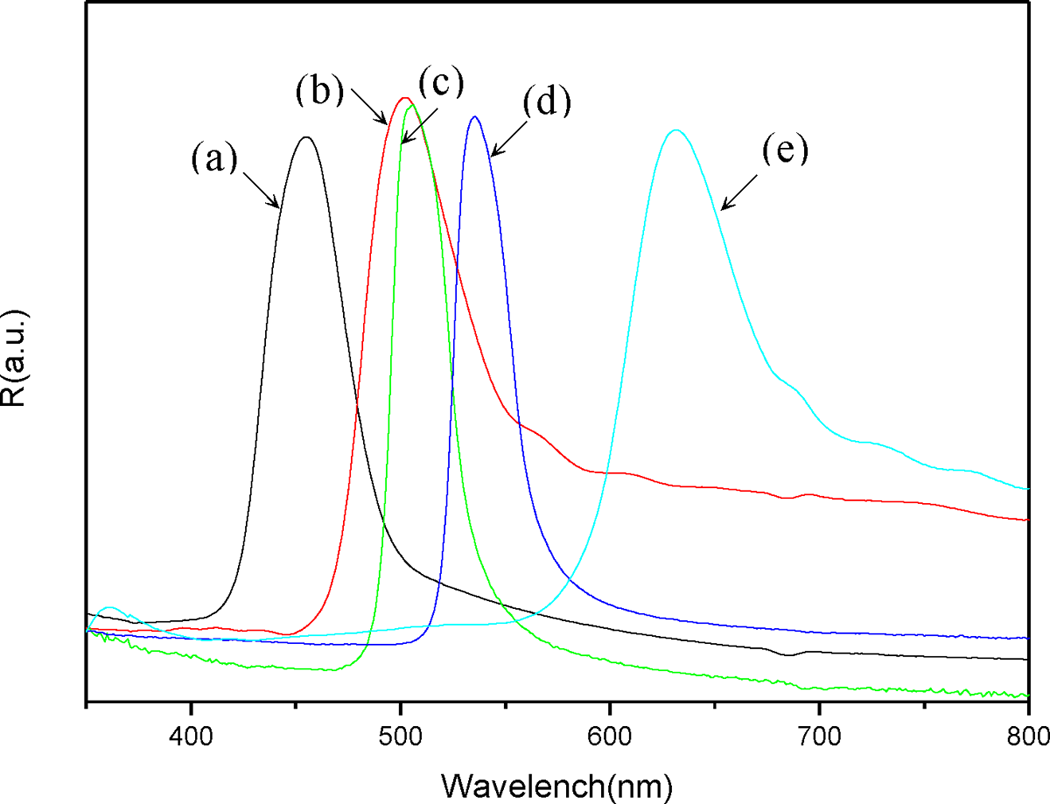
UV–visual spectra of (a) poly(styrene), (b) S10C8B6, (c) S10C8B8, (d) S10C8B10, (e) S10C8B15, and submicrospheres dried at 30°C.
Characteristics of opal film self-assembled from poly(St)/poly(St-co-BMA) submicrospheres.
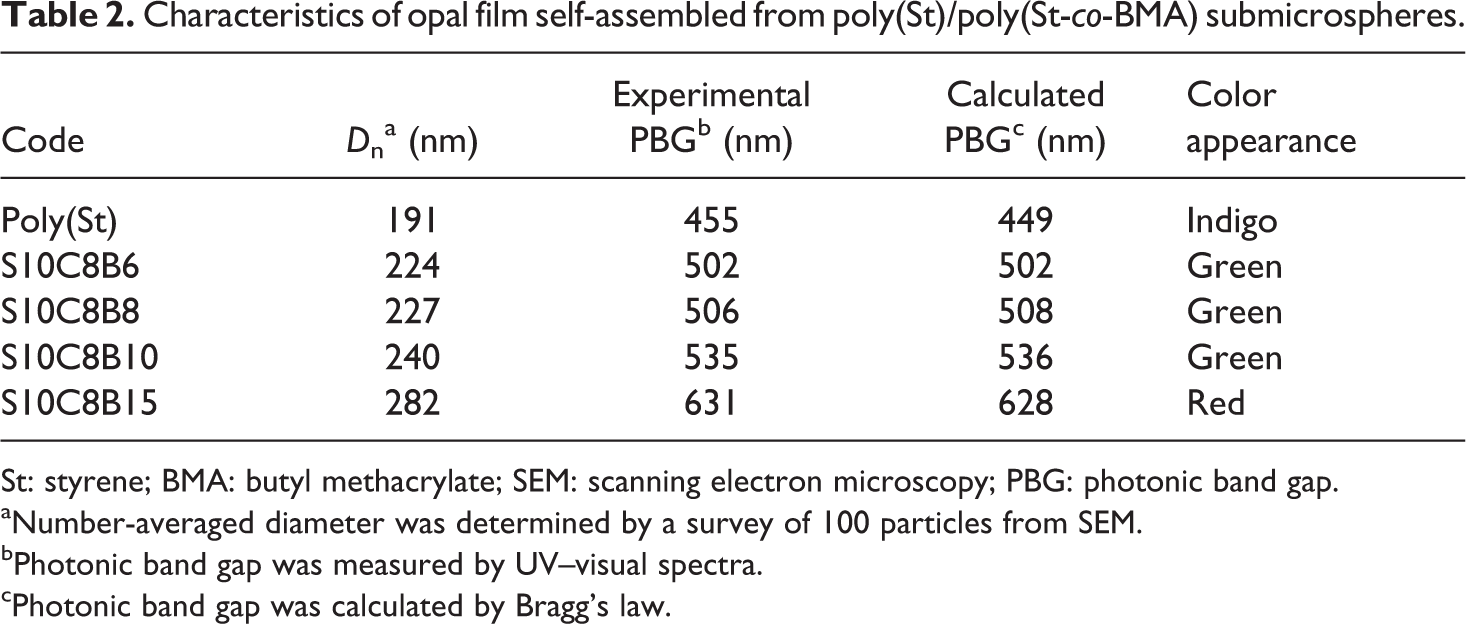
St: styrene; BMA: butyl methacrylate; SEM: scanning electron microscopy; PBG: photonic band gap.
aNumber-averaged diameter was determined by a survey of 100 particles from SEM.
bPhotonic band gap was measured by UV–visual spectra.
cPhotonic band gap was calculated by Bragg’s law.
The environmental temperatures of 30, 50, and 80°C were employed in the self-assembly of the series of spheres, and the solid content of the latex was fixed at 5 wt%. It was significant that opal films could be formed from all of the samples by self-assembly of the spheres at 30 and 50°C. However, increasing the environmental temperature to 80°C caused only a weak structure color to be observed in the opal film made with S10C8B10 and S10C8B15; these opal films also exhibited cracking. As discussed above, the quantity of poly(St-co-BMA) in the spheres was enhanced by increasing the amount of BMA monomer; thus, the periodic structure of these two samples containing higher quantities of BMA collapsed because of the thicker poly(St-co-BMA) shell in the opal film made by self-assembly in air. The weak structure color was attributed to the collapsed structure of the opal film, and cracking of the opal film was due to the quick evaporation of solvent during the self-assembly process. Pencil hardness is a generally accepted method for measuring the resistance of an opal film. Pencil hardness is determined as the hardest pencil grade that does not mark the opal film when pressed firmly against it at 45°; the results of these tests are shown in Table 3. Regardless of the self-assembly environment temperature employed, opal films made with poly(St) exhibited less than satisfactory mechanical resistance (lower than 6B). In contrast, the opal films consisting of core–shell spheres exhibited increasing pencil hardness with increasing self-assembly environment temperature and increasing thickness of the poly(St-co-BMA) in the core–shell sphere. It was well known that the mobility level of the polymer chain on the molecular scale is reflected in the T g value. The environment temperatures (30, 50, and 80°C) employed in the self-assembly process were lower than the T g of poly(St) (107.7°C). This means that the polymer chains of poly(St) remained in the glassy state and the poly(St) spheres were simply arranged on the surface of the glass slide without any binding interaction between the spheres and the surface. Consequently, the poly(St) opal films exhibited unsatisfactory mechanical hardness. Contrastingly, the structure of the core–shell sphere was designed to enhance the properties of the opal film by combining the periodic structure derived from the glassy state of the poly(St) core and the binding force between the spheres resulting from the rubbery state of the poly(St-co-BMA) shell. The T gs of poly(St-co-BMA) in the series of core–shell spheres were in the range of 42–57°C. The mobility of the polymer chain was expected to increase with increasing self-assembly environment temperature. Self-assembly of the core–shell spheres at 80°C produced high mobility of the poly(St-co-BMA) polymer chain, and the soft shell generated a binding interaction force between the sphere surfaces. Consequently, HB pencil hardness could be achieved with the opal film self-assembled from core–shell spheres.
Pencil hardness of opal films made with core–shell spheres.

In order to investigate the surface morphology of the opal film, S10C8B6 was chosen for self-assembly at various environmental temperatures; the SEM images are shown in Figure 9. It was difficult to observe the deformation of the poly(St-co-BMA) shell for the core–shell spheres that were self-assembled below 70°C. When the self-assembly temperature was raised to 80°C, an indistinct boundary was observed, which could be attributed to the deformation of the poly(St-co-BMA) shell at higher temperature. This deformation generated a binding force between the surfaces of the spheres, thus making the periodic structure self-assembled in air resistant to collapse.

Scanning electron microscopy images of S10C8B6 submicrospheres self-assembled at (a) 30°C, (b) 50°C, (c) 60°C, (d) 70°C, and (e) 80°C.
Finally, the color photograph and reflection spectra of a 2 × 2 cm2 opal film constructed using S10C8B6 spheres and dried at 80°C are shown in Figure 10(a). The opal film exhibited an evident bright-green color and a monopeak at 506 nm could be observed in the reflection spectra (shown in Figure 10(b)). These results indicated that the high quality optical properties of the film could be achieved because of the core–shell structure spheres without any deleterious effects to the photonic band gap of the opal film.
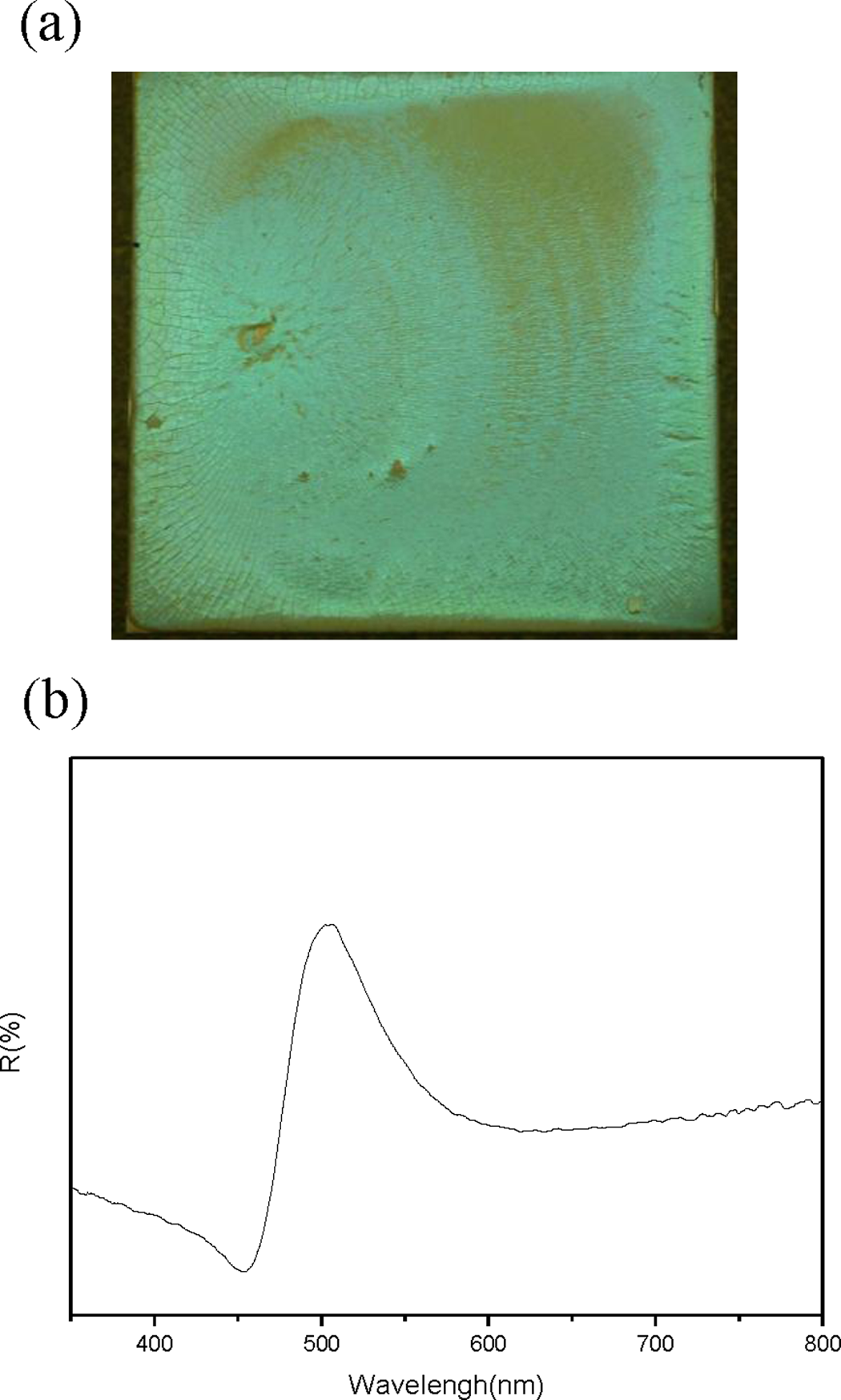
Color photograph (a) and reflection spectra of opal film constructed with S10C8B6 (b) dried at 80°C.
Conclusions
Monodisperse poly(St)/poly(St-co-BMA) spheres were successfully prepared by the two-step, soap-free emulsion polymerization of St and BMA monomers in the boiling point. Furthermore, the relationship between conversion of poly(St) and reaction time was evaluated and used to determine the optimal conversion point for the introduction of the BMA monomer in the second step. Distinctive monodisperse core–shell sphere structures were obtained by introducing the BMA monomer into the reactor when the conversion of poly(St) was higher than 40%. Core–shell spheres of various diameters ranging from 224 to 282 nm were obtained by varying the quantities of BMA monomer in the feed. A range of shell T g values from 56.7 to 41.9°C were also observed from the DSC curves of these spheres. Opal films constructed using the fabricated spheres exhibited brilliant colors in the visible region, which could be controlled by varying the particle diameter. The low occurrence of defects and disordered areas in the opal films was evidenced by the harmony of the PBG values from measurement with the calculated (from Bragg’s law) values. Additionally, comparison of the mechanical hardness of the film fabricated using poly(St) and poly(St)/poly(St-co-BMA) spheres, showed a significant improvement in the mechanical properties of the latter (from lower than 6B to HB). The results of this study provide a simple approach for the synthesis of core–shell spheres and provide an account of useful information for fabrication of robust opal films. Furthermore, this method can be employed for the synthesis of various core–shell structure spheres comprising various monomer functionalities and should therefore be widely applicable in various fields.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.