
Abstract
This study presents a simple approach to the preparation of hollow silica spheres via template-sacrificial techniques. The hard poly(styrene) (PS) template was prepared by soap-free emulsion polymerization in the boiling state for a specified period, followed by introduction of silane into the reaction system to generate the core–shell PS/silicon dioxide (SiO2) spheres without the use of structure-directing agents or surface modification. The SiO2 shell was constructed by co-hydrolysis/condensation of mixed silane containing various weight ratios of tetraethyl orthosilicate (TEOS) and triethoxymethylsilane. The degree of compatibility between PS and SiO2 was shown to be affected by the composition of the silane mixture, and increasing the proportion of TEOS in the silane mix enhanced the mechanical robustness of the SiO2 shell. Based on the residual weight percentage from thermogravimetric analysis, the portion of SiO2 in the PS/SiO2 spheres prepared using various compositions of silane was about 10 wt%. Furthermore, hollow SiO2 spheres were obtained by calcination of the PS/SiO2 spheres prepared using an appropriate formulation of silane at 400°C. Scanning electron microscopy analysis indicated that the size of the hollow spheres was about 200 nm, and the spheres displayed a high degree of monodispersity. Observation of certain cavities on the surface of the spheres also demonstrated the hollow structure of the SiO2 spheres.
Introduction
Recently, hollow nano-/microspheres with well-defined structures have been the focus of considerable attention due to their low density, high specific surface area, high stability, and so on. 1,2 Based on these properties, numerous potential applications of these spheres in various fields have been derived, including use in drug delivery, and as lightweight fillers, chemical catalysts, and photonic band gap materials. 3 –10 Recent studies have presented a variety of methods for the construction of these hollow spheres including self-assembly techniques, 11,12 layer-by-layer (LBL) techniques, 13–15 template-sacrificial techniques, 16,17 and spray-drying techniques. 18,19 Two of these techniques are particularly interesting and are generally used to prepare homogeneous, dense, hollow spheres. The LBL technique pioneered by Caruso et al. is an attractive approach 20 in which electrostatic association between alternately deposited layers of the shells is facilitated by oppositely charged species. The multilayered shells are assembled onto submicrometer-sized colloidal particles by sequentially depositing inorganic precursors or nanoparticles with opposite charges. By subsequent calcination of the obtained core/shell particles, inorganic hollow spheres of various diameters and wall thicknesses can be prepared from a variety of inorganic materials. Another approach to the preparation of hollow spheres is the template-sacrificial technique, in which the size of the sphere cavity can be easily controlled by selecting a template of the appropriate size. 10,21 The sacrificial template may be a soft template 12,22–24 (e.g. micelles, emulsions, vesicles, or gaseous species) or a hard template 12,25,26 (e.g. polymer colloid or metal oxide particles). In order to maintain the structural integrity of soft templates, relatively sensitive reaction environments and strict reaction conditions are required, thereby increasing the difficulty of the experiment. 9,10,27 Consequently, hard templates such as gold, cadmium sulfide, lead, zinc sulfide, or polymer beans 9,10,24 are generally employed in the synthesis of hollow spheres of prescribed sizes and morphologies. In a typical template-sacrificial process, hollow spheres are prepared by coating a metal oxide layer onto the surface of a polymer template. The cavity of the spheres is then formed after removing the polymer template cores by dissolution in an appropriate solvent or by calcination at elevated temperatures in air. 28–31 A number of methods such as soap-free emulsion polymerization, 32,33 dispersion polymerization, 34,35 Pickering emulsion polymerization, 36,37 and miniemulsion polymerization 38,39 may be used to synthesize the polymer template depending on the monomer species and the size of the polymeric core. The soap-free emulsion polymerization and dispersion polymerization techniques are generally used to synthesize respective submicron-sized and micron-sized polymeric cores. Furthermore, various inorganic hollow spheres such as silicon dioxide (SiO2), 20,40 nickel, 41 titanium dioxide (TiO2)/SiO2, 42 iron oxide, 43 and TiO2 44 have been prepared using polymer templates. 45 Among these, hollow SiO2 spheres have garnered significant attention due to their low toxicity, highly biocompatible nature, and high mechanical stability. 10
In order to coat the SiO2 onto the template surface, a template with a certain charge is required to attract the oppositely charged inorganic molecular precursor or colloidal SiO2. For example, Zhang et al.
46
indicated that a positively charged template can be synthesized using azodiisobutyramidine dihydrochloride as a cationic initiator and polyvinylpyrrolidone (PVP) as a stabilizer. The positively charged polymer templates are capable of attracting the negatively charged silica precursor, which precipitates on the surface of the template via electrostatic interactions.
28
A number of molecules such as poly(vinyl alcohol),
47
cetyltrimethylammonium bromide,
29,47
PVP,
14
and poly-
Although numerous reports relating to the use of charged templates or functionalized surfaces have been published, the stepwise construction of multiple layers used in these methods is necessarily time consuming and requires complex procedures such as repeated adsorption/centrifugation/water wash/redispersion cycles for the LBL method 21,48 or surface modification 14,49 or the use of structure-directing agents 7,14,29,47 and exchange of solvent for the template-sacrificial technologies. 16,17 Therefore, the preparation of hollow silica spheres by a simple route is worth investigating.
Recently, a new method for fabricating monodispersed hollow silica spheres was reported by Chen et al., 50 in which monodispersed positively charged PS particles employing 2-(methacryloyl)ethyltrimethylammonium chloride as the comonomer were initially prepared by dispersion polymerization, followed by the hydrolysis and condensation of silane in aqueous ammoniacal alcohol medium in which the PS particles were subsequently, even synchronously, dissolved. Neither additional dissolution nor a calcination process was required. This technique was particularly attractive given its simplicity relative to conventional synthesis methods, and it also represents a simple approach to the construction of hollow, micrometer-sized SiO2 spheres. Notwithstanding the elegance of this technique, the size of the hollow spheres was limited to a distribution within the micrometer range of the dispersion polymerization, and precipitation of the SiO2 precursor on the PS surface still functioned by electrostatic interactions. To our knowledge, no other ideal interaction force between the silica and the template has been investigated to date. Therefore, a novel approach is presented herein for the synthesis of submicron-scale hollow spheres to obtain the smaller core–shell spheres, in which the complex procedural requirements are eliminated. The basis of this technique involves polymerization of the PS particle by soap-free emulsion polymerization for a specified period combined with the use of boiling liquid medium and addition of the silane into reaction system while the polymerization of PS is still in progress. Control of the polarity of the silane mixture is achieved by adjusting the ratio of tetraethyl orthosilicate (TEOS) and triethoxymethylsilane (MTES). In this manner, silane is introduced onto the surface of the PS particle without the need for structure-directing agents or surface modification, due to the simple active force of diffusion. The SiO2 shell structure was obtained by a hydrolysis–condensation process, and hollow SiO2 spheres were generated subsequent to calcination.
Experimental methods
Materials
The reagent-grade monomers, styrene (St) and methacrylic acid (MAA), were purchased from SHOWA (Showa Chemical Co., Ltd., Japan) and purified by vacuum distillation. The silanes, TEOS, and MTES were obtained from Shin-Etsu Polymer Co. Ltd. (Japan) Potassium persulfate (KPS), used as an initiator, was acquired from SHOWA.
Preparation of PS core
Monodisperse PS cores were synthesized via soap-free emulsion polymerization of St at the boiling point. Initially, 85 g of deionized water was mixed with 10 g St and 490 µl MAA in a 250-ml three-necked flask equipped with a reflux condenser. The reaction mixture was mechanically stirred at 420 r/min, and the reaction temperature was then increased to reach the boiling point. After 5 min, 0.0876 g of KPS dissolved in 5 g of deionized water was added to the reaction mixture. The polymerization reaction was terminated after 2 h. Besides, latex of 2 ml was withdrawn from the reactor during the polymerization at set intervals, and the St conversions were determined by gravimetric analysis.
Preparation of PS/SiO2 spheres
PS/SiO2 spheres were fabricated by introducing the silane into the reactor, while the core polymerization was still ongoing. The typical synthesis procedure is as follows: 85 g of deionized water, 10 g St, and 490 µl of MAA were combined in a 250-ml three-necked flask. The reaction mixture was mechanically stirred at 420 r/min, and the reaction temperature was increased to reach the boiling point. After 5 min, 0.0876 g of KPS is dissolved in 5 g of deionized water was added to the reaction mixture. The reaction mixture was polymerized for 30 min, after which 4 g of silane was added into the latex mixture. The polymerization was terminated after 2 h. The SiO2 was further aged by adding 1 g hydrochloric acid (HCl, used as a catalyst) to the PS/SiO2 latex. The aging process was carried out for 6 h, and the PS/SiO2 latex was purified by centrifuging four times with water prior to storage.
Fabrication of hollow SiO2 spheres
Hollow SiO2 spheres with confirmed spherical shape were obtained after heat treatment of the PS/SiO2 latex in a programmed muffle furnace. Then, 1 ml of the PS/SiO2 latex was dropped onto the surface of a slide and heated to the predetermined temperature for 30 min. The hollow SiO2 spheres were produced by heating at the intended temperature for 10 min in air atmosphere.
Characterization
The particle diameter was estimated using field-emission scanning electron microscopy (SEM) (model S80, Hitachi, Tokyo, Japan) observation of more than 100 particles to ensure accuracy. The monodispersity of the spheres was determined on the basis of the coefficient of variation (C
v) which can be derived using the following equations:




where the terms D n, D i, n i, D v, and σ represent the number-averaged diameter, the particle diameter, amount of particles, volume-averaged diameter, and SD, respectively. In addition, the thermogravimetric analysis (TGA, TGA 7, Perkin Elmer) was applied to detect the thermostability and the attached mass of silica in the PS/SiO2 spheres at air atmosphere in the temperature range of 25–800°C.
Results and discussion
Characteristics of monodisperse PS core
Monodisperse PS/SiO2 structure spheres were fabricated herein by two-step polymerization. Initially, monodisperse PS spheres were synthesized by soap-free emulsion polymerization in the boiling state. After a fixed period, silane monomer was added to generate the core–shell structure microsphere, and HCl used as an acid catalyst was either added or not added in order to confirm the further reaction of the silane monomer. Hollow SiO2 spheres were subsequently obtained by calcination of the PS/SiO2 spheres.
The diameter of the PS core as a function of the degree of St conversion is shown in Figure 1. The diameter of the PS core increased with increasing PS conversion. As the PS conversion increased from 3 to 95%, the average particle size of the PS core increased from 55 to 188 nm. The relationship between the volume of the PS core (calculated from the diameter of the PS core) and the conversion is also shown in Figure 1; a positive linear trend was observed with increasing St conversion.
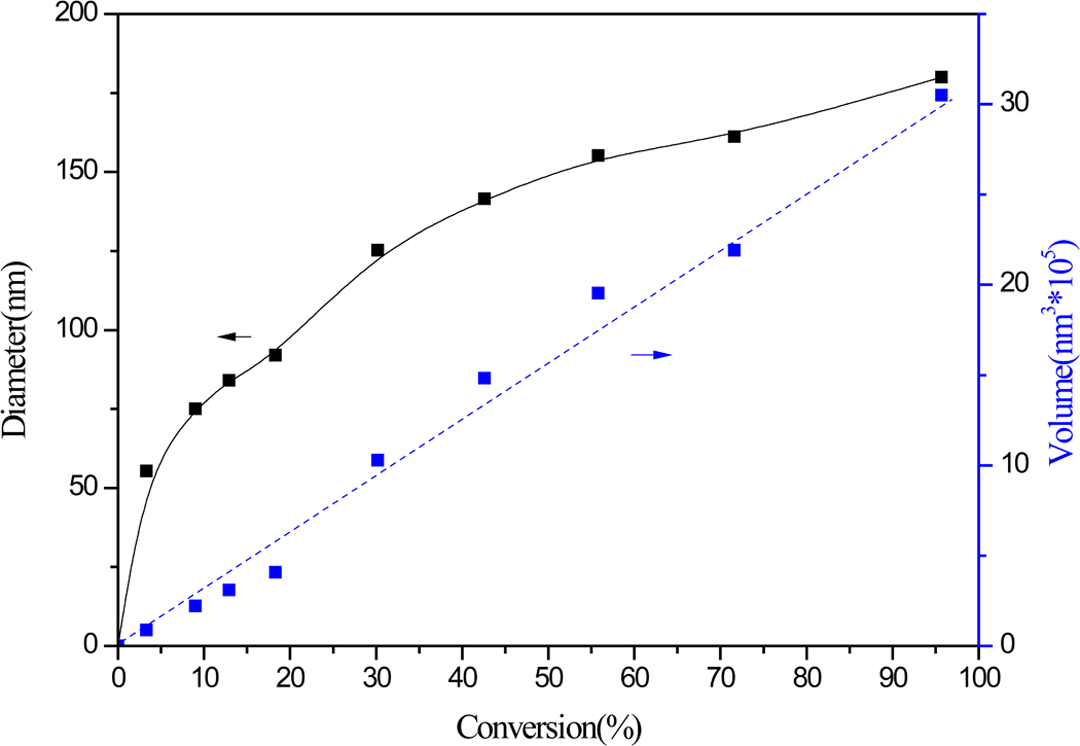
Relationship between PS diameter, volume, and PS conversion. PS: poly(styrene).
The surface morphologies of the PS core at various degrees of conversion are shown in Figure 2. The value of C v is below 8%, which indicates that the PS spheres are all monodispersed. However, at 3% conversion of the PS spheres, irregular aggregation is observed in the SEM image, where it can be considered that the PS spheres were prepared in 97% residual St monomer.

SEM photographs of PS core at different degrees of conversion: (a) 3%, (b) 30%, (c) 70%, and (d) 95%. SEM: scanning electron microscopy; PS: poly(styrene).
The relationship between PS conversion and the polymerization time is shown in Figure 3. The PS conversion increased dramatically with increasing polymerization time during the first 40 min. The conversion remained constant at 95% after 50 min of polymerization time. According to the literature, 51 three distinct intervals of the polymerization process have been defined in the plot depending on the conversion of PS; the first interval where the conversion of St is below 10 wt% is referred to as the particle nucleation stage. The St dissolved in the aqueous phase undergoes waterborne free radical polymerization to generate particle nuclei, and the number of particles and degree of monodispersity can be determined in this interval. In the second interval (particle growth stage), at which the conversion of St is between 20and 90%, the process of free-radical propagation takes place primarily in monomer-swollen particles, and these particles grow continuously by the supply of monomer from monomer droplets. In the final interval, all the monomer droplets disappear, and the monomer concentration can no longer be maintained in the latex particle. Thus, the polymerization rate decreases toward the end of polymerization. In order to ensure the stability of the PS nuclei and a high degree of monodispersity, the addition of silane should be performed in second interval (conversion 20–90%).
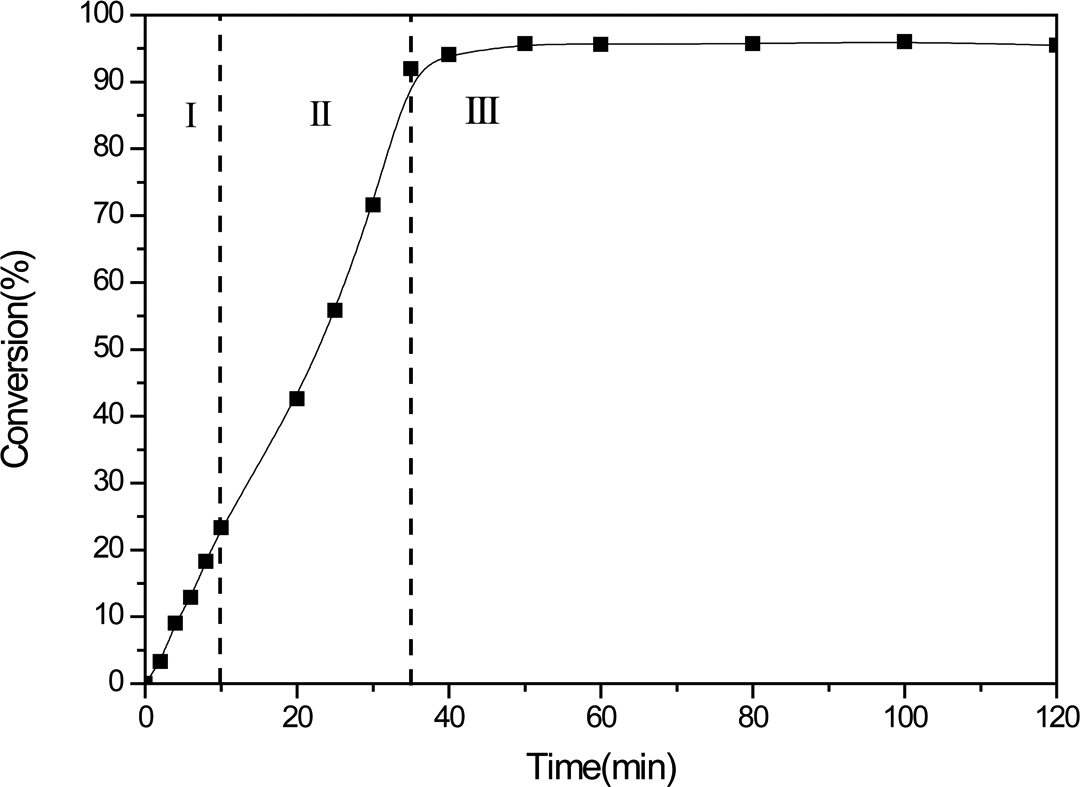
Poly(styrene) conversion as a function of polymerization time.
Preparation of monodisperse PS/SiO2 submicrospheres
Based on the description presented in the previous section, monodisperse PS/SiO2 can theoretically be obtained by adding the silane during the particle growth stage of the PS polymerization. TEOS was selected for the preparation of the core–shell structure spheres. At this stage, TEOS can diffuse into the PS spheres containing the St monomer. The conversion of the St monomer was constant at 95% for polymerization times exceeding 50 min. The particle growth stage was operative at PS conversion and polymerization reaction times between 20 and 90% at 15 and 50 min, respectively. Therefore, the PS polymerization reaction time of 30 min was chosen for the addition of TEOS, that is, 10 g of St was polymerized for 30 min (conversion of St was about 70%), after which 4 g of TEOS was added to the reaction system. After 5.5 h, the PS/SiO2 spheres were obtained by washing with water for four times. The TGA profile of the PS/SiO2 spheres is shown in Figure 4. TGA of the PS/SiO2 spheres generated in the absence of catalyst up to 600°C indicated that the weight of the residual was nearly 0 wt%, which indicates that no SiO2 was present. In order to enhance the reaction rate of the sol–gel process, HCl was added as an acid catalyst to promote the reaction of the silane monomer. TGA up to a temperature of 600°C (Figure 4) showed almost no residual for the PS/SiO2 system treated with the acid catalyst. Based on these results, no SiO2 was present in the so-called PS/SiO2 microspheres. In order to further confirm the residual of the above so-called core–shell structure spheres, the range of temperature scan between 500 and 650°C was enlarged in attached small figure (Figure 4), which was indicated that the deviation of residual was limited in 1 wt%, also confirmed the statements of “almost no residual.” Based on the above results, it is proposed that the polarity of TEOS and its precursor were much higher than that of PS and St; thus, TEOS and its precursor did not readily diffuse into the PS sphere.
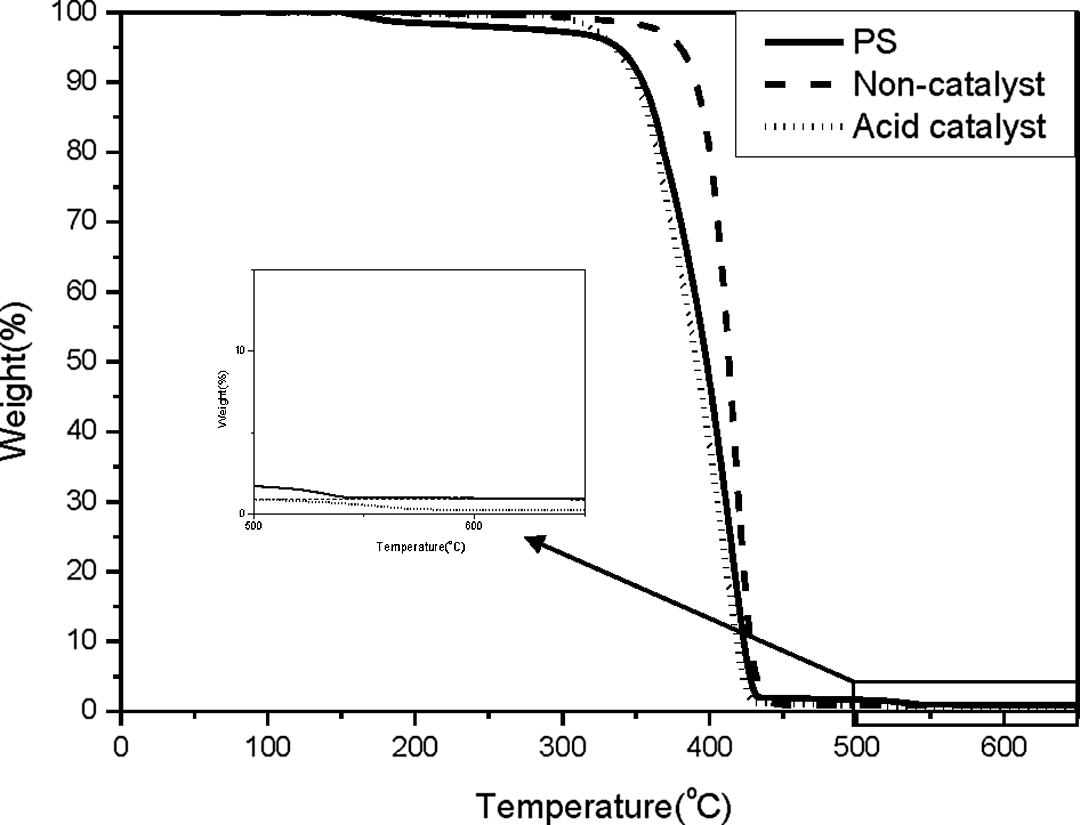
TGA profiles of PS core and PS/SiO2 submicrospheres prepared from TEOS. TGA: thermogravimetric analyzer; PS: poly(styrene); SiO2: silicon dioxide; TEOS: tetraethyl orthosilicate.
The surface morphology of the PS/SiO2 (TEOS) spheres synthesized in the absence of catalyst is shown in the SEM image presented in Figure 5(a). The spherical shape of the particles was accompanied by a high degree of monodispersity and uniform arrangement as well as distinct borders between the surfaces of the spheres. The SEM image of the PS/SiO2 spheres treated with the acid catalyst is shown in Figure 5(b). Comparison with Figure 5(a) shows that with acid treatment, the edges of the spheres become more indistinct, and the spheres appear to be immersed within some type of matrix. It was well known that the addition of HCl leads to enhanced reactivity of the sol–gel process. Consequently, a significant amount of SiO2 was generated in the system. Based on the TGA results presented in Figure 4, most of the SiO2 tended to react out of the spheres (after drying the latex, the portion of SiO2 that was not removed by washing was further reacted, leading to the indistinct boundaries between the surfaces of the spheres).

SEM photographs of PS/SiO2 spheres prepared from TEOS: (a) no catalyst and (b) acid catalyst. SEM: scanning electron microscopy; PS: poly(styrene); SiO2: silicon dioxide; TEOS: tetraethyl orthosilicate.
In order to increase the extent of diffusion of silane into the PS sphere, MTES was used in the preparation of the PS/SiO2 spheres in lieu of TEOS. Figure 6 shows the TGA profile of the PS/SiO2 (MTES) spheres prepared using the same synthesis technique as mentioned before. Residual of 10 wt% was clearly evident after calcination at 600°C, irrespective of whether HCl was added. This demonstrates that the core–shell PS/SiO2 spheres were obtained when MTES was employed as the silane, and the portion of SiO2 in the PS/SiO2 spheres was about 10 wt%. Furthermore, this proportion of SiO2 corresponds to the initial nominal MTES ratio in the mixture. The polarity of MTES is much lower than that of TEOS due to the methyl group adjunctive in the former. Consequently, MTES was much more compatible with the St monomer and the PS core than TEOS, which facilitated the effective diffusion of MTES into the PS core. Subsequent to the sol–gel treatment, the SiO2 derived from MTES remained within the PS core and successfully hybridized with the PS chains. For this reason, SiO2 was not readily removed by washing and was incorporated into the core–shell structure spheres, accounting for the residual observed in the TGA evaluation.
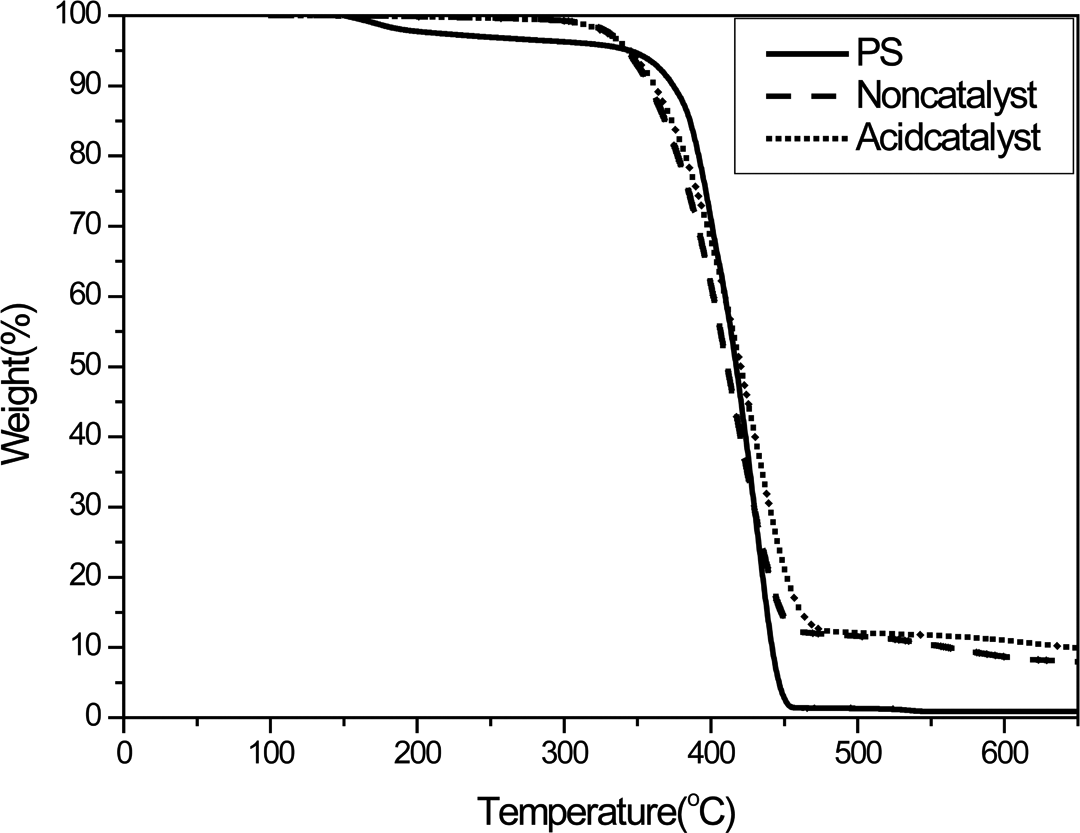
TGA profiles of PS core and PS/SiO2 submicrospheres prepared from MTES. TGA: thermogravimetric analyzer; PS: poly(styrene); SiO2: silicon dioxide; MTES: triethoxymethylsilane.
The surface morphology based on SEM images of the PS/SiO2 (MTES) spheres generated in the absence of the catalyst is shown in Figure 7(a). The spheres were evidently monodispersed with smooth surfaces and distinct boundaries. Similar features were observed for the PS/SiO2 spheres treated with an acid catalyst (Figure 7(b)). These observations indicated that the high compatibility between MTES and PS led to the smooth diffusion of MTES into the PS core. The SiO2 tended to remain within the spheres thereby precluding further reaction of SiO2 outside the spheres. Consequently, the distinct boundaries between the surfaces of the individual spheres were observed, which corroborates the 10 wt% residual ascribed to SiO2 from the TGA evaluation.

SEM photographs of PS/SiO2 spheres prepared from MTES: (a) no catalyst and (b) acid catalyst. SEM: scanning electron microscopy; PS: poly(styrene); SiO2: silicon dioxide; MTES: triethoxymethylsilane.
In general, hollow spheres can be prepared by calcination of PS/SiO2 spheres at 400°C. The organic portion is decomposed, and the residual of the inorganic portion comprises the hollow spheres. Hollow spheres were generated by calcination of the PS/SiO2 (MTES) spheres at 400°C for 2 h; the SEM image is shown in Figure 8. It was difficult to identify spheres in the SEM image after calcination, and a number of subcircular holes were embedded within the network structure of the material. This confirms that MTES indeed diffused into the PS core and further reacted to form SiO2. However, the observed surface morphology also demonstrates that the mechanical stability of the SiO2 generated from MTES was insufficient to maintain the spherical shape constructed from the PS component. The subcircular holes might be due to the lower degree of cross-linking in the SiO2 shell.
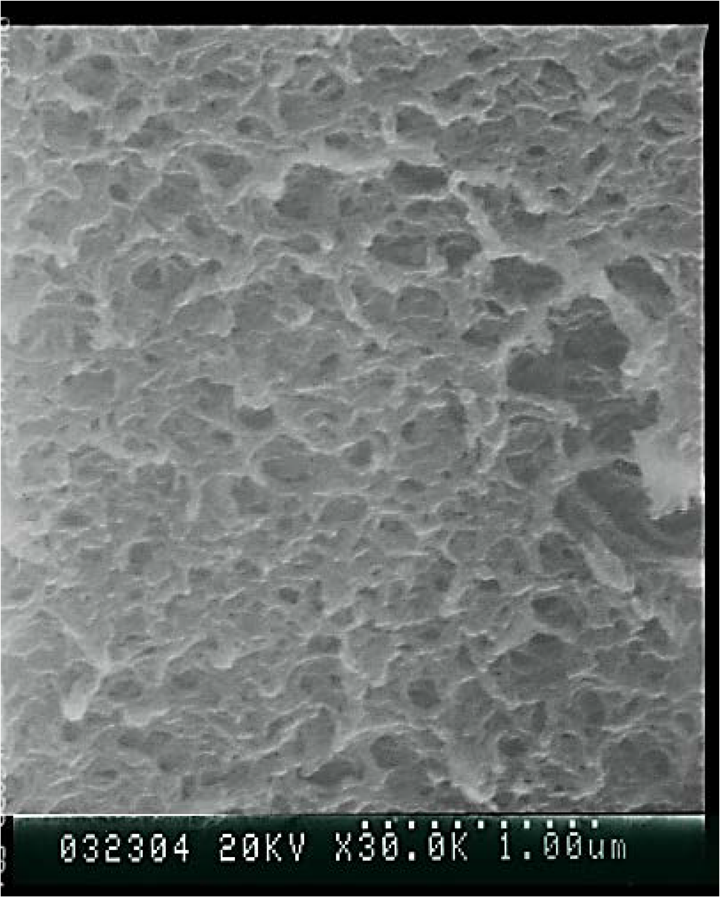
SEM photographs of PS/SiO2 (MTES) spheres calcined at 400°C. SEM: scanning electron microscopy; PS: poly(styrene); SiO2: silicon dioxide; MTES: triethoxymethylsilane.
Preparation of monodisperse hollow SiO2 submicrospheres
The previous sections illustrated that PS/SiO2 spheres could be successfully prepared by adding MTES within the particle growth stage of PS polymerization. However, the mechanical stability of the SiO2 framework derived from MTES was insufficient to maintain the spherical shape after calcination. In order to increase the mechanical stability of the PS/SiO2 sphere, MTES and TEOS were used in combination as the silane in various compositions. Though the polarity of MTES is much lower than that of TEOS, they are readily mixed. Therefore, TEOS could diffuse into the PS core in conjunction with MTES thereby forming the PS/SiO2 sphere. Moreover, the four reactive functional groups of TEOS could improve the mechanical stability of the SiO2 structure. The TGA profiles of the PS/SiO2 spheres prepared using various silane compositions are shown in Figure 9. Irrespective of the composition of the silanes, a residual of about 11 wt% is observed after thermal treatment at 600°C. These values are highly consistent with the theoretical weight percentages based on the nominal composition of the reaction mixture, which confirms that the PS/SiO2 core–shell spheres could be prepared from the various compositions of silane and that silane was converted to form the PS/SiO2 spheres and did not remain in the reaction solution. In more detail, the TGA residual of PS/SiO2 prepared from 33 wt% MTES at 800°C was 11.4 wt%, which was higher than the PS/SiO2 prepared from 50 and 25 wt% MTES (10.4 and 10 wt%, respectively). As the composition of MTES was 25 wt%, the polarity of silane was insufficiently low to fully diffuse into PS core. Therefore, after gelation, the SiO2 out of spheres was removed by centrifuging exhibited lowest residual. However, as the PS/SiO2 was prepared by appropriate composition of 33 wt% MTES, the highest residual indicated that the great compatibility between PS core and silane was performed. Moreover, by further increasing the MTES composition to 50 wt%, the enhancement compatibility among PS and silane was predictable, while also caused the lower degree of SiO2 cross-linking and lower residual of TGA result.
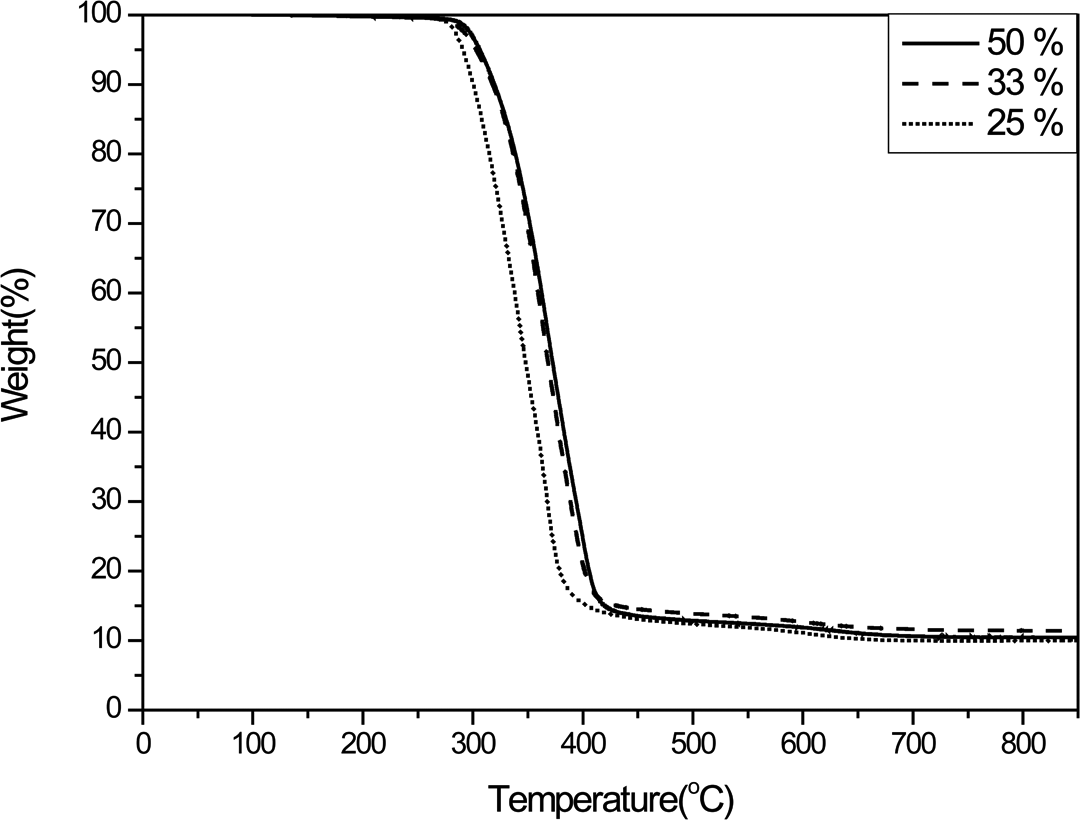
TGA profiles of PS/SiO2 spheres prepared using different MTES/TEOS ratios (i.e. 50, 33, and 25 wt% MTES). PS: poly(styrene); SiO2: silicon dioxide; MTES: triethoxymethylsilane; TGA: thermogravimetric analyzer; TEOS: tetraethyl orthosilicate.
The surface morphology of the PS/SiO2 spheres prepared using the various compositions of silane are shown in Figure 10. Monodispersed spheres were observed for all silane compositions, which indicated that monodispersed core/shell structure spheres were easily prepared by this method. In addition, for the MTES compositions of 50 and 33 wt%, distinct boundaries of the spheres were observed. In contrast, indistinct sphere boundaries and aggregation was observed when the MTES composition decreased to 25 wt%. The compatibility between the PS core and the silane decreased with decreasing concentration of MTES. On the other hand, a lower concentration of MTES represented a higher concentration of TEOS in the silane, which is desirable in terms of furnishing more reactive sites from the hydroxyl groups. The precursors did not readily diffuse into the PS core and remained on the outer layer of the spheres at a composition of 25 wt% MTES. For this reason, the SiO2 was more prone to undergo further reaction leading to the indistinct spherical shape (Figure 10(c)). Besides, the 193 nm diameter of PS/SiO2 spheres (33 wt% MTES) was calculated from SEM image (Figure 10(b)), while the SiO2 thickness was able to roughly determine by subtracting the diameter of PS core (Figure 2(d), 188 nm). Thus, the SiO2 shell thickness of 2.5 nm was rough obtained. Based on the volume ratio between polymer and SiO2, the density of SiO2 from sol–gel process 52 was assumed to be 1.66 g/cm3, theoretical calculated residual of SiO2 was about 12.6 wt%, which was roughly corresponded to the residual from TGA (11.4 wt%). On the other hand, the observations based on the SEM image were also consistent with the TGA results (Figure 9). The TGA curves shifted to higher temperatures when the composition of MTES was higher than 25 wt%. It was deemed that the compatibility between PS and SiO2 was improved by MTES; thus, decreasing the MTES concentration to 25 wt% lowered the compatibility of SiO2 with PS, thereby effecting a shift in the TGA curve to a lower temperature.

SEM images of PS/SiO2 spheres prepared at 25°C using various MTES/TEOS ratios: (a) 50 wt%, (b) 33 wt%, and (c) 25 wt% MTES. PS: poly(styrene); SiO2: silicon dioxide; MTES: triethoxymethylsilane; SEM: scanning electron microscopy; TEOS: tetraethyl orthosilicate.
In general, the glass transition temperature of PS is about 100°C. The PS core of the PS/SiO2 spheres becomes flexible (rubber-like state) and no longer maintains its shape at temperatures exceeding 100°C. At this point, the spherical shape is supported by the SiO2 shell structure. The influence of varying the silane composition on the shell structure was evaluated by calcination of the PS/SiO2 spheres prepared with various silane compositions at 200°C for 2 h; the surface morphologies are shown in Figure 11. A definitive spherical morphology with distinct boundaries and a high degree of monodispersity was observed only for the MTES composition of 33 wt%. This demonstrates that the spherical shape could be supported by the SiO2 shell structure when the appropriate MTES/TEOS composition was used. However, the corresponding decline in the proportion of TEOS when the proportion of MTES was increased to 50 wt% resulted in collapsed structures given that the structural stability is largely derived from the functional groups of TEOS (Figure 11(a)). At an MTES weight ratio of 25 wt%, the proportion of TEOS is sufficient to support the shape of the shell, but the compatibility between PS and SiO2 decreased and the outer layer of the spheres reacted readily to destroy the spherical shape as shown in Figure 11(c). The difference in the surface morphologies of these samples is attributed to the various degrees of SiO2 cross-linking. Enhanced compatibility of SiO2 and the PS core is achieved at an MTES composition of 50 wt%, but this benefit is offset by the lower degree of cross-linking. During calcination at 200°C, the mechanical stability of the sphere was not sufficient to support the structural integrity of the SiO2 shell, resulting in the rough surface. On the other hand, lowering the proportion of MTES to 25 wt% led to lower compatibility between PS and SiO2. After calcination, a rough surface was observed due to further reaction of the SiO2. Importantly, at the MTES composition of 33 wt%, where the corresponding composition of TEOS in the silane was increased to 67 wt%, the spherical shape could be maintained (Figure 11(b)). This higher TEOS content is thought to result in a higher degree of cross-linking, which furnishes sufficient mechanical integrity to support the spherical shape during calcination at 200°C. Therefore, the MTES composition of 33 wt% was regarded as apposite for the construction of the PS/SiO2 spheres given that the structural integrity of the sphere was preserved intact after calcination at 200°C.

SEM images of PS/SiO2 spheres prepared at 200°C using various MTES/TEOS ratios: (a) 50 wt%, (b) 33 wt%, (c) 25 wt% MTES, and (d) 33 wt% MTES sample calcined at 400°C. PS: poly(styrene); SiO2: silicon dioxide; MTES: triethoxymethylsilane; SEM: scanning electron microscopy; TEOS: tetraethyl orthosilicate.
In order to fabricate SiO2 hollow spheres, the PS/SiO2 core–shell spheres prepared from 33 wt% MTES were calcined at the higher temperature of 400°C for 2 h. In general, the PS core can be decomposed at 400°C, and the residual is regarded as the surface structure constructed from SiO2. The surface morphologies of the calcined spheres are shown in Figure 11(d). Spherical structures with holes were observed in the SEM image. The holes in the spheres are due to the channels resulting from the decomposed PS. The SEM image demonstrates that the hollow spherical shape could be supported by the SiO2 shell structure when the appropriate MTES composition was used. The construction of these hollow spheres also indicated that sufficient compatibility between PS and SiO2 could be achieved using the appropriate composition of MTES, thereby preventing further reaction of the SiO2. Moreover, in the presence of sufficient TEOS in the silane, the degree of cross-linking of SiO2 was enough for the construction of a shell structure with sufficient mechanical stability to maintain the spherical shape after calcination at 400°C. The MTES composition of 33 wt% was regarded as the appropriate composition for construction of hollow SiO2 spheres given that the spherical shape remained intact after calcination at 400°C.
Conclusion
In this study, a novel and feasible approach for the synthesis of hollow SiO2 spheres was developed based on the template-sacrificial technique. Core–shell PS/SiO2 spheres were prepared from hard PS templates that were prepared by soap-free emulsion polymerization in the boiling state for a specified period. Subsequent introduction of silane into the reaction system furnished the core–shell PS/SiO2 spheres. The active interaction force between PS and SiO2 (or the silane precursor) was simple diffusion of the silane. The compatibility between PS and SiO2 could be controlled by tuning the polarity of the silane in response to variation of the relative compositions of TEOS and MTES with a synchronous effect of the silane composition on the mechanical properties of the SiO2 shell. Based on this technique, PS/SiO2 spheres could be synthesized by a simple approach without the requirement for structure-directing agents or surface modification. Hollow SiO2 spheres were generated by calcination of the PS/SiO2 spheres at 400°C in air atmosphere. This technique represents a new paradigm in the preparation of hollow SiO2 submicrospheres.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.