
Abstract
Poly(lactic acid) (PLA) and polypropylene (PP) were comparatively investigated as matrices for injection-moulded composites containing small (1–3 wt%) amounts of short sisal fibre. The morphology, thermal and dynamic mechanical properties, as well as degradation characteristics were investigated. The scanning electron microscopy (SEM) micrographs of the composites show more intimate contact and better interaction between the fibres and PLA, compared to PP. This improved interaction was confirmed by the Fourier-transform infrared (FTIR) spectroscopy results which showed the presence of hydrogen bonding interaction between PLA and the fibres. The thermal stability (as determined through thermogravimetric analysis [TGA]) of both polymers increased with increasing fibre content, with a more significant improvement in the case of PP. The differential scanning calorimetry (DSC) results showed a significant influence of the fibres on the cold crystallization and melting behaviour of PLA, even at the low fibre contents of 1–3%. The influence of the fibres on the melting characteristics of the PP was negligible, but it had a significant influence on the nonisothermal crystallization temperature range. Both the storage and loss moduli of the PLA decreased with increasing fibre content below the glass transition of PLA, but the influence on the loss modulus was more significant. The dynamic mechanical analysis (DMA) results clearly show cold crystallization of PLA around 110°C, and the presence of fibres gave rise to higher modulus values between the cold crystallization and melting of the PLA. The presence of fibres also had an influence on the dynamic mechanical properties of PP. This article further describes basic biodegradation observations for the investigated samples.
Introduction
Natural fibre-reinforced polymer composites have outstanding potential as an alternative for synthetic fibre composites. 1 Due to their structural properties and the fact that they are relatively cost effective, biodegradable, and lightweight, research is now based on the utilisation of natural fibres as load-bearing constituents in composite materials. Fibres provide the strength and stiffness and act as reinforcement in a fibre-reinforced composite material. 2
There have been several publications on PP composites reinforced with short/long sisal fibres (SFs).3–8 It was generally found that SFs enhanced the mechanical properties of the composites. The improvement was mainly observed for composites containing low fibre contents, while higher fibre contents normally gave rise to reduced mechanical properties. This was explained as being due to the incompatibility between the fibres and the matrix, which promoted microcrack formation at the interface as well as nonuniform stress transfer due to fibre agglomeration in the matrix. Reports also indicated an increase in the thermal stability of the PP in the presence of SF. Other thermal properties such as melting temperature, crystallization temperature, heat of fusion, and percentage of crystallinity were also influenced by the presence of SF, due to the nucleating ability of these fibres. Although PP/sisal composites are called biocomposites, they are still not completely biodegradable.
One of the most promising biodegradable polymers is poly(lactic acid) (PLA), and PLA can be manufactured from renewable resources, most commonly from corn.9–12 Most of the research on PLA composites ultimately seeks to improve the mechanical properties to a level that satisfies the particular applications, where PLA could be a replacement for synthetic polymers like PP. 13 PLA has better mechanical properties than PP, with a tensile strength of 62 MPa and a modulus of 2.7 GPa, in contrast to 36 MPa and 1.2 GPa for PP. Moreover, PLA can be processed by injection moulding, blow moulding, 14 and film extrusion because the glass temperature of PLA is 50–60°C and the melting temperature is 168–172°C. 15 Limited research has been published on the use of SF as filler or reinforcement in the PLA matrix.7,15–17 Transcrystallinity of PLA on SF was observed, and it was found that both treated and untreated SFs act as efficient nucleating agents in PLA and that nucleation occurs preferentially along the fibre axis. The results obtained from literature suggests that SF surface modification using alkali or silane has little or no influence on the nucleation ability of SF in a PLA matrix. 18 No reports could be found on the mechanical and thermal properties for PLA/SF composites in the available literature.
There is limited information on the use of low contents of short natural fibre in polymer composites.4,19 In these publications, it was reported that there is a reduction in the mechanical properties of these composites. At low fibre contents (≤10 wt%), the fibres may act as flaws or have a plasticization effect in the matrix, reducing the tensile strength of the composite. 4
In this article, SF-reinforced PLA and PP composites, containing low fibre contents and prepared through extrusion and injection moulding, were studied. The morphology and biodegradability, as well as the thermal and thermomechanical properties, of the two systems were investigated with the aim to understand whether the PLA composites will be a good substitute for the PP composites in some applications.
Materials and methods
SF was obtained from the National Sisal Marketing Committee in Pietermaritzburg, South Africa. It has a diameter range of 100–300 μm, average tensile strength of 490 MPa, average modulus of 11,350 MPa, and elongation at break of 5%.
Polylactide (PLA) polymer 3051D was supplied by Nature-Works LLC in Europe. The density of the PLA was 1.25 g cm−3, melt flow index ([MFI] 210°C/2.16 kg) of 10–25 g/10 min, relative viscosity of 3.0–3.5, crystalline melting temperature of 150–165°C, glass transition temperature of 55–65°C, tensile strength at a yield of 48 MPa, and tensile elongation at a yield of 2.5%.
Polypropylene (PP) TIPPLEN H 116F homopolymer, as opaque crystalline pellets, was supplied by the Tiszai Chemical Group Plc, Hungary. This is a high melt flow polymer that was designed for fibres of medium to high spin speeds, and it offers good homogeneity, stable extrusion, and excellent processability. It has the following properties: tensile strength at yield of 34.5 MPa, MFI (230°C/2.16 kg) of 25 g/10 min, and tensile elongation at yield of 10%.
The sisal was cut into 5-mm long fibres and dried in an oven at 130°C for 6 h to eliminate the absorbed moisture. The PLA was also dried in an oven at 85°C for 6 h before it could be used. PP was used as supplied without drying. The PLA and PP composites with SF content of 1–3 wt% were extruded at 190°C. After the extrusion, the pelletized PLA/sisal composites were placed in the oven at 120°C for 3 h to recrystallize. After the annealing process, the PLA samples were dried in the oven at 85°C followed by injection moulding at 190°C. The PP and its composites were injection moulded immediately after they were pelletized. An Arburg 320C Allrounder 600-250 injection moulding machine was used to prepare 2-mm thick and 80 × 80 mm flat sheet samples, and the injection moulding parameters were as follows:

After processing the fibres were well dispersed in the polymers and the fibre lengths were in the range of 2–5 mm.
A Shimadzu ZU SSX-550 Superscan scanning electron microscope (SEM) was used to study the fracture surfaces of the samples. The samples were fractured after insertion in liquid nitrogen and sputter-coated with gold under an argon gas flow for 20 min. The coated samples were left to dry at room temperature for 2 h before SEM could be performed. The SEM analyses were done at 15 kV and at a magnification of ×300.
A Perkin-Elmer Spectrum 100 Fourier-transform infrared (FTIR) spectroscope fitted with an attenuated total reflection (ATR) detector, equipped with a diamond crystal, and a Perkin Elmer Multiscope FTIR microscope was used to study the chemical bonding and molecular structure of the composites and polymers. The infrared (IR) spectra were recorded between 4000 and 650 cm−1 at a resolution of 8 cm−1. A clean, empty diamond crystal was used for the collection of the background spectrum.
A Perkin-Elmer DSC7 differential scanning calorimeter (DSC) from Waltham, MA, USA, was used for the DSC analyses. All the analyses were performed under nitrogen flow (20 ml min−1). The PP samples (5–10 mg) were analysed at 25–180°C at a rate of 10°C min−1, while the PLA samples were analysed at 25–160°C at the same rate. The samples were heated, cooled, and reheated under the same conditions mentioned above. Three samples from each composition were analysed. Only the second heating scan was used to determine the melting enthalpies and temperatures. The crystallization enthalpies and temperatures were determined from the first cooling scan.
The thermogravimetric analyses (TGAs) were carried out in a Perkin-Elmer TGA7 thermogravimetric analyser, from Waltham, MA, USA. Nitrogen was used as the purge gas at a flow rate of 20 ml min−1. The samples (5–10 mg) were heated at 30–600°C at a rate of 10°C min−1.
A Perkin-Elmer Diamond dynamic mechanical analyzer (DMA) was used in determining the thermomechanical properties of all the samples. The samples, with the dimensions 50 × 12 × 1 mm, were analysed in the bending mode and the parameters were as follows:

The biodegradability (degradation through hydrolysis) of the samples was tested by monitoring the mass loss during insertion in water at 80°C, as well as by taking SEM pictures of the samples that were periodically removed from the hot water. The higher temperature was used to shorten the time of exposure in order to speed up the experiment.
Results and discussion
Morphology
The SEM micrographs in Figure 1 show that for the PLA composites there are some fibre breakages and a minimal number of fibre pull-outs. It seems as if some matrix material may be covering the fibres. The presence of matrix coverage on the fibre surfaces and the fibre breakages indicate good interaction between the fibre and the matrix, which results in better interfacial adhesion. The gaps between the fibre and the PLA matrix in the pictures clearly indicate that the fibre was trying to pull-out but it could not, which also indicates good fibre/matrix interaction. Huda et al. 20 investigated the mechanical and thermomechanical properties of wood fibre-reinforced PLA, and compared them with wood fibre-reinforced PP composites which were processed in the same way by a micro-compounding moulding system. They reported that there was a good adhesion between the wood fibre and the PLA, and the maleic anhydride-grafted PP (MAPP) coupling agent had no effect on the morphological properties of the PLA/wood fibre composites. This is in line with our own observations.

Scanning electron microscopy (SEM) micrograph of the fracture surface of a poly(lactic acid) (PLA)/sisal composite (×300 magnification).
The SEM micrographs in Figure 2 seem to show surprisingly good adhesion between the hydrophilic SF and the hydrophobic PP polymer, which does not reflect in the properties that will be discussed later in this article. Most of the fibres at the fracture surface are still attached to the PP. The fibre breakages and the seeming lack of fibre pull-outs in all the pictures seem to indicate good matrix/fibre adhesion. The reason for this may be that, at the high temperature used for composite preparation, the viscosity of the PP (because of its high MFI) was low enough for it to penetrate the pores on the fibre surfaces.

Scanning electron microscopy (SEM) micrograph of the fracture surface of a polypropylene (PP)/sisal composite (×300 magnification).
To examine the existence and type of interfacial interaction in the composites, FTIR experiments were performed and compared with those of the neat PP and PLA. The regions of interest for PLA and the composites are 1780 and 1680 cm−1 for the C=O stretch, and 3600–3000 cm−1 for the O–H stretch. The peaks at about 1750 and 1180 cm−1, which belong to the C=O stretching and the C–O–C stretching of PLA, are clearly visible in all the PLA spectra (Figure 3). The O–H band for the PLA-based samples became more pronounced and broader and shifted to slightly lower wave numbers, as the fibre content was increased. This is probably due to the ‘free’ hydroxyl groups that are now engaged in hydrogen bonding. 21 There is a development of a small peak just below the carbonyl peak at 1650 cm−1. This is an O–H peak that originated from bending of the unresolved hydroxyl group of the absorbed water usually carried by cellulose.22,23 As expected, the FTIR spectra of the PP and the PP/sisal composites did not show any evidence of interfacial interaction between the polymer and the fibre.
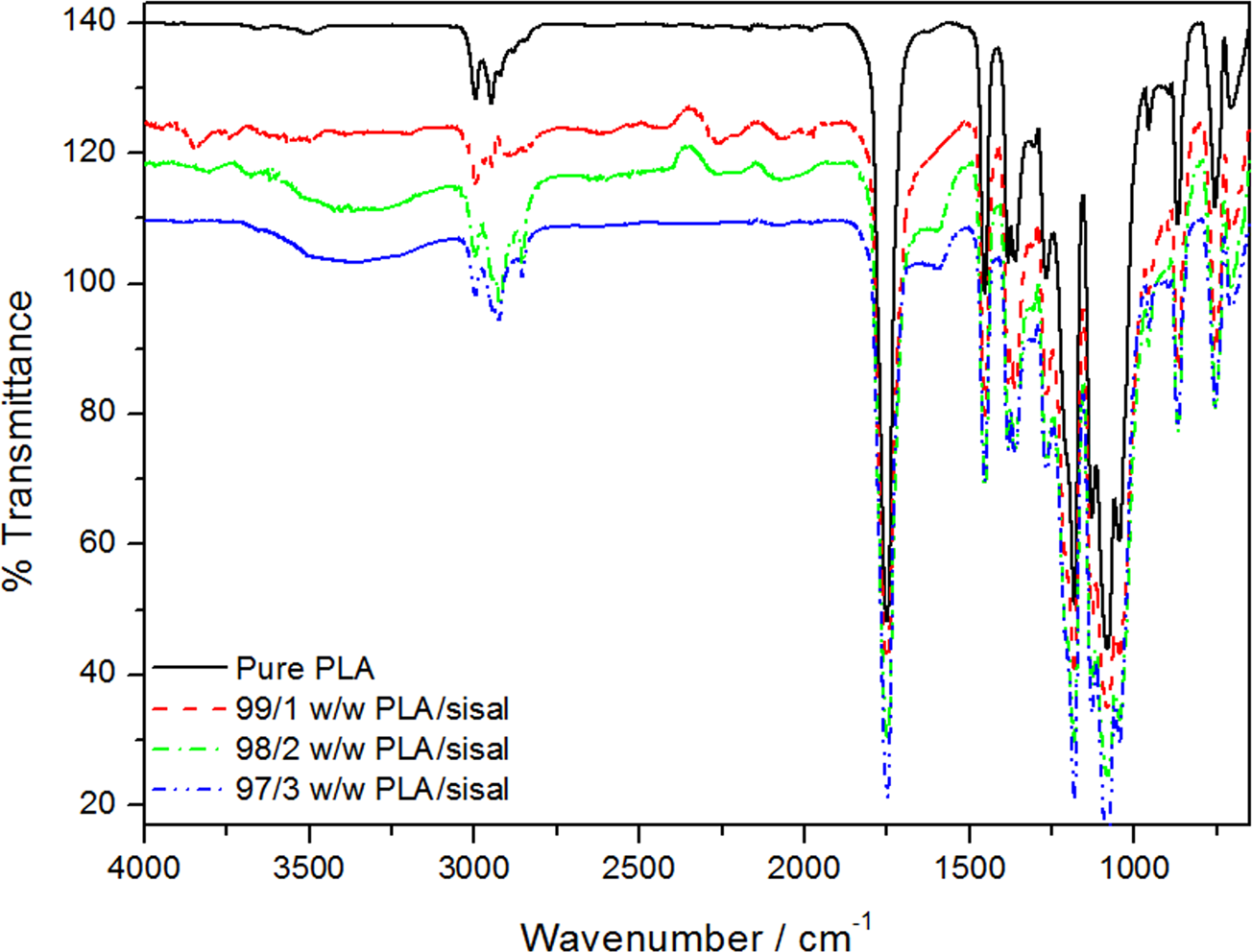
Fourier-transform infrared (FTIR) spectra of poly(lactic acid) (PLA) and the PLA/sisal fibre composites.
Thermogravimetric analysis
The TGA curves for all the samples are shown in Figures 4 and 5. One of the goals when incorporating SF into the two polymers was to increase the temperature region in which PLA and PP can be useful. 24 SF loses moisture around 100°C. The degradation of the SF is a two-step degradation process as indicated by the two mass loss steps at 285 and 357°C. The first step is due to the thermal depolymerisation of hemicellulose and the glycosidic linkages of cellulose. The second step is due to the cellulose decomposition which produces a relatively high char residue.25,26 Only one degradation step was observed for the neat polymer and for the composites, and the thermal stability was better for the composites but did not change significantly with increasing fibre content. The surrounding polymer seems to have protected the fibre from lower-temperature degradation, and the interaction between the fibre and the polymer seems to have retarded the degradation process and/or restricted the diffusion of volatile degradation products out of the sample. The degradation of sisal in the composites was not observed as a separate step (Figure 4), and we assume that this was due to the low fibre contents and the good interaction between the fibre and PLA.
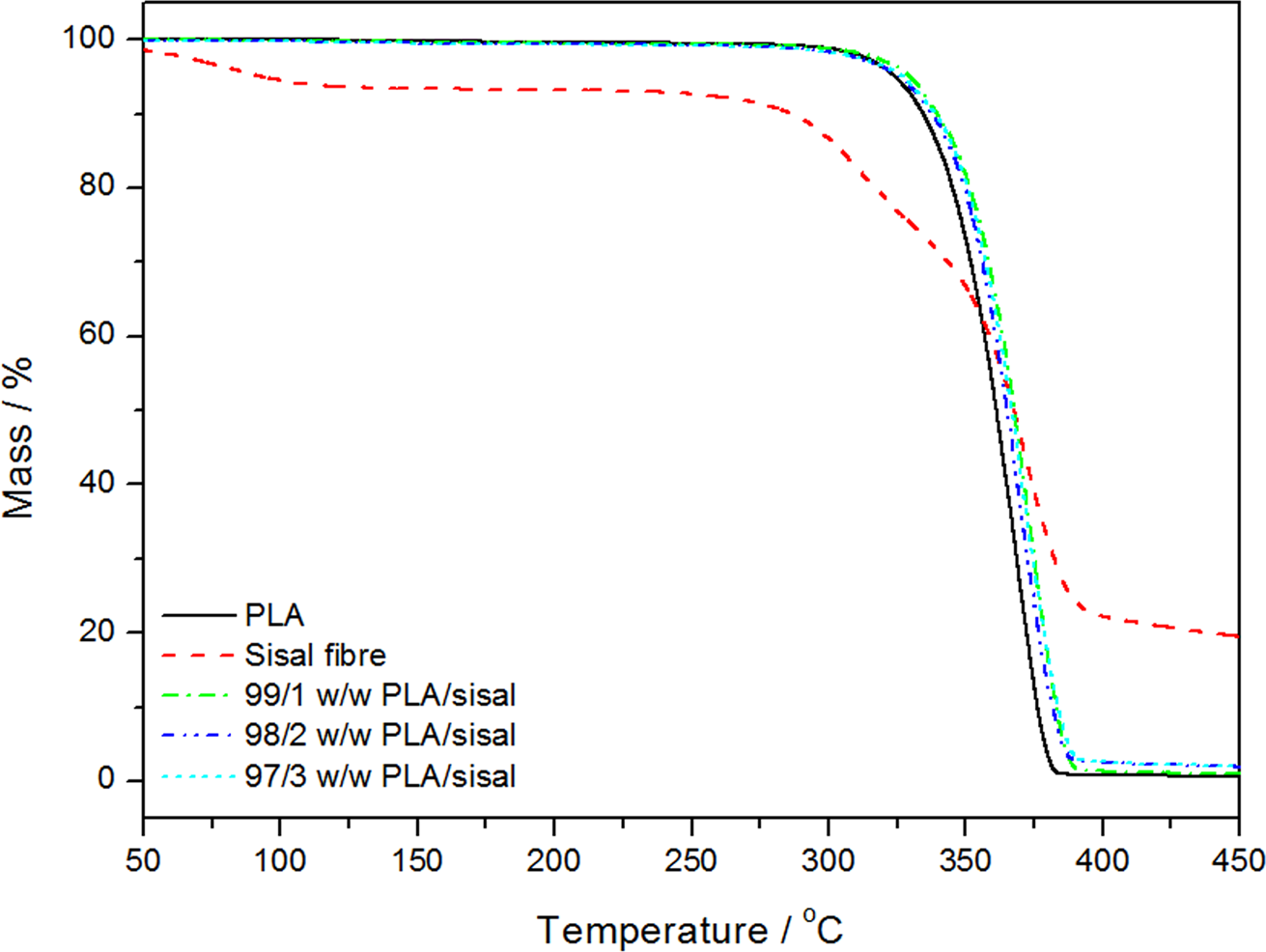
Thermogravimetric analysis (TGA) curves of poly(lactic acid) (PLA) and the PLA/sisal fibre composites.
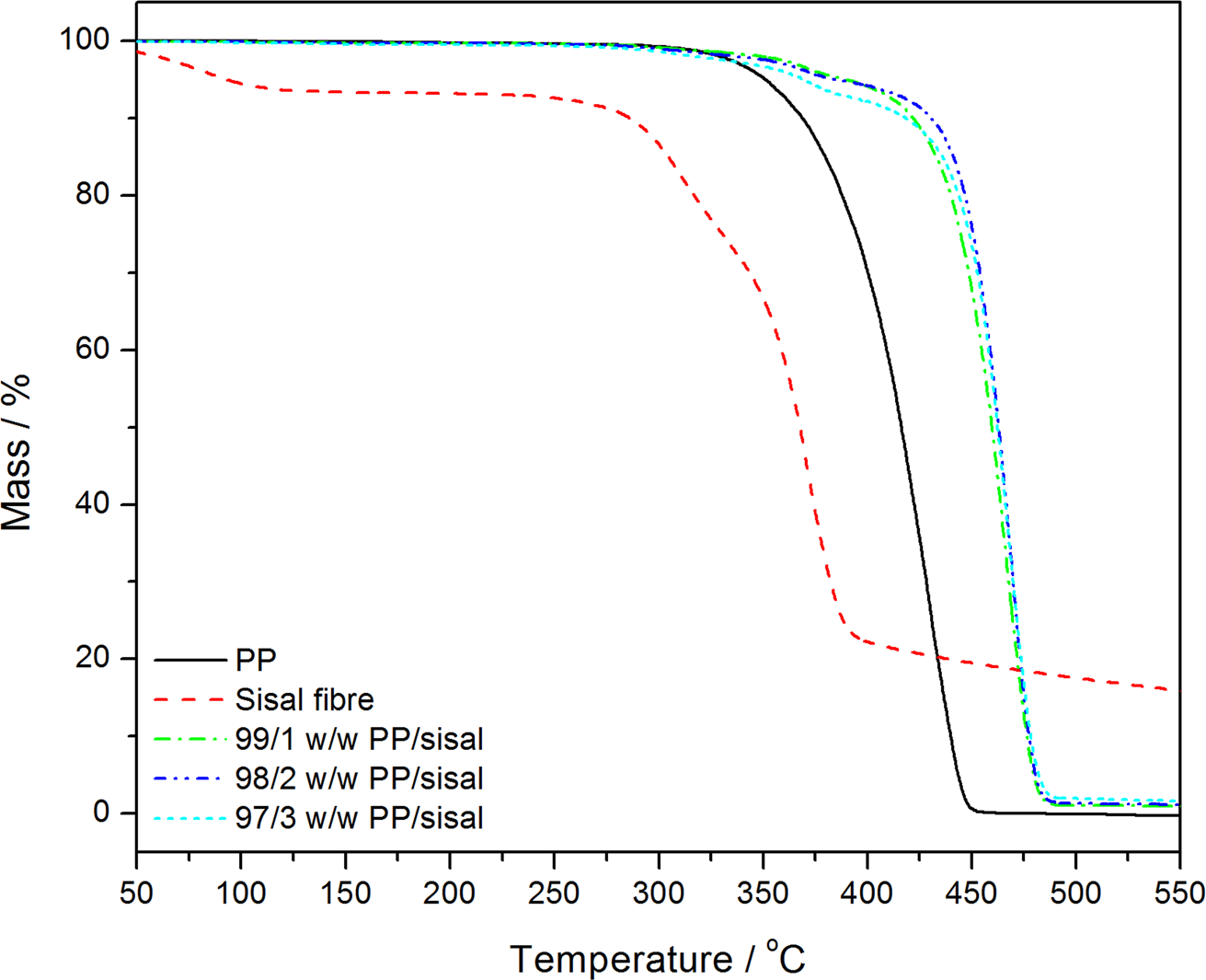
Thermogravimetric analysis (TGA) curves of polypropylene (PP) and the PP/sisal fibre composites.
Pure PP shows a single degradation step, while the composites show two degradation steps (Figure 5). The low mass loss around 330°C is due to the degradation of SF, while the major step around 440°C is due to the degradation of PP. The degradation temperatures of both the fibre and PP in the composites are significantly higher than those of pure fibre and pure PP. Salemane et al. 27 reported the same behaviour when PP was filled with wood flour (WF) of different WF particle sizes. The fibre probably started degrading at higher temperatures because of the thermal protection by the thermally more stable PP that surrounded the fibre. The increased thermal stability of the PP is probably because the presence of the fibre might have (i) immobilized the free radicals formed during the initiation of the degradation of the polymer chains and (ii) inhibited the diffusion of volatile degradation products because of the interaction with the fibre.
Differential scanning calorimetry
The DSC results of the annealed PLA samples are shown in Figure 6. The glass transition, cold crystallisation, and melting of the samples are clearly observed in the curves. The glass transition of the annealed neat PLA shows a sharp transition called a hysteresis peak. This happens mostly with annealed semicrystalline polymers. The reason for this endothermic hysteresis peak is not a lowering of the enthalpy of the polymer due to annealing but a change in the kinetics of unfreezing. 28 The sharpness and the intensity of the hysteresis peak decrease in the presence of the SF. This is an indication that the presence of fibre in some way changed the morphology of the amorphous fractions in the polymer during the cooling process. The glass transition temperature of the PLA did not change for the fibre composites (Table 1). The reasons for this is that (i) the annealing produced more, and more perfect, crystals that acted as physical crosslinks and reduced the mobility of the amorphous fraction in the polymer and (ii) the fibre content was too low and any anticipated influence that it may have had on the polymer chain mobility was overshadowed by the influence of the PLA crystals.
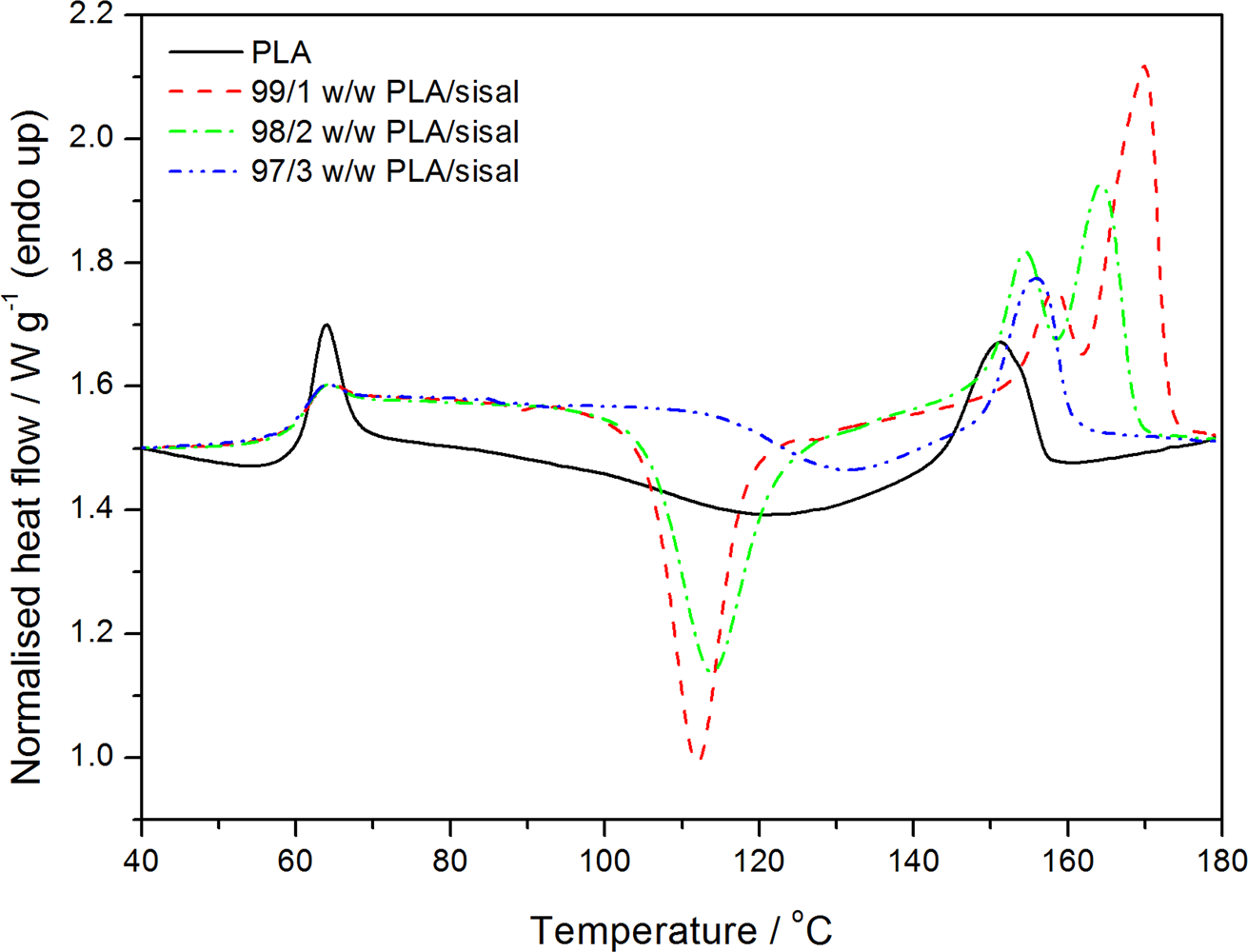
Differential scanning calorimetry (DSC) heating curves of poly(lactic acid) (PLA) and the PLA/sisal fibre composites.
DSC results of all the investigated PLA and PLA/sisal composite samples
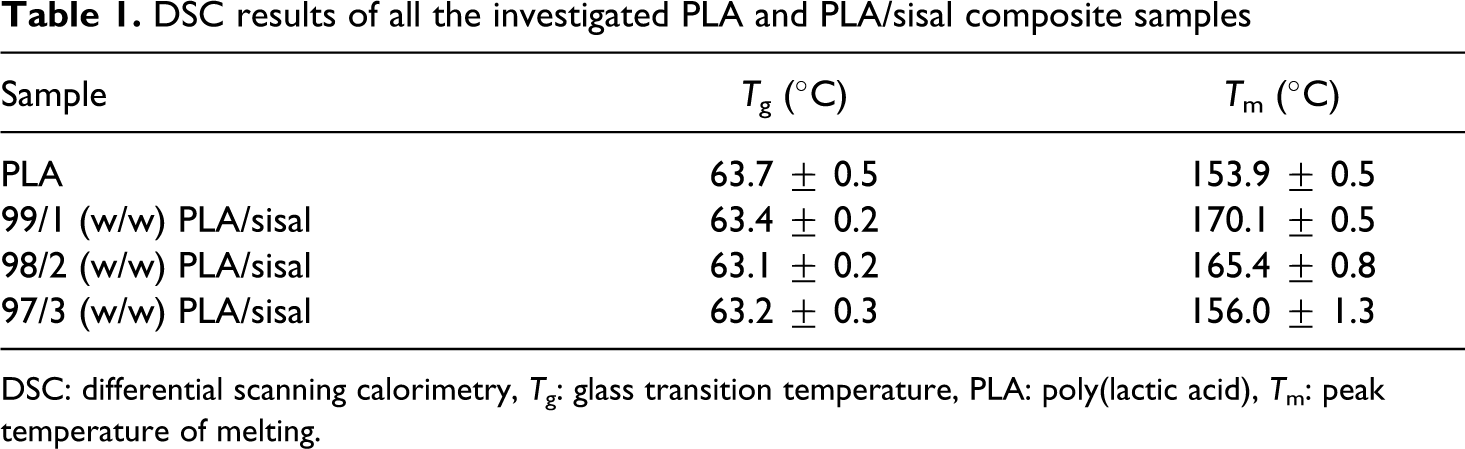
DSC: differential scanning calorimetry, T g: glass transition temperature, PLA: poly(lactic acid), T m: peak temperature of melting.
The exothermic transition around 110°C (Figure 6) is due to the cold crystallization of PLA, and it is sharp and well resolved for the sample containing 1 wt% of SF. This peak shifts to higher temperatures and also decreases in intensity with the increase in the SF content. The fibres can either act as nucleation sites for the crystallization of the polymer or restrict the mobility of the polymer chains. These two actions may always be present when a polymer is cooled in the presence of well-dispersed short fibres. When only 1% of SF was present, the nucleation by the short fibres was probably dominant, giving rise to a large extent of cold crystallization and the formation of two melting peaks, indicative of two crystal fractions (meta-stable and perfected crystals 28 ) formed during the original cooling of the sample and the subsequent cold crystallization. As the fibre content increased (2 wt%), the cold crystallisation peak intensity decreased and it shifted to higher temperatures. The intensity of the second melting peak also decreased, and both melting peaks shifted to lower temperatures. This indicates that the immobilization effect became more dominant. At higher fibre content (3 wt%), the cold crystallisation peak shifted to even higher temperatures and its intensity decreased significantly, and the second melting peak completely disappeared. This shows that the immobilization effect was even more dominant, but the fact that the melting temperature for this composite was still higher than that of neat PLA shows that some fibres still acted as nucleating agents.
Figures 7 and 8 illustrate the DSC heating and cooling curves, and Table 2 summarizes the DSC results for PP and the PP/SF composites. Figure 7 and Table 2 show that the melting temperatures of the composites are higher than that of pure PP, and the experimental melting enthalpies are higher than the calculated ones, indicating a higher total crystallinity for the composites. The crystallisation temperature (T c) values of PP in the PP/SF composites are higher than that of pure PP. This indicates that the fibres acted as nucleating agents, hence the PP in the composites started crystallizing at higher temperatures. Normally, upon cooling, the polymer will crystallize at a given temperature, and an effective nucleating agent will cause the T c of the polymer to increase, because crystallization will start earlier during the cooling process. This behaviour can be explained by the fibre surfaces acting as nucleation sites for the crystallization of the polymer matrix. 29
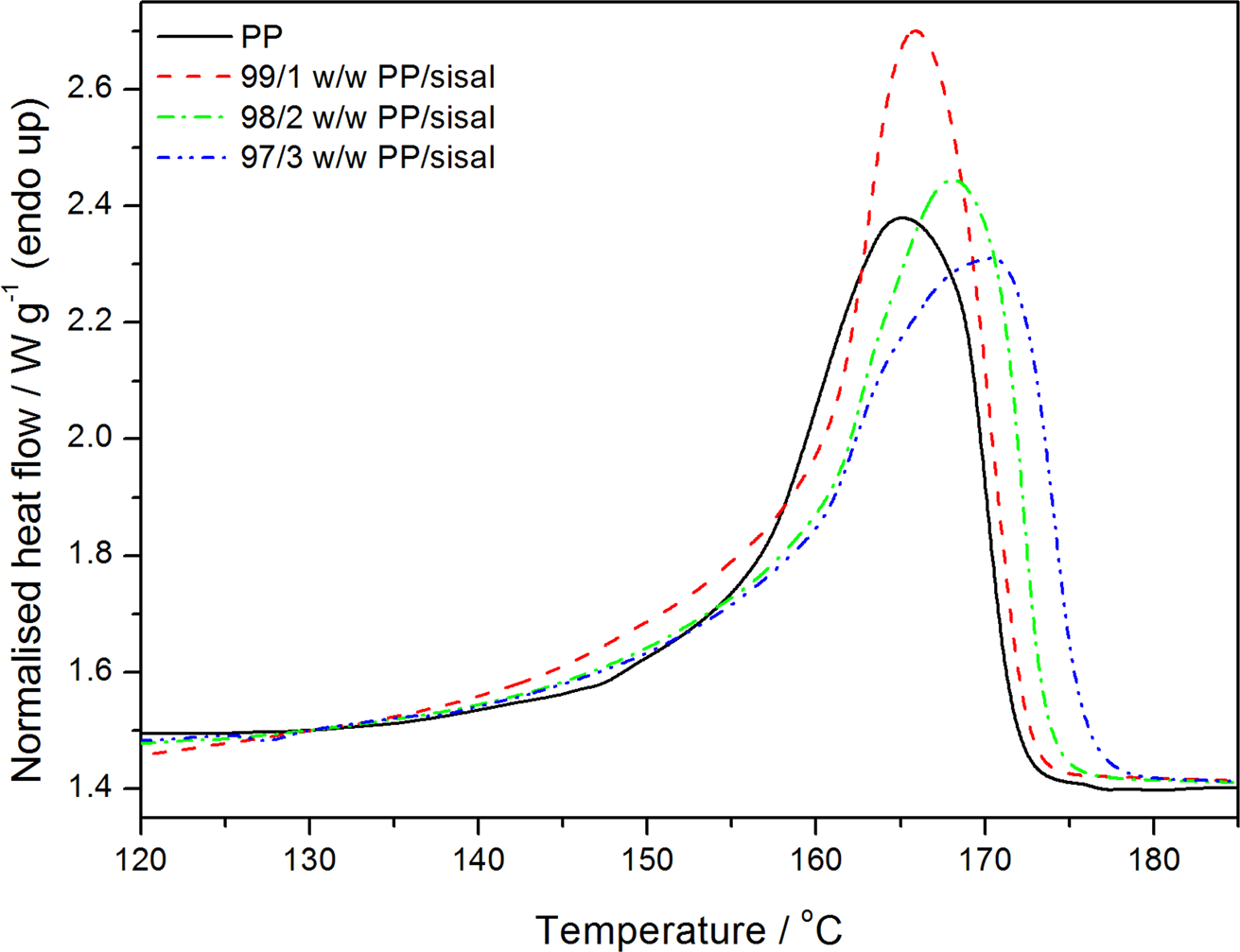
Differential scanning calorimetry (DSC) heating curves of polypropylene (PP) and the PP/sisal fibre composites.
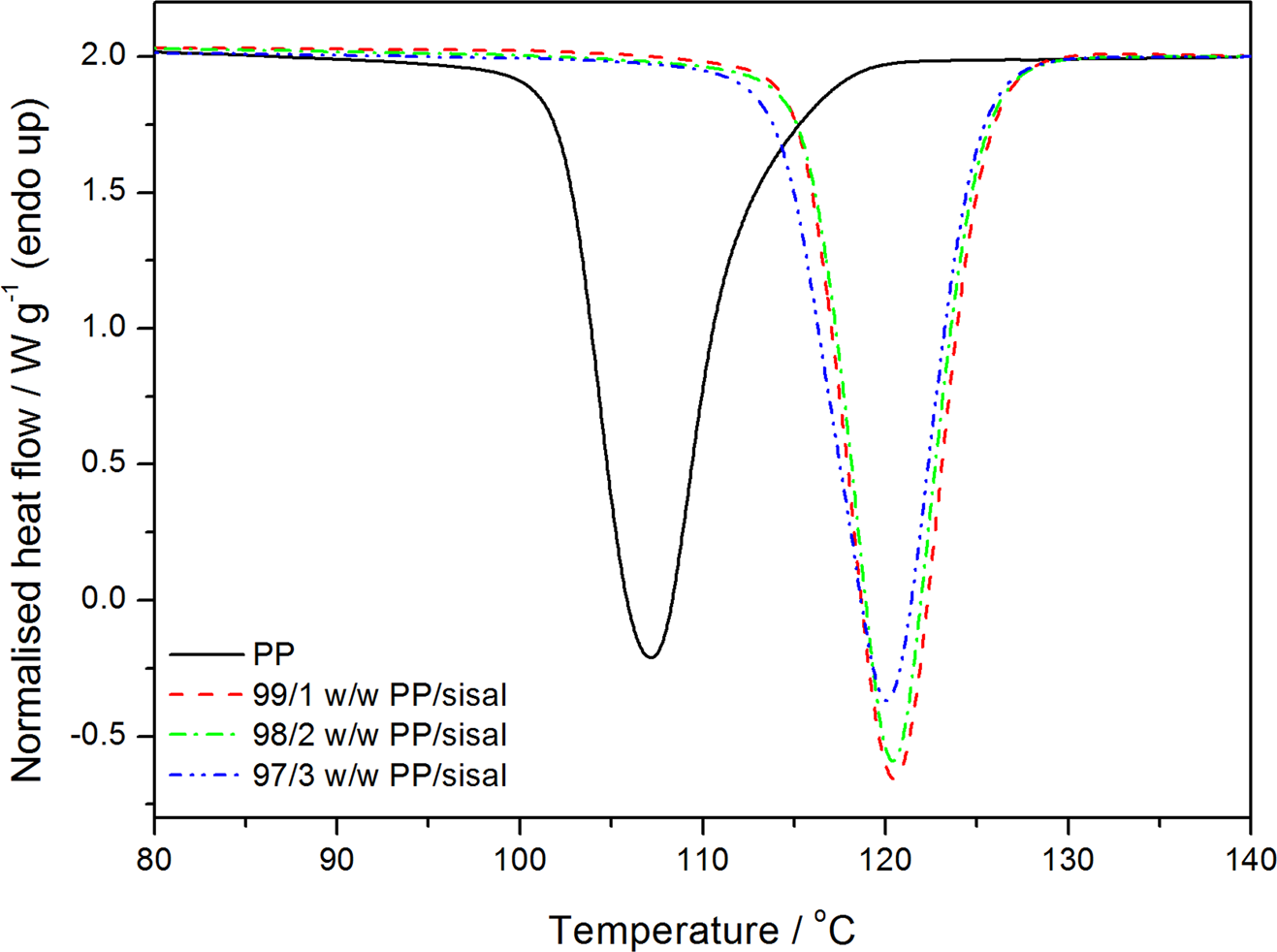
Differential scanning calorimetry (DSC) cooling curves of polypropylene (PP) and the PP/sisal fibre composites.
DSC results of all the investigated PP and PP/sisal composite samples
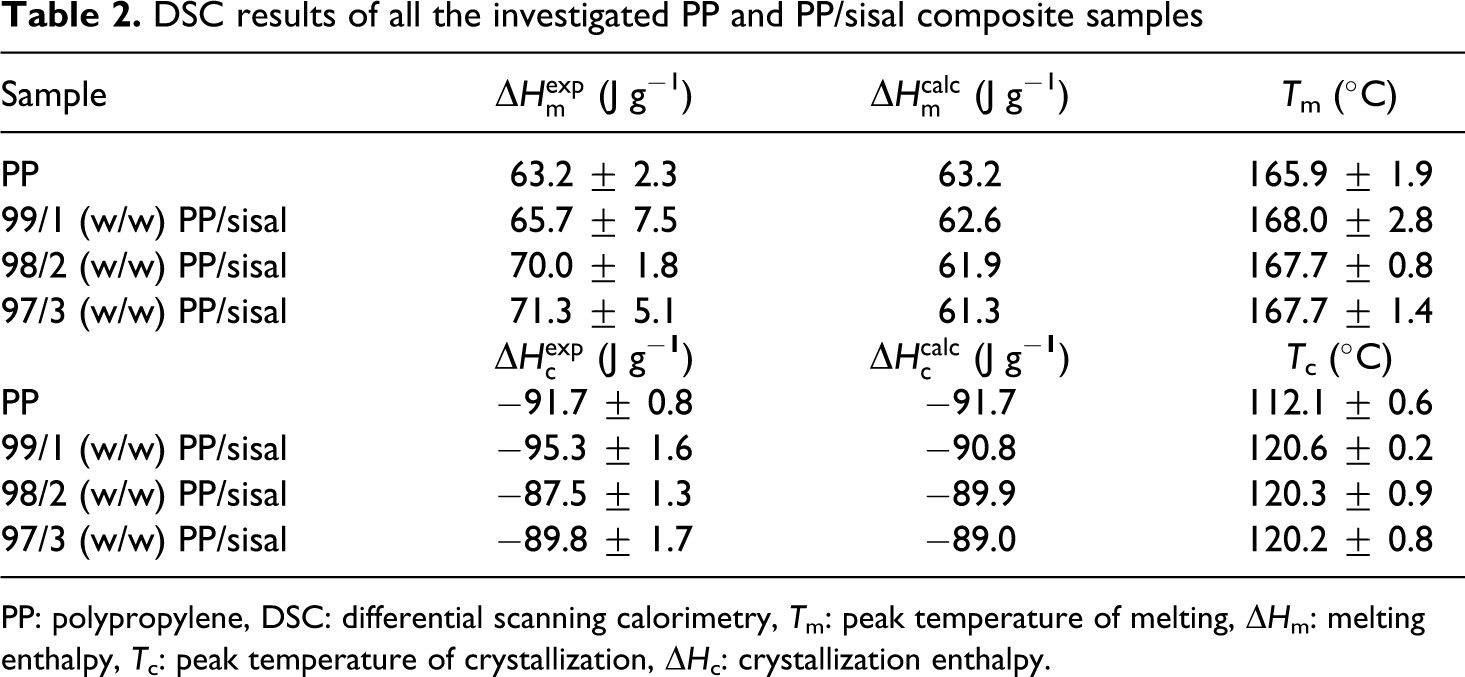
PP: polypropylene, DSC: differential scanning calorimetry, T m: peak temperature of melting, ▵H m: melting enthalpy, T c: peak temperature of crystallization, ▵H c: crystallization enthalpy.
Dynamic mechanical analysis
Figures 9–14 show how the addition of different contents of SF influenced the storage modulus (E′′), loss modulus (E′), and damping factor (tan δ) of PLA, PP, and their respective composites. The storage modulus (E′) below the glass transition of PLA slightly decreased with increasing fibre content. Mathew et al. 30 studied the crystallisation of PLA in the presence of different cellulose-based reinforcements. They reported that the heat treatment was not expected to result in 100% crystallinity but that the crystalline regions might restrict the chain mobility. Therefore, the introduction of fibres might decrease the stiffness by possibly acting like plasticizers.
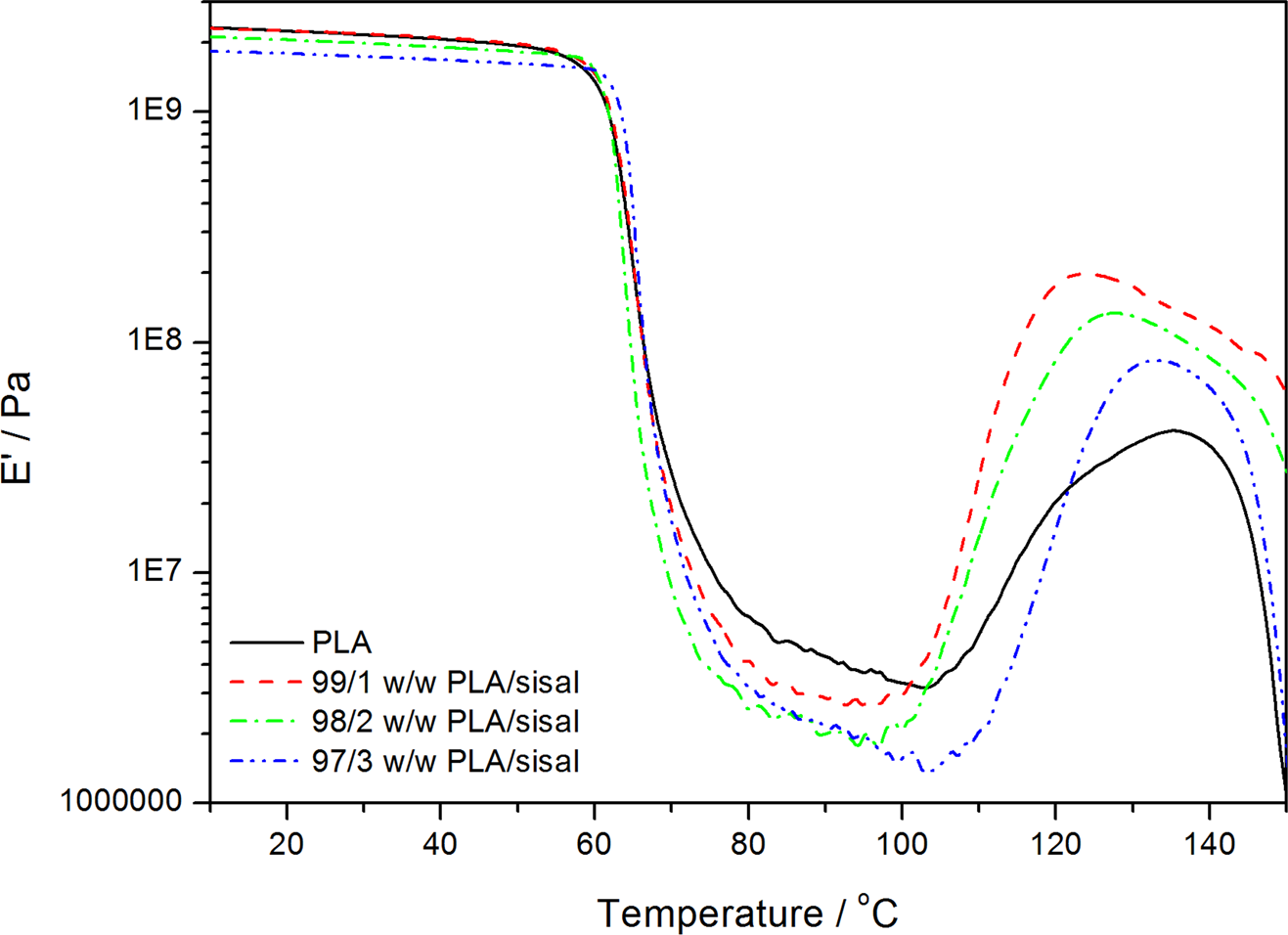
Dynamic mechanical analysis (DMA) storage modulus curves of poly(lactic acid) (PLA) and the PLA/sisal fibre composites.
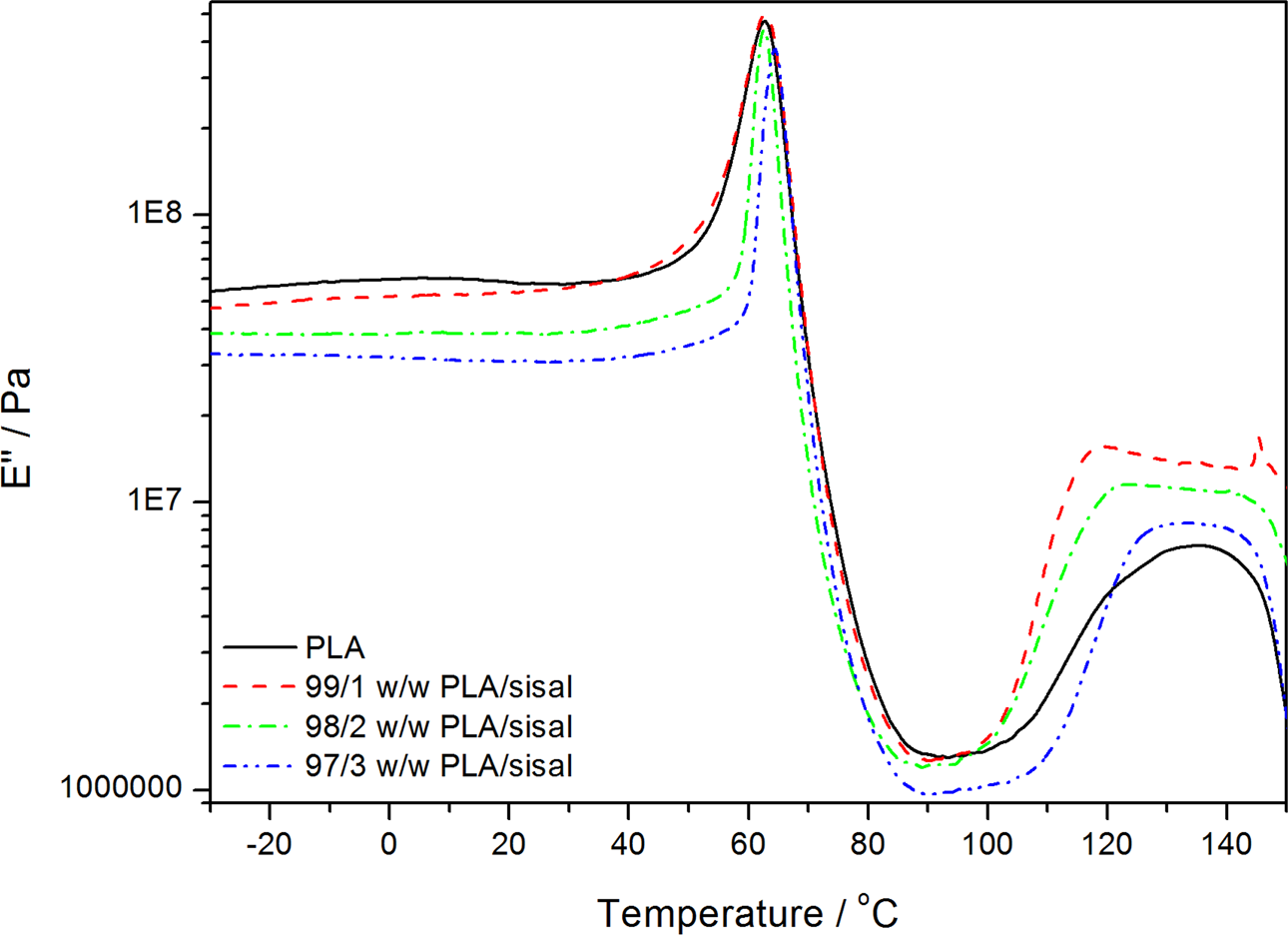
Dynamic mechanical analysis (DMA) loss modulus curves of poly(lactic acid) (PLA) and the PLA/sisal fibre composites.
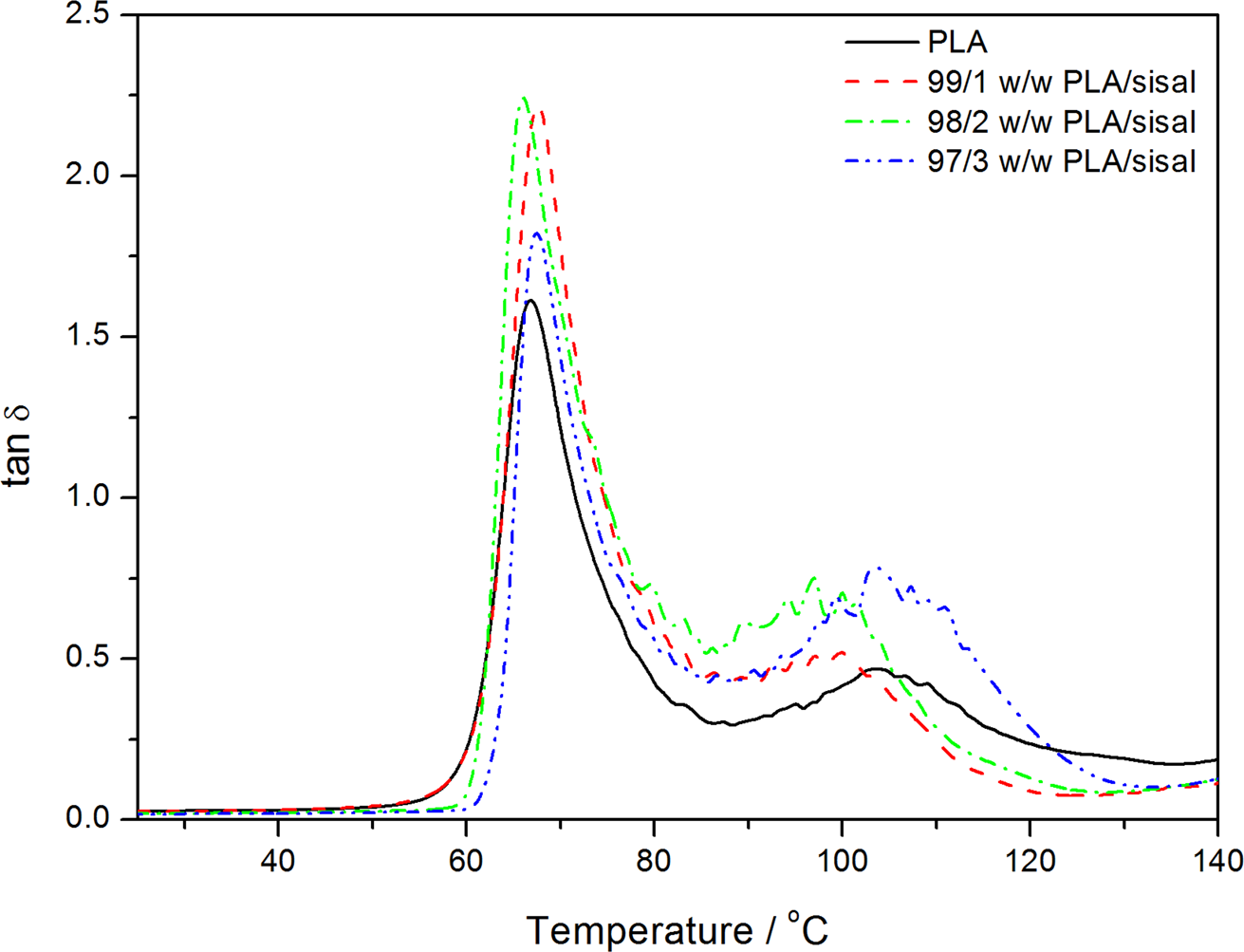
Dynamic mechanical analysis (DMA) damping factor curves of poly(lactic acid) (PLA) and the PLA/sisal fibre composites.
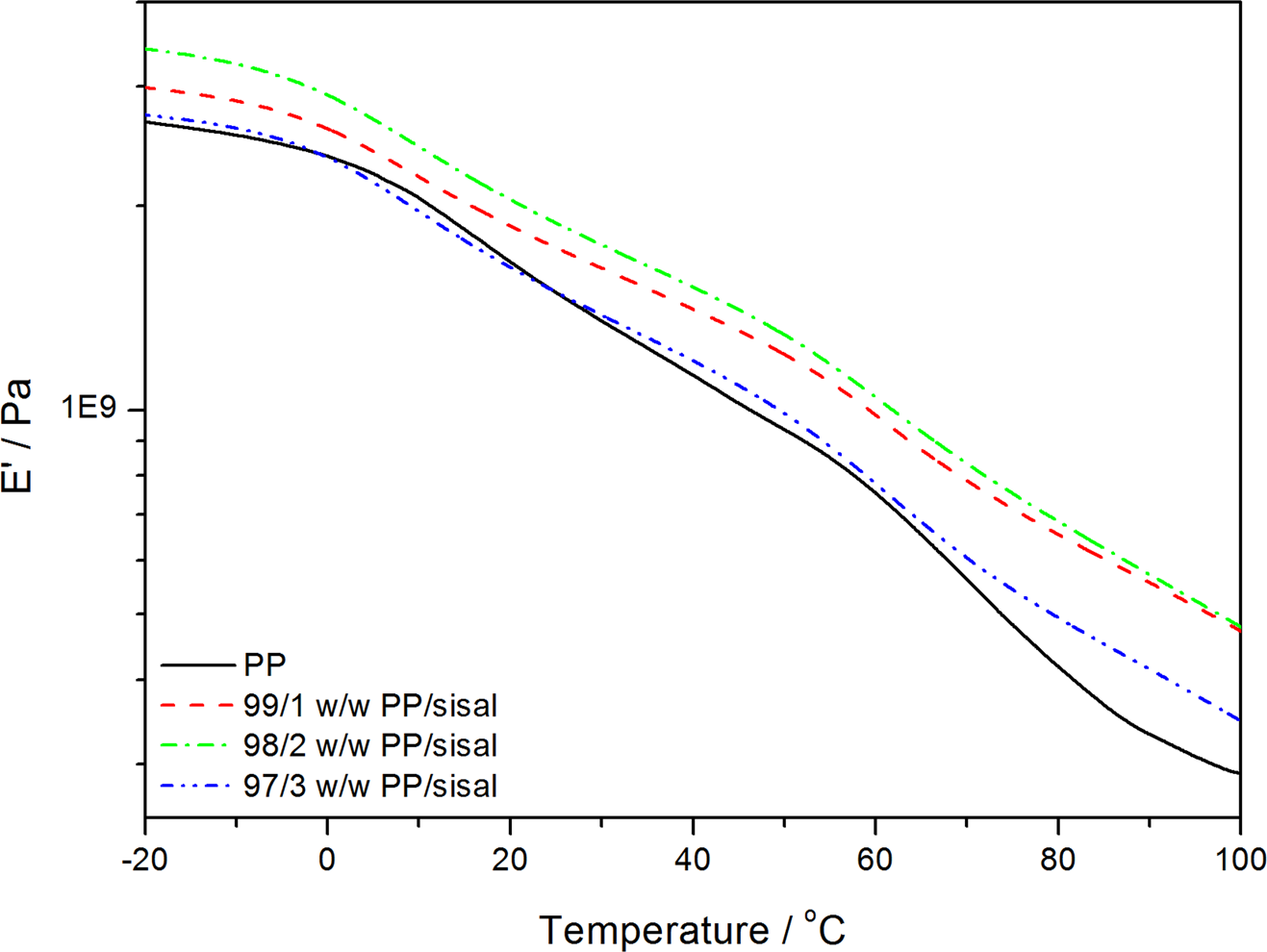
Dynamic mechanical analysis (DMA) storage modulus curves of polypropylene (PP) and the PP/sisal fibre composites.
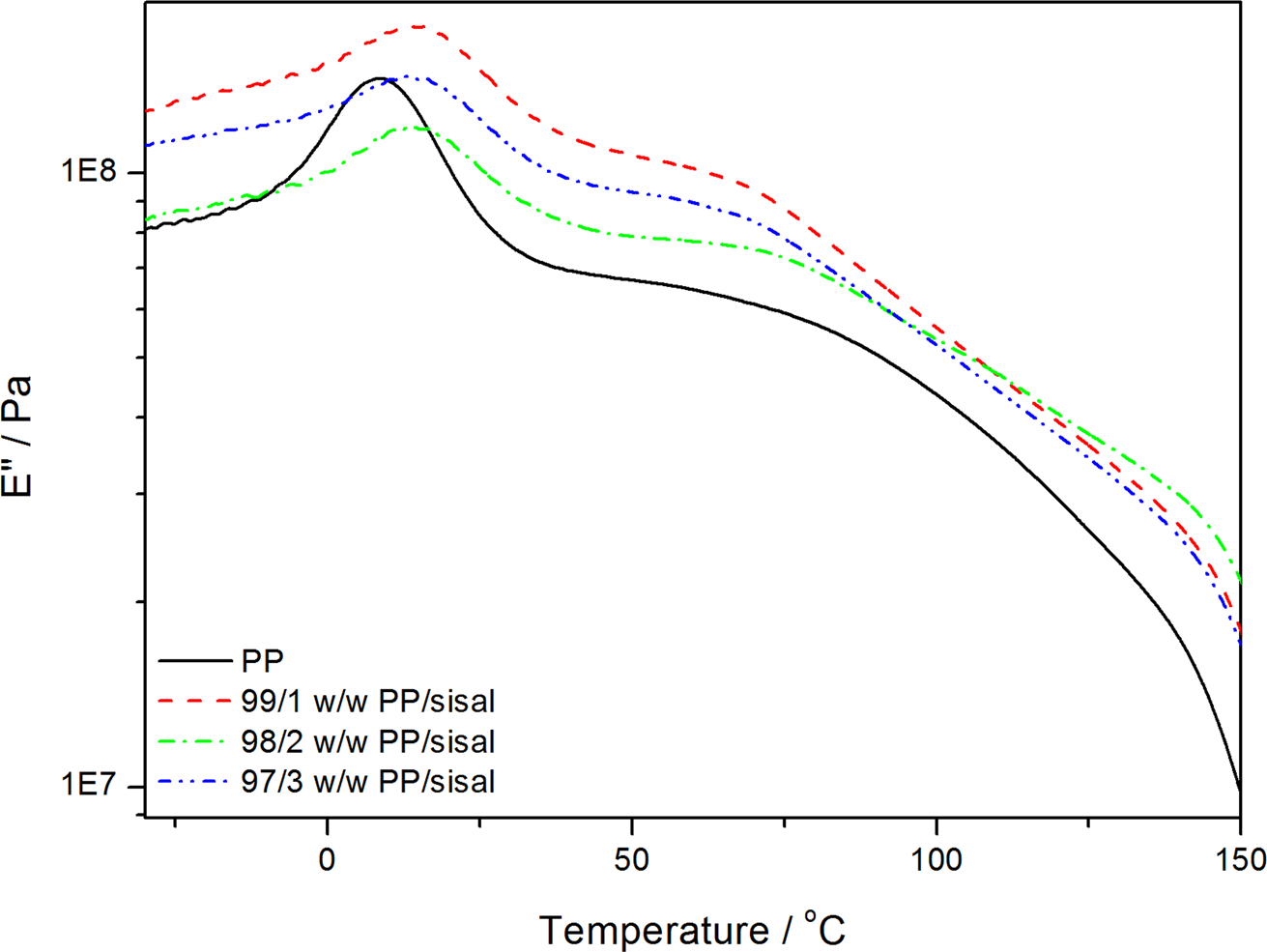
Dynamic mechanical analysis (DMA) loss modulus curves of polypropylene (PP) and the PP/sisal fibre composites.
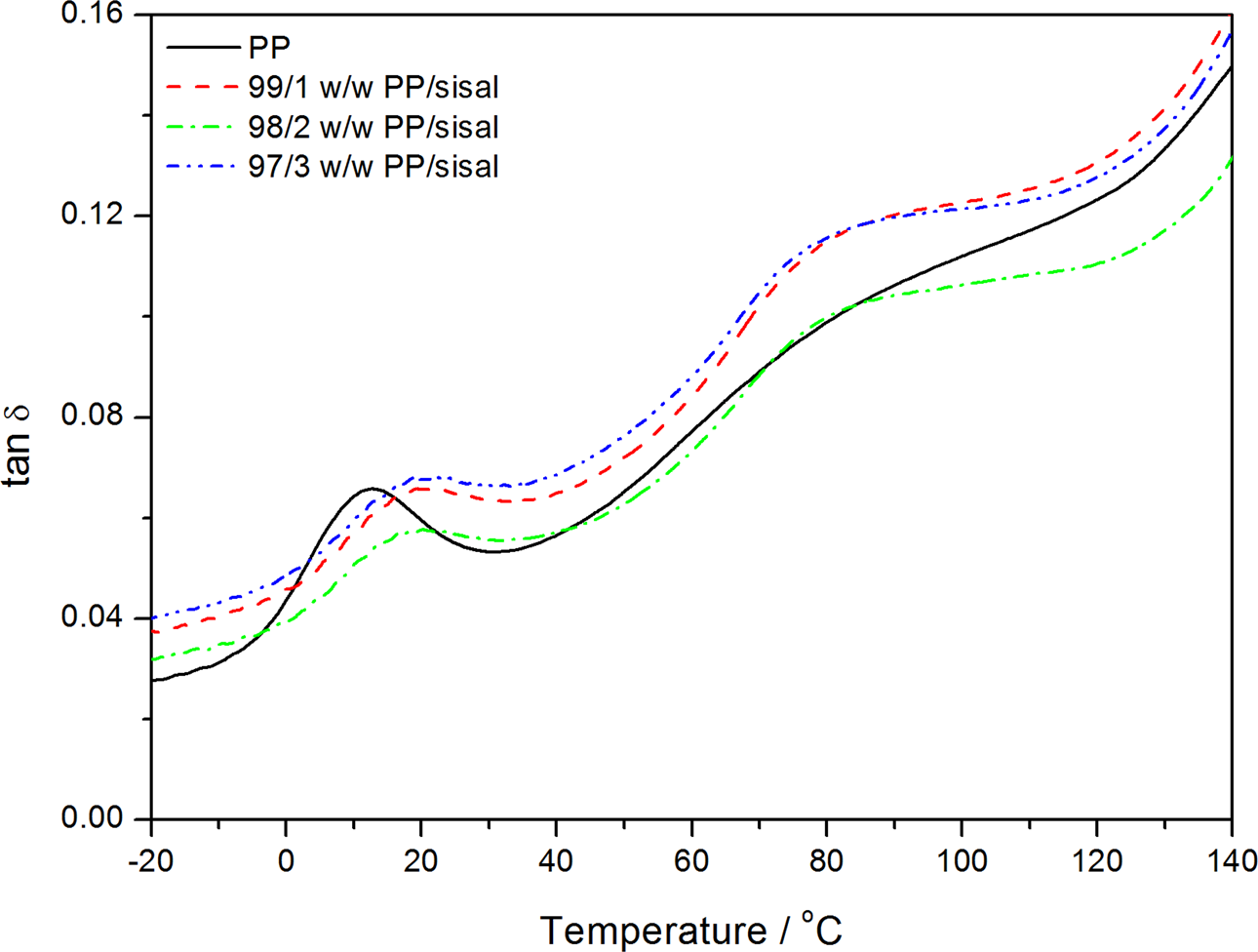
Dynamic mechanical analysis (DMA) damping factor curves of polypropylene (PP) and the PP/sisal fibre composites.
The sharp decrease in the storage modulus (around 59–63°C for most of the samples) corresponds to the α-relaxation of the amorphous regions in PLA. 30 The glass transition temperature (T g) seems to increase with increasing SF content. This shows that the presence of fibre restricts the segmental motion of the polymer chains. The loss modulus curves in Figure 10 confirm this slight increase in the glass transition temperature. The storage and loss moduli started to increase again at temperatures around 100°C, which is the result of the cold crystallization which is typical for PLA, 31 and which was also observed in the DSC curves. The decrease in modulus at temperatures around 140°C indicates the softening of the sample before the onset of melting, which was observed from 140°C in the DSC curves.
A larger area under the α-relaxation peak in the tan δ curves of a polymer indicates that the molecular chains exhibit a higher degree of mobility, thus better damping properties. 32 The area under this peak (at about 67°C) for the PLA composites (Figure 11) seems to be slightly larger for the fibre composites and shows no specific trend with increase in the fibre content. There is very little difference between the peak temperatures of this peak for the PLA and the composite samples, and there is no increasing or decreasing trend with increasing fibre content. It therefore seems as if the fibre did have some influence on the PLA chain mobility, but that the fibre contents were too low to conclusively establish such influence. The broad transition (between 90 and 130°C) for all the samples relates to the cold crystallisation of PLA in this temperature region.
Figure 12 shows the temperature dependence on the E′ for neat PP and its composites. The E′ increased when SF was introduced into PP. The increase in the E′ indicates an increase in the stiffness of the polymer, which may be the result of a restriction in the segmental motion. This is confirmed by the observably higher T g of the fibre-containing samples (Figure 13). The PP and PP/sisal composite curves show two relaxation peaks at about 15 and 70°C (Figure 14). The low-temperature peak is related to the β-transition of the amorphous fractions of PP and is considered the glass transition. The higher temperature peak corresponds to the α-transition related to the PP crystalline fractions. 33 The composite β-transitions are observed at a slightly higher temperature than that of the pure PP, which indicates an immobilization effect of the fibre on the PP chains. The α-relaxation peak is observed for pure PP and all the composite samples, indicating that the fibre was more concentrated in the pure amorphous phase of the polymer, because it had very little influence on the interlamellar chain mobility.
Degradability through hydrolysis
The mass loss results of the PLA samples after being immersed in distilled water at 80°C for 10 days are shown in Figure 15. The mass loss of neat PLA and its composites increased almost linearly with time of immersion. A total mass loss of almost 80% was observed for neat PLA after 10 days. At this point, the neat PLA sample was very soft and it was difficult to decant water and rinse the sample without losing some material, therefore the experiment was discontinued after 10 days. This mass loss was due to the removal of PLA from the surface upon biodegradation. In PLA biodegradation, moisture susceptibility is the primary driving force and involves the following four steps: water absorption, ester cleavage forming oligomers, solubilisation of oligomer fractions, and diffusion of bacteria into soluble oligomers.31,34 The mass loss of the composites followed the same trend and show mass loss values within the same order of magnitude as those of neat PLA. The observed mass loss is due to the removal of the PLA matrix on the surface as a result of biodegradation, 35 together with the removal of some substances from the SF during the immersion. Chow et al. 36 studied the effect of water absorption (at 90°C) on the mechanical properties of SF/PP (SF/PP) composites. They reported that the mass loss of the composites was due to the removal of certain fractions from the SF during the water immersion. They analysed the samples’ morphology before and after immersion using SEM. The results of the tensile broken samples before immersion showed that the fibre was intact in the PP matrix. For the samples immersed in the hot water for 72 h, serious debonding between the fibre and the matrix was observed, but the SF did not show any signs of degradation. After 216 h immersion, the interface between the SF and PP was totally removed, a significant portion of the lignin and hemicellulose had been leached out, and the SF showed microfibrillation. Lee and Wang 37 studied the biodegradation of PLA/bamboo fibre (BF), and they reported that the degradation of PLA was slow but enhanced in the presence of BF. The SEM results indicated that most of the matrix was degraded even though the degradation time was short. Shibata et al. 38 studied the biodegradation of PLA/abaca fibre and reported higher mass loss of the untreated fibre composites compared to the neat polymer. They could not conclude whether the biodegradation of the matrix polymer was promoted by the presence of abaca fibre. However, they did not exclude the possibility that the presence of the highly hydrophilic fibre promoted the biodegradation of the matrix, probably by an action of hydrolytic depolymerisation.
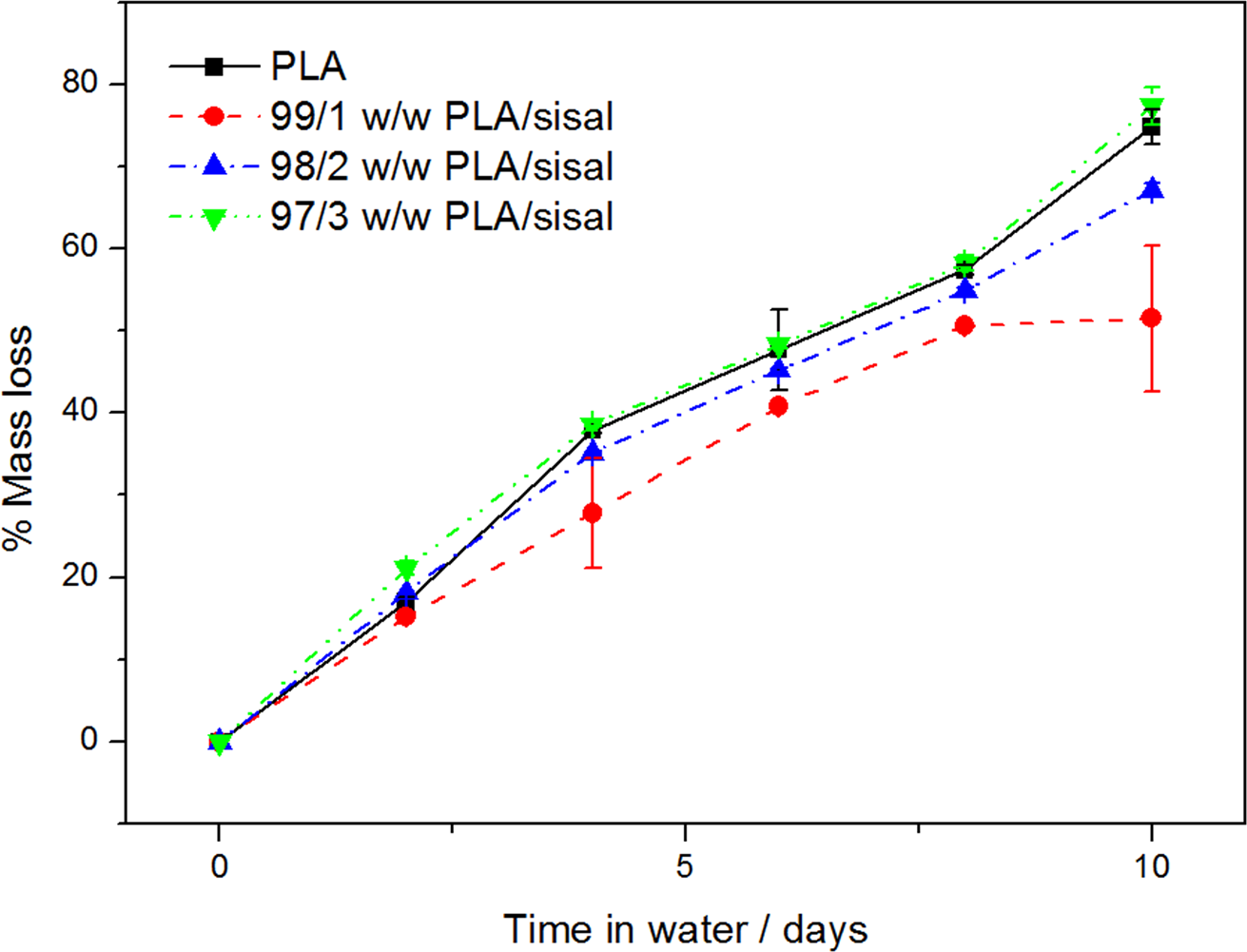
Mass loss as function of time for poly(lactic acid) (PLA) and PLA/sisal fibre composites during immersion in water at 80°C.
No degradation was anticipated for PP during the immersion in water at 80°C. The neat PP showed no mass loss up to 10 days of immersion, and all the PP/SF composites showed less than 1% mass loss after 10 days, which is probably due to limited degradation of the fibre on the sample surface. The fibre in the bulk would have been protected by the surrounding PP. This mass loss of the PP composites was significantly lower than those of the PLA and its composites.
The SEM micrographs of the surface of the biodegraded samples immersed in water at 80°C for 2, 8, and 10 days are displayed in Figures 16 and 17. The neat PLA shows only some small cracks after 2 days, but the cracks become larger when the SF content increases (Figure 16). This shows that the fibre enhanced the biodegradability of the PLA. Figure 17 shows the samples that were immersed for 10 days, and visible lines and pores were formed on the surface of neat PLA after 10 days of immersion (Figure 17a). The composites with fibre content of 2 and 3 wt% (Figure 17b) show the collapsed surface of the samples. This indicates that the fibre itself degrades in the process of immersion in water at 80°C, resulting in further biodegradation of the PLA matrix.

Scanning electron microscopy (SEM) micrographs (×200 magnification) of the biodegraded surfaces of (a) poly(lactic acid) (PLA) and (b) 98/2 (w/w) PLA/sisal after 2 days of immersion.

Scanning electron microscopy (SEM) micrographs (×200 magnification) of the biodegraded surfaces of (a) PLA and (b) 98/2 (w/w) PLA/sisal after 10 days of immersion.
Conclusions
PLA and PP were comparatively investigated as matrices for injection-moulded composites containing low (1–3 wt%) short SF contents. The SEM micrographs of the composites show more intimate contact and better interaction between the fibres and PLA, compared to PP, although surprisingly good adhesion was observed for the PP composites. The improved interaction in the case of the PLA composites was confirmed by the FTIR spectroscopy results that showed the presence of hydrogen bonding interaction between PLA and the fibres. The thermal stability (as determined through TGA) of both polymers increased with increasing fibre content, with a more significant improvement in the case of PP. The DSC results showed a significant influence of the presence of the fibres on the cold crystallization and melting behaviour of PLA, even at the low fibre contents of 1–3%. The influence of the fibres on the melting characteristics of the PP was negligible, but it had a significant influence on the nonisothermal crystallization temperature range. Both the storage and loss moduli of the PLA decreased with increasing fibre content below the glass transition of PLA, but the influence on the loss modulus was more significant. The DMA results clearly show cold crystallization of PLA around 110°C, and the presence of fibres gave rise to higher modulus values between the cold crystallization and melting of the PLA. The damping properties of PLA seem to have increased in the presence of SF, but the fibre did not seem to have a significant influence on the T g of PLA. The presence of fibres increased the T g of PP, but had almost no influence on the α-relaxation, which is related to the crystalline fraction of PP. Although the presence of SFs had little influence on the mass loss of PLA during degradation in water at 80°C, the degradation mechanism seems to have been influenced by the presence of fibres.
Footnotes
Acknowledgements
The National Research Foundation and the University of the Free State in South Africa are acknowledged for their financial support of this project. The work reported in this article was also supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. The authors would further like to thank Arburg Hungaria Ltd., for the injection moulding machine. This work is connected to the scientific program of the ‘Development of quality-oriented and harmonized R+D+I strategy and functional model at BME’ and was supported by the New Hungary Development Plan (Project ID: TÁMOP-4.2.1/B-09/1/KMR-2010-0002).
Funding
This research received funding from agencies as mentioned under ‘Acknowledgements’.