
Abstract
Daily biological rhythms are fundamental to retinal physiology and visual function. They are generated by a local circadian clock composed of a network of cell type/layer-specific, coupled oscillators. Animal models of retinal degeneration have been instrumental in characterizing the anatomical organization of the retinal clock. However, it is still unclear, among the multiple cell-types composing the retina, which ones are essential for proper circadian function. In this study, we used a previously well-characterized mouse model for autosomal dominant retinitis pigmentosa to examine the relationship between rod degeneration and the retinal circadian clock. This model carries the P23H mutation in rhodopsin, which induces mild rod degeneration in heterozygous and rapid loss of photoreceptors in homozygous genotypes. By measuring PER2::LUC bioluminescence rhythms, we show that the retinal clock in P23H/+ heterozygous mice displays circadian rhythms with significantly increased robustness and amplitude. By treating retinal explants with L-α aminoadipic acid, we further provide evidence that this enhanced rhythmicity might involve activation of Müller glial cells.
Twenty-four-hour biological rhythms are a fundamental hallmark of every cellular and physiological process, running in synchrony with the light/dark (LD) cycle (Albrecht, 2012). These rhythms are generated by molecular clocks widely distributed in tissues and orchestrated by the principal pacemaker in the suprachiasmatic nuclei (SCN). The main synchronizing agent to the SCN is environmental light, perceived by classical retinal photoreceptors (PRs: rods, cones) and melanopsin-containing photosensitive retinal ganglion cells. Intracellular circadian clocks generate timing through transcriptional/translational feedback loops involving transcription factors encoded by clock genes (e.g. Bmal1, Clock, Per1-3, Cry1-2). This leads to rhythmic expression of clock factors with a ≈24-h period, driving gene expression programs and cellular functions along with the LD cycle. In the vertebrate retina, many rhythmic processes have been documented. Examples that optimize vision and retinal survival include electroretinogram (ERG) amplitudes, melatonin and dopamine release, and shedding and phagocytosis of PR outer segments (McMahon et al., 2014). Daily retinal rhythms are controlled by a self-sustained circadian clock system distributed across the distinct cellular layers, including PR and pigmented epithelium, and that is entrained by light (Jaeger et al., 2015; Milicevic et al., 2021; Tosini and Menaker, 1996). We recently reported that rod-specific deletion of Bmal1 blunts the difference in dark-adapted ERG amplitudes between subjective day and night, indicating that a circadian clock located in rods regulates visual light processing in a cell autonomous manner (Gegnaw et al., 2021). However, how individual cell types are connected within the retinal clock network and contribute to the overall rhythmic retinal function is not yet understood.
Animal models of retinal degeneration (rd) have been widely used to understand light entrainment mechanisms in SCN and retina (Calligaro et al., 2019; Foster et al., 2020). Indeed, retinal degenerations are genetically and clinically heterogeneous, affecting various cell types. This provides the opportunity to investigate distinct aspects of rod/cone function in the circadian system. Photoreceptor degeneration also triggers substantial changes in the inner retina, in particular in Müller glial cells (MGC) that modify their gene expression programs as a cellular attempt to slow down photoreceptor death and to promote tissue repair. This activation of MGC, or gliotic response, includes metabolic alterations such as increased glial fibrillary acidic protein (GFAP) expression and altered glutamate uptake, and release of neuroprotective factors and antioxydants, among others (Bringmann and Wiedemann, 2012; Fletcher, 2000; Jones et al., 2016; Nagar et al., 2009; Roesch et al., 2012; Tomita et al., 2021). In this study, we questioned the role of rods in the retinal circadian network using a mouse knock-in model of the human P23H rhodopsin mutation, associated with autosomal dominant retinitis pigmentosa (Sakami et al., 2011). We show that mildly degenerating retinal explants carrying the heterozygous P23H mutation display enhanced circadian rhythmicity of PER2 protein expression that might involve glial cell activation.
Materials and Methods
All experimental protocols were carried out according to the European Parliament and The Council of the European Union Directive (2010/63/EU) and institutional ethical guidelines (APAFIS#10213-2017060920001367-v3). Animals were maintained in the Chronobiotron animal facility (UMS 3415, Strasbourg) under 12-h/12-h LD cycle (300 lx and <5 lx dim red light during light and dark, respectively) at a 22 ± 1 °C ambient temperature, with free access to food and water. Mice carrying the P23H mutation knock-in in the rhodopsin gene (Sakami et al., 2011) and the PER2::Luciferase reporter gene (Yoo et al., 2004) (Jackson Laboratories, USA) were bred together to generate following genotypes: P23H/P23H homozygotes (RhoP23H/P23H), P23H/+ heterozygotes (RhoP23H/+) and their wild-type (WT) littermates, all on the C57BL/6J and Per2Luc/+ backgrounds. Animals were genotyped by polymerase chain reaction (PCR) on tail genomic DNA (Yoo et al., 2004); primers for P23H knock-in detection: 5′-GCCTGTTTAGCTGAGAAAAC-3′ and 5′-GACCACGTAACAAACTTCTG-3′. The retinal status in the distinct genotype/age groups was assessed by morphological analysis using hematoxylin/eosin staining (Supplementary Figure S1). The results are in agreement with what was previously described for mice carrying heterozygous and homozygous versions of the P23H mutation at various ages (Sakami et al., 2011).
Whole retina circadian rhythms were assessed by PER2::Luciferase bioluminescence recordings carried out at Postanatal Days 36 (P36) and 70 (P70) as previously described (Jaeger et al., 2015). Oscillations were analyzed using the Lumicycle Analysis software (Actimetrics, USA). Raw data (counts per second, cps) were baseline-subtracted (24-h running average). Periods and amplitudes (height of the three first peaks) were determined by using LMFit (damped sine). The relative rhythmic power was determined using Periodogram function of Lumicycle Analysis to evaluate robustness of rhythms (Buonfiglio et al., 2014; Herzog et al., 2015; Ruan et al., 2012). Analyses were performed on 3 complete cycles after exclusion of the first oscillation. Glial cell involvement was assessed using the metabolic inhibitor of glial cells, L-α aminoadipic acid (L-AAA), on retinal explants from 3-month-old mice (Jablonski and Iannaccone, 2000; Thangaraj et al., 2015). After three complete cycles of control recording (baseline), medium was replaced with a fresh medium containing L-AAA (0.4 mM; Brown and Kretzschmar, 1998) or vehicle (treatment; three complete cycles) and finally the medium was refreshed (washout). L-AAA effects were evaluated by calculating the ratio of first peak amplitudes for treatment or washout versus baseline. The lack of effect of the gliotoxin on cell survival was assessed by TUNEL assay on WT retina explants (Supplementary Figure S4).
To assess central clock rhythms, 1 mm2 SCN-containing explants were dissected from fresh brain coronal sections and cultured as described (Salaberry et al., 2017). Period, amplitude, phase were determined with the Sin Fit (damped) function (Lumicycle Analysis).
General locomotor activity of 8-month-old mice was recorded using infrared motion captors placed above cages, connected to a Circadian Activity Monitoring System (CAMS, INSERM, France), 12 days in LD, then 12 days under constant darkness conditions (DD). Data were analyzed using ClockLab (Actimetrics).
Results are presented as means ± SEM. Statistical analysis was performed as described in figure legends (Sigma Plot 13, Systat Software, USA). When required, ANOVAs were followed by post hoc Holm-Sidak multiple comparisons tests. Significance level was set at p < 0.05.
Results
We characterized the effects of distinct degrees of rod degeneration on the retinal clock activity in WT, P23H/+, and P23 H/P23 H mice sampled at two ages. First, we measured at P36, the time point at which P23H/+ mice display minor visual impairment (Supplementary Figure S2) and retain ≈50% of their photoreceptors (Sakami et al., 2011). Interestingly, retinal explants from all genotypes displayed sustained oscillations in PER2::Luciferase activity (Figure 1a-1c), with similar periods close to the one previously reported in mPer2Luc mice (Jaeger et al., 2015) (WT 22.79 ± 0.07 h, P23H/+ 22.76 ± 0.15 h, P23H/P23H 23.08 ± 0.26 h) (Figure 1d). Unexpectedly, retinas from P23H/+ heterozygous mutants showed the most robust oscillations, as exemplified by their rhythmic power (1.96 ± 0.02) that was significantly higher than in WT (1.75 ± 0.05) (Figure 1e). This result was corroborated by the amplitude values, significantly higher in P23H/+ retinas than in WT (Figure 1f). The P23H/P23H mutants, which lose 90% photoreceptors by P36 (Sakami et al., 2011), showed a relative rhythmic power comparable to WT retinas (1.79 ± 0.05) (Figure 1e).
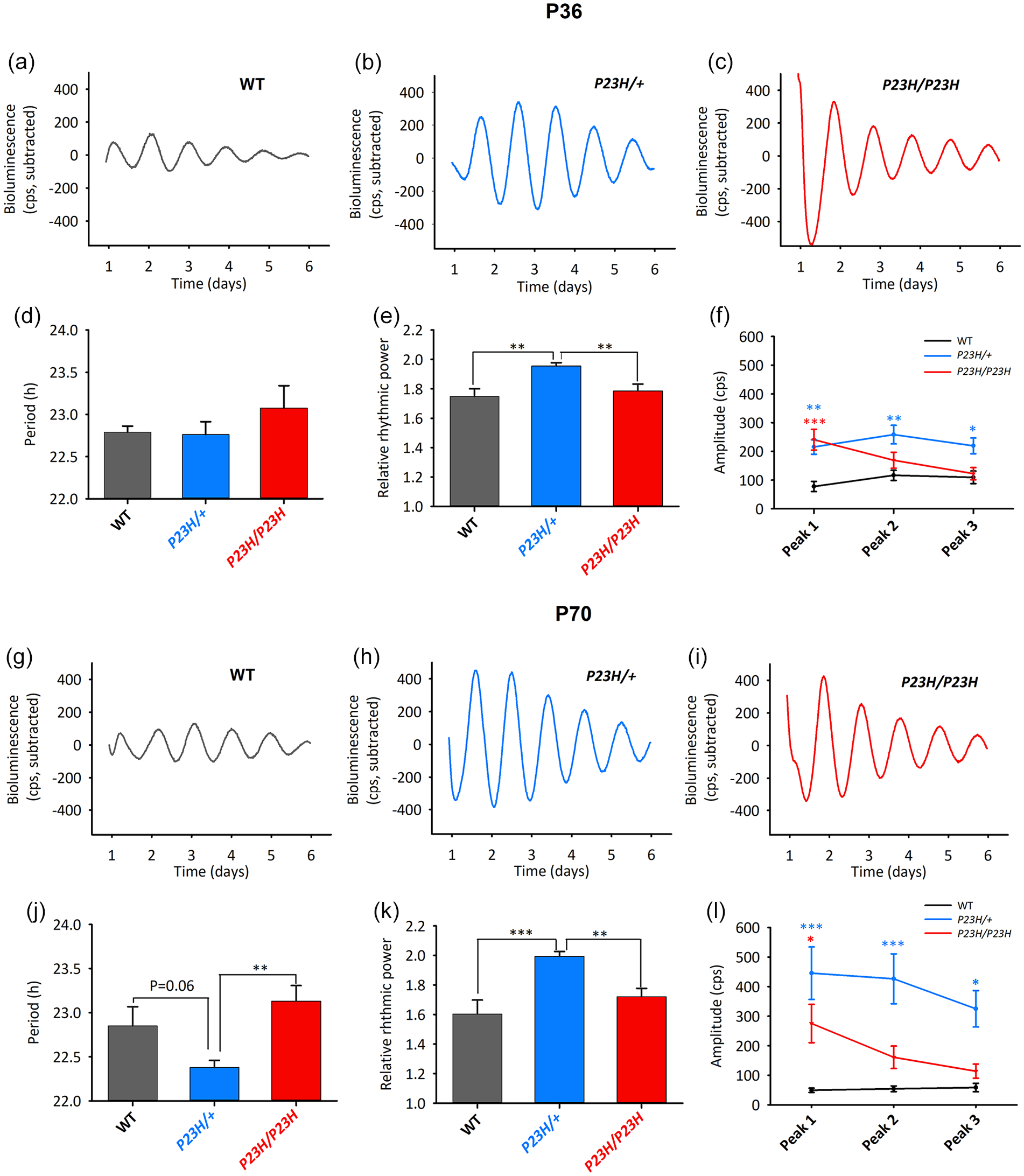
Rod degeneration affects robustness of the retinal circadian clock. PER2::Luciferase activity was analyzed in retinal explants sampled at P36 (a-f) and P70 (g-l). Representative bioluminescence traces from WT (a, g), P23H/+ (b, h), and P23H/P23H (c, i) retinas. At P36 (N = 11-16 per genotype) explants from all groups exhibited similar periods (p = 0.406, one-way ANOVA) (d) but P23H/+ samples showed highest relative rhythmic power (e) (genotype effect, p < 0.001). (f) Genotype effects were also observed for amplitudes of the three first peaks (genotype, p = 0.003; Genotype × Peak, p < 0.001; two-way repeated measures ANOVA). At P70 (N = 13-17 per genotype), there was a genotype effect on the period (p = 0.002, one-way ANOVA) (j), the relative rhythmic power (p < 0.001) (k), and the amplitudes at the distinct peaks (genotype, p = 0.001; Genotype × Peak, p < 0.001) (l). WT = wild-type.
We next analyzed bioluminescence activity in P70 retinas (Figure 1g-1i). Compared to P36, visual ERG response at P70 is further impaired in P23H/+ mutants (Supplementary Figure S2). Nonetheless, at this age the P23H/+ retinas displayed circadian oscillations similar to those at P36, yielding the largest robustness. This was shown by relative rhythmic power values (1.99 ± 0.03) which were significantly higher than in WT (1.71 ± 0.06) and homozygotes (1.79 ± 0.03) (Figure 1k) and by amplitudes being significantly larger in heterozygotes than in WT retinas (Figure 1l).
At both P36 and P70, amplitudes of the P23H/P23H samples were decreasing from high at peak 1 (similar to P23H/+) to low (similar to WT) at Peaks 2 and 3 (Figure 1f and 1l). Uniquely at P70, the heterozygous mutant retinas also displayed a trend for shortened period (22.38 ± 0.08 h) with respect to WT (22.65 ± 0.19 h) and were significantly shorter with respect to homozygous retinas (23.10 ± 0.15 h) (Figure 1j). Interestingly, retinas carrying another type of severe rod degeneration induced by the rd10 recessive mutation of the rod phosphodiesterase beta subunit (Chang et al., 2002; Gargini et al., 2007), display similar circadian activity as the P23H/P23H retinas; for example, increased amplitudes but similar periods and rhythmic power with respect to their controls (Supplementary Figure S3). Taken together, the above results suggest that the retinal circadian clock becomes more robust upon ongoing rod degeneration.
We hypothesized that activation of Müller glial cells induced by ongoing PR degeneration, might be involved in the enhanced oscillating capacity of the P23H/+ mutant. We treated retinal explants after 3 days of baseline oscillations with the L-AAA (0.4 mM) gliotoxin. During the 3 days of drug-exposure oscillations of WT and P23H/P23H explants displayed dramatic amplitude reduction and, accordingly, period could not be determined (Figure 2a, 2c, 2d, and 2f). In contrast, rhythms of P23H/+ retinas were barely affected by the drug, suggesting that activation of MGC in the degenerating retina, might compensate for the inhibitory effect of L-AAA (Figure 2b, 2d, and 2f). Likewise, P23H/+ retinas mostly behaved like vehicle controls upon drug washout, whereas WT and P23H/P23H samples retained reduced amplitudes and showed altered periods (Figure 2e and 2 g).
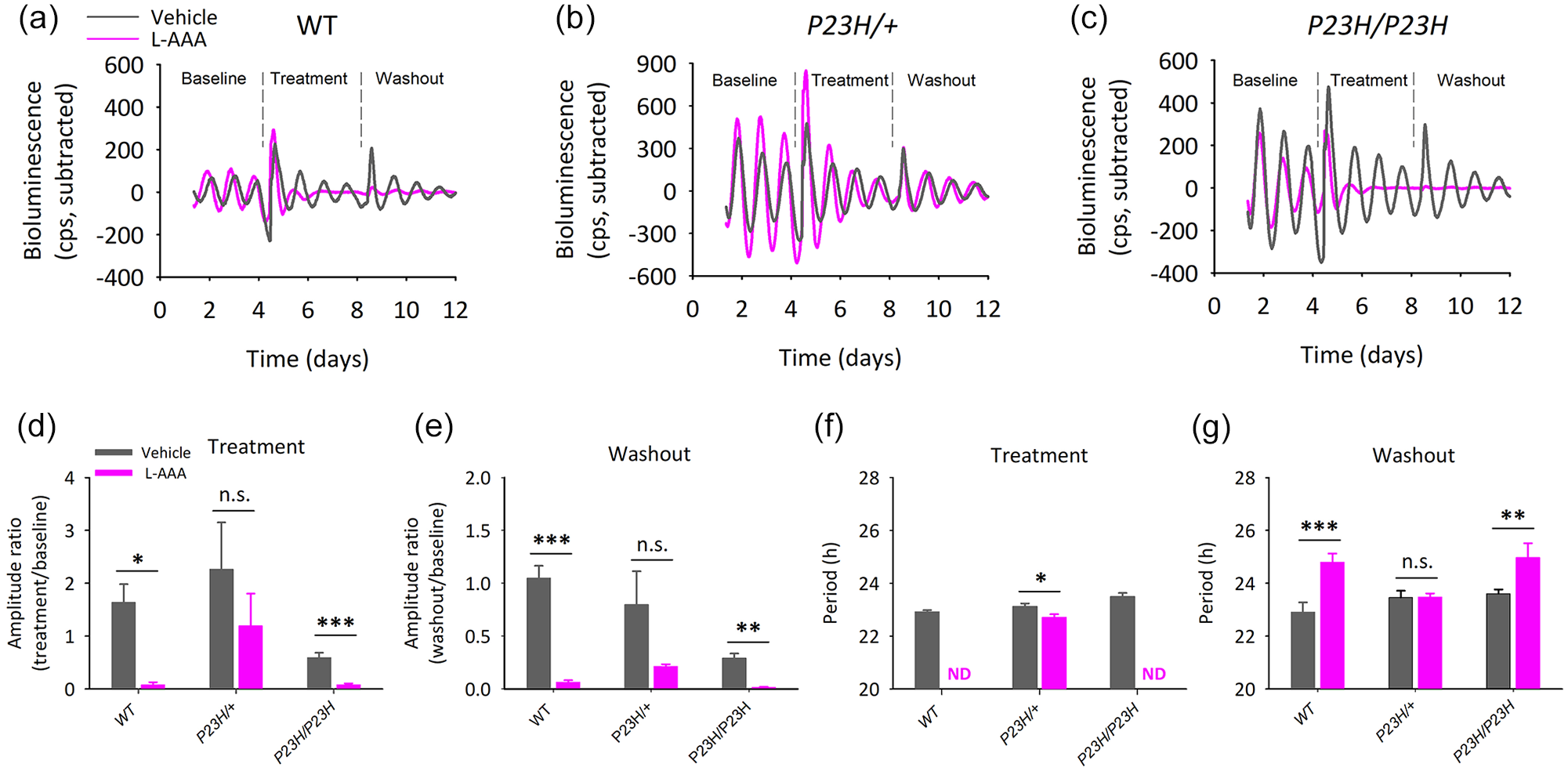
PER2::Luciferase oscillations are not affected by L-AAA treatment in P23H/+ retinas. (a-c) Representative bioluminescence profiles in retinal explants from 3-month-old WT (a), P23H/+ (b) and P23H/P23H (c) mice upon baseline, treatment with 0.4 mM L-AAA and washout (N = 5-6 per genotype and treatment group). (d) L-AAA strongly affected oscillation amplitudes (effect of treatment, p = 0.019; genotype, p = 0.026, two-way ANOVA). It induced marked amplitude reduction in WT and P23H/P23H retinas but had no significant effect in P23H/+ samples (p = 0.362) (Student t test). (e) Differential effects of L-AAA on the three genotypes persisted upon washout (effect of treatment, p < 0.001; genotype, p = 0.015, two-way ANOVA and Student t tests). (f) Oscillations were abolished in WT and P23H/P23H samples during L-AAA treatment, but were retained in P23H/+ retinas with minor period alteration (p = 0.041, Student t test). (g) Upon washout PER2 oscillations were measurable in retinas from all genotypes but were differentially affected by previous L-AAA treatment (treatment effect, p < 0.001; Treatment × Genotype, p = 0.029, two-way ANOVA). n.s. = not significant; ND = not determined; WT = wild-type; ANOVA = Analysis of variance; L-AAA = L-α aminoadipic acid.
We finally evaluated whether a retina with increased clock robustness would affect the SCN clock functionality. We first analyzed locomotor activity rhythms in WT and P23H/+ mice (Figure 3a and 3b). Animals from both genotypes were synchronized to the LD cycle, and showed similar periods of circadian locomotor activity under DD conditions (WT 23.92 ± 0.00 h; P23H/+ 23.95 ± 0.02 h). Likewise, SCN explants did not show any significant difference in period, amplitude, or phase of PER2::Luciferase oscillations (Figure 3c-3f). Thus, increased robustness of the retinal clock does not influence the central clock and the downstream rest/activity cycle.
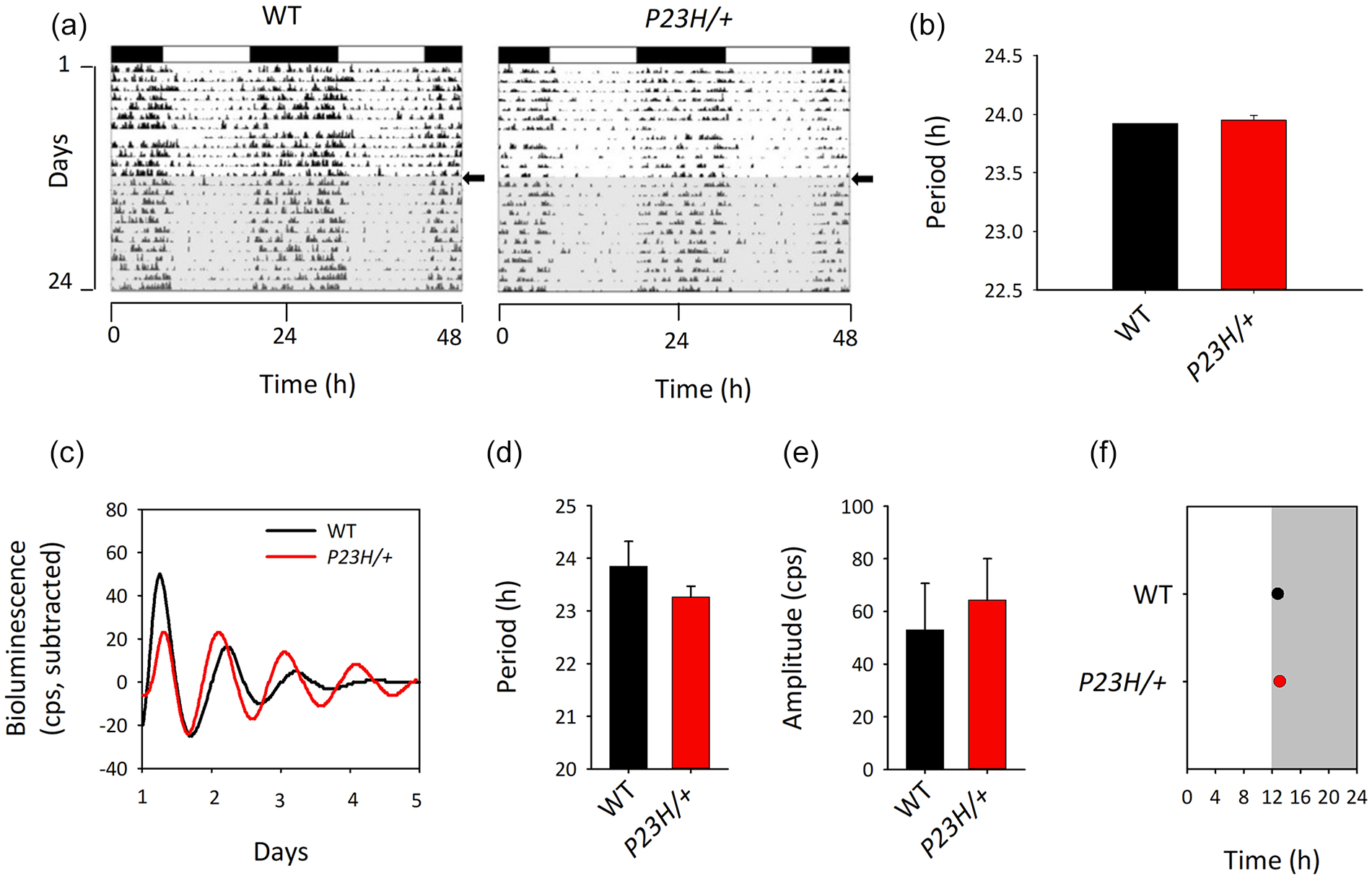
The P23H/+ mutation does not induce alterations of the central clock. (a) Representative actograms of P23H/+ and WT littermates in LD (white/black bars:light/dark cycle) and DD (gray background) showing similar light-entrained behavior and endogenous rhythmic activity, as confirmed by periods in DD (p = 0.730, N = 4-5) (b). (c-f) SCN sampled at the end of the behavioral experiment showed no significant difference in bioluminescence profile (c), period (d), amplitude (e) and phase (f) (N = 3-6). WT = wild-type; DD = constant darkness; SCN = suprachiasmatic nuclei; LD = light/dark.
Discussion
We recently reported evidence indicating that rods harbor a circadian clockwork that regulates daily visual light processing in a cell autonomous manner, since rod-specific Bmal1 disruption abolishes the circadian day–night differences in dark-adapted ERG a- and b-wave amplitudes (Gegnaw et al., 2021). By contrast, absence of clock in rods did not exert major effect on the global retinal clock, as assessed by PER2::Luciferase bioluminescence in explants. To get further insight into the contribution of rods to the clock network, we here addressed the potential relationship between degeneration of these cells and retinal circadian clock activity. We provide evidence that mice with a heterozygous P23H rhodopsin mutation, that induces slow rod degeneration, display increased robustness and amplitude of the retinal clock at both P36 and P70. Previous histological analyses in this model showed that degeneration at these ages is progressing but not complete (Sakami et al., 2011), as currently confirmed by our histological analysis (Supplementary Figure S1) and ERG recordings (Supplementary Figure S2). It was reported that PR affect retinal clock activity, but the underlying mechanisms remain unknown. In another animal model, the RCS/N-rdy rats displaying extensive PR degeneration, clock gene expression measured in whole retinas was perturbed but no major change was observed for Per2 mRNA rhythms (Tosini et al., 2007b). In yet another study, retinal explants from rd1 homozygous mice displayed sustained rhythms in PER2::Luciferase activity, whereas their heterozygous littermates (with normal retinas) were reported not to show retinal rhythms (Ruan et al., 2006). Our results provide a more complex picture of PR contribution to the retinal clock network, pointing at a large, likely transient, enhancing effect when their degeneration is ongoing. The enhanced robustness of the P23H/+ retinal clock (higher rhythmic power than in the other genotypes) might be due to a stimulatory effect induced by dying rods on gene expression in inner retinal cells, at this early stage of retinal remodeling (likely Phase 1 of retinal degeneration) (Pfeiffer et al., 2020). The moderate phenotype of the homozygous mutant retinas indicates that this enhancing effect might fade once most PR are gone (likely Phase 2).
It is indeed well-known that remodeling of the retinal (clock) network occurs upon PR death (Marc et al., 2003), which might also affect rhythms in the remaining cells. MGC are first-line reactive cells following PR degeneration, with notably gliosis and early phagocytosis of dying cells (Bringmann and Wiedemann, 2012; Sakami et al., 2019). Acute PR degeneration in zebrafish triggers MGC to regenerate neurons (Hamon et al., 2016). This has been associated with induction of a specific gene expression program supporting the regenerative capacity, that includes activation of clock genes (Sifuentes et al., 2016). Basing on these results, we here questioned whether the enhancement of retinal PER2 oscillations in the P23H/+ mutant, at least in part, might be due to activation of MGC and of their clock. We reasoned that MGC stimulation might counterbalance the deleterious effects of the L-AAA gliotoxin. Indeed, L-AAA treatment drastically reduced oscillating capacity in WT retinas without inducing their death, but did not affect rhythms in P23H/+ retinas. The effect of L-AAA on rhythms in WT is in agreement with the fact that MGC contain a circadian clock and supports the idea that this clock contributes to the overall oscillating capacity of the retina (Xu et al., 2016). By contrast, retinas from P23H/+ mice appear resilient to 0.4mM L-AAA treatment. This corroborates our hypothesis that early rod degeneration induces early stage retinal remodeling with glial cell activation that can counterbalance the deleterious effects of the drug. Strikingly, retinas from homozygous P23H/P23H mice were affected by L-AAA treatment similar to WT. One explanation for this finding is that in P23H/P23H retinas, that have lost almost all photoreceptors at 3 months, the early glial cell activation is over and the drug can further exert its deleterious effect on glial cells and the global retinal clock. A limitation of our study is that, besides MGC stimulation, rod degeneration also activates more global phenotypic changes in the retina, in particular inflammatory/immune response and activation of microglial cells (Olivares-Gonzalez et al., 2021) and rewiring inside the retinal network (Marc et al., 2003). Thus, we cannot exclude that enhanced rhythmicity in P23H/+ retinas also involves activation of other clock-carrying retinal cell type(s).
We also assessed whether this enhanced rhythmicity of P23H/+ retinas affects the circadian system more generally but we did not find any difference with controls, neither in locomotor activity rhythms nor in PER2 oscillations in the SCN. Previously, Storch and coworkers reported that behavioral rhythms are normal in mice with retina-specific Bmal1 deletion (Storch et al., 2007). Thus, even if cellular content of the retina can profoundly impact the SCN (Lupi et al., 1999; Tosini et al., 2007a), its circadian clock itself does not appear to play an overt role. In agreement with this result, our preliminary data indicate minor effect of the P23H/+ mutation on melanopsin expression (data not shown), which confirms previous conclusion from investigation of rodless/coneless mice (Semo et al., 2003).
In conclusion, retinal clock rhythmicity is enhanced when rods are degenerating, which likely involves activation of glial cells and suggests that rods contribute minorly to the robustness of the retinal clock network. These results provide new insights into the cellular/molecular events that are turned on in mice, when PR degenerate.
Supplemental Material
sj-pdf-1-jbr-10.1177_07487304221112845 – Supplemental material for Enhanced Robustness of the Mouse Retinal Circadian Clock Upon Inherited Retina Degeneration
Supplemental material, sj-pdf-1-jbr-10.1177_07487304221112845 for Enhanced Robustness of the Mouse Retinal Circadian Clock Upon Inherited Retina Degeneration by Shumet T. Gegnaw, Cristina Sandu, Nadia Mazzaro, Jorge Mendoza, Arthur A. Bergen and Marie-Paule Felder-Schmittbuhl in Journal of Biological Rhythms
Footnotes
Acknowledgements
The authors thank Dr. Dominique Sage, Dr. Sophie Reibel, and Nicolas Lethenet for animal care and Dr. Jérôme Roger for rd10 mice. This project was funded with support from the NeuroTime Erasmus + Program of the European Commission and the Center National pour la Recherche Scientifique.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Note
Supplementary material is available for this article online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.