
Abstract
NEUROLIGIN-1 (NLGN1) is a postsynaptic adhesion molecule involved in the regulation of glutamatergic transmission. It has been associated with several features of sleep and psychiatric disorders. Our previous work suggested that transcription of the Nlgn1 gene could be regulated by the transcription factors CLOCK and BMAL1 because they bind to the Nlgn1 gene promoter in vivo. However, whether CLOCK/BMAL1 can directly activate Nlgn1 transcription is not yet known. We thus aimed to verify whether CLOCK/BMAL1, as well as their homologs NPAS2 and BMAL2, can activate transcription via the Nlgn1 promoter by using luciferase assays in COS-7 cells. We also investigated how Nlgn1 expression was affected in Clock mutant mice. Our results show transcriptional activation in vitro mediated by CLOCK/BMAL1 and by combinations with their homologs NPAS2 and BMAL2. Moreover, CLOCK/BMAL1 activation via the Nlgn1 gene fragment was repressed by GSK3β. In vivo, Nlgn1 mRNA expression was significantly modified in the forebrain of Clock mutant mice in a transcript variant–dependent manner. However, no significant change in NLGN1 protein level was observed in Clock mutant mice. These findings will increase knowledge about the transcriptional regulation of Nlgn1 and the relationship between circadian rhythms, mental health, and sleep.
Keywords
Neuroligins (NLGNs) are postsynaptic adhesion molecules interacting with presynaptic Neurexins (NRXNs) to mediate cell adhesion (Nguyen and Südhof, 1997; Song et al., 1999). Neuroligin-1 (NLGN1) is a member of this system that is mostly localized at excitatory glutamatergic synapses, where it determines the number of functional N-methyl-
The literature suggests regulation of Nlgn1 expression by epigenetic mechanisms. For instance, the gene expression of Nlgn1 is altered in the brain of mice lacking methyl-CpG-binding protein 2 (MeCP2), a transcriptional regulator binding to methylated genomic sequences (Runkel et al., 2013). In addition, gene repression via DNA methylation has been reported for Nlgn1 following treatment with amyloid beta fibrils (Bie et al., 2014). In parallel, we have shown that the expression of a specific transcript variant of Nlgn1 varies with time of day in the mouse forebrain (El Helou et al., 2013), which could suggest transcriptional regulation by clock proteins.
Among clock proteins, CLOCK (circadian locomotor output cycles kaput) and BMAL1 (brain and muscle Arnt-like protein-1; also known as aryl hydrocarbon receptor nuclear translocator-like protein 1 [Arntl]) are core elements of the transcriptional-translational feedback loop that governs circadian rhythms in mammals (Ko and Takahashi, 2006). CLOCK and BMAL1 (or their homologs NPAS2 and BMAL2, respectively) dimerize and generally bind to a gene sequence of 6 nucleotides called an E-box to activate transcription (Gekakis et al., 1998; Hogenesh et al., 1998; Dardente et al., 2007). We have reported the presence of several E-boxes in the mouse Nlgn1 gene, some of these being conserved across different species (El Helou et al., 2013). Furthermore, we found that CLOCK and BMAL1 bind rhythmically to a sequence of the Nlgn1 gene that contains a consensus (i.e., CACGTG) E-box (El Helou et al., 2013). These observations suggest a transcriptional regulation of Nlgn1 by core clock transcription factors.
Posttranslational modifications, and in particular phosphorylation, are of particular relevance to the functioning of the molecular circadian clock by, notably, shaping trafficking in and out of the nucleus as well as protein degradation (Duguay and Cermakian, 2009). Glycogen synthase kinase 3 beta (GSK3β) is a multifunctional serine/threonine kinase, which has been reported to phosphorylate clock components such as PER2 (Period 2), CRY2 (Cryptochrome 2), and REV-ERBα in mammals, leading to different effects, including protein degradation and stabilization (Harada et al., 2005; Yin et al., 2006). GSK3β also phosphorylates serine 17 and threonine 21 of BMAL1, which leads to its degradation (Sahar et al., 2010), and genetic downregulation of GSK3β modifies the length of the endogenous circadian period of locomotor activity in mice (Lavoie et al., 2013). This supposes a modulatory role of GSK3β on the activity of the CLOCK/BMAL1 heterodimer and thus on the regulation of its demonstrated and putative target genes, such as Nlgn1.
In this study, in vitro luciferase assays were performed to investigate the transcriptional activation of Nlgn1 by CLOCK, BMAL1, and their homologs, as well as the modulatory role of GSK3β. Luciferase assays were also performed on a fragment of the Nlgn2 promoter to test whether this regulation by CLOCK/BMAL1 is specific for Nlgn1 within the NLGN/NRXN system. In addition, to understand which nucleotides of the Nlgn1 gene are specifically involved in transcriptional activation by CLOCK/BMAL1, site-directed mutagenesis was used. Finally, in vivo support for transcriptional regulation of Nlgn1 by clock transcription factors was investigated using mRNA and protein quantifications in ClockΔ19/Δ19 mutant mice. Our results support a contribution of core clock factors to the regulation of Nlgn1 gene expression.
Materials and Methods
Molecular Cloning
A 969 bp DNA fragment, including 1 canonical and 3 noncanonical E-boxes, corresponding to the region (-1159/ -190) of the mouse Nlgn1 promoter (Figure 1A) was PCR-amplified from mouse ear genomic DNA (50 ng of genomic DNA in 25 µL of PCR mixture). E-box mutations were generated by overlapping PCR mutagenesis as previously described (Guillaumond et al., 2005; Mongrain et al., 2008) using the 969 bp Nlgn1 promoter cloned into pGL3-Basic plasmid (see hereafter) as a template. The first and second rounds of PCR were carried out with a primer pair having the mutation in the Reverse primer and the Forward primer, respectively. A 1167 bp DNA fragment, including 5 noncanonical E-boxes, corresponding to the region (-1387/ -220) of the mouse Nlgn2 promoter (Figure 2A) was PCR-amplified from mouse ear genomic DNA (50 ng of genomic DNA in 25 µL of PCR mixture). Oligonucleotide sequences used in PCR reactions are listed in Table 1. Wild-type (WT) and mutant Nlgn1 promoter sequences (Figure 1A) were cloned between XhoI and HindIII sites in a pGL3-Basic vector (Promega Corporation, Madison, WI) that encodes luciferase protein and ampicillin resistance gene. Nlgn1 and Nlgn2 (Figure 2A) WT promoter sequences were cloned, respectively, between XhoI and HindIII sites and between SmaI and XhoI sites in a pGL2-Basic vector (Promega Corporation) that encodes luciferase protein and ampicillin resistance gene. Plasmids were sequenced at McGill University and Genome Quebec Innovation Centre (Montreal, Canada).
Primer and probe sequences used for PCR and qPCR.
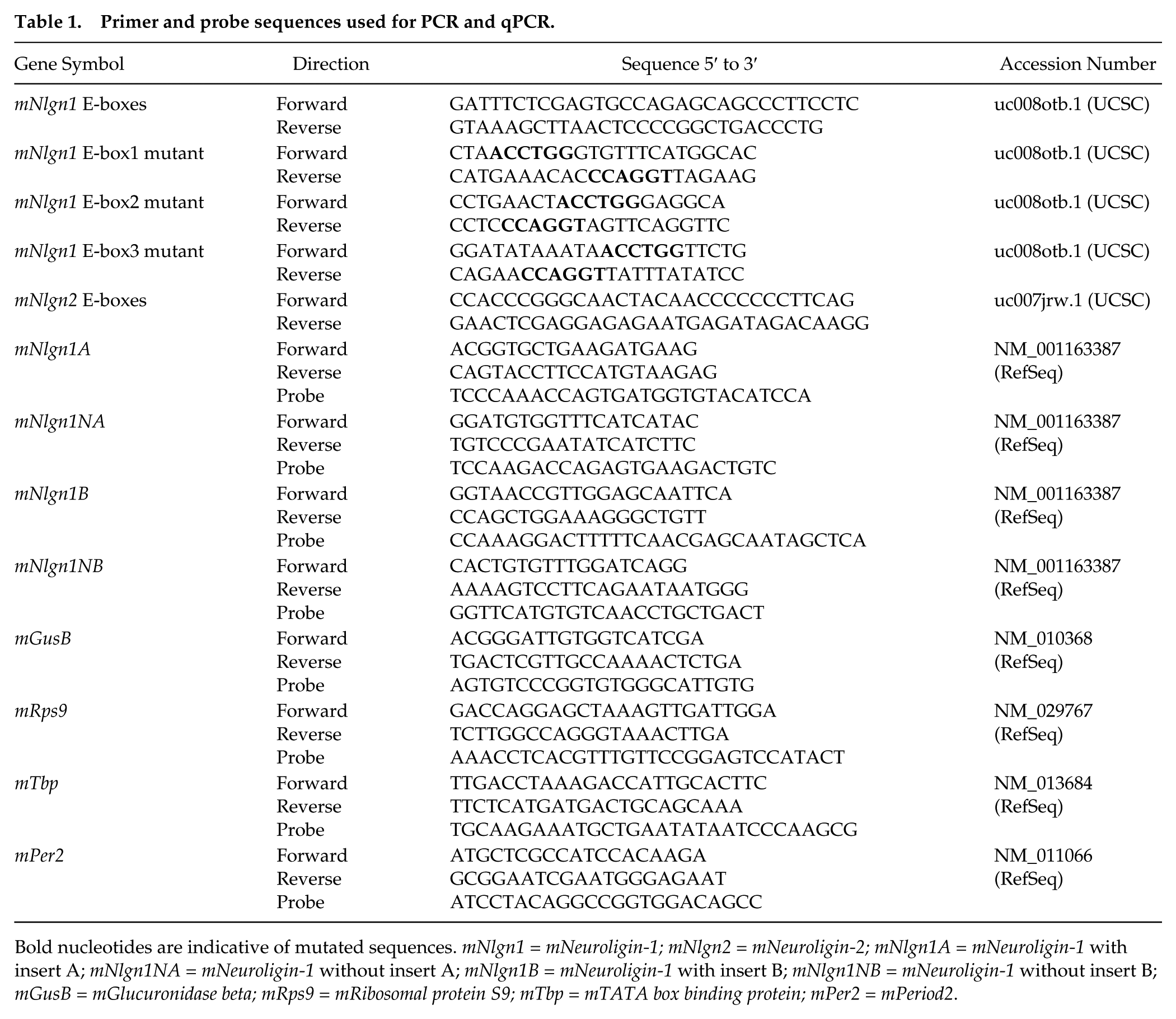
Bold nucleotides are indicative of mutated sequences. mNlgn1 = mNeuroligin-1; mNlgn2 = mNeuroligin-2; mNlgn1A = mNeuroligin-1 with insert A; mNlgn1NA = mNeuroligin-1 without insert A; mNlgn1B = mNeuroligin-1 with insert B; mNlgn1NB = mNeuroligin-1 without insert B; mGusB = mGlucuronidase beta; mRps9 = mRibosomal protein S9; mTbp = mTATA box binding protein; mPer2 = mPeriod2.
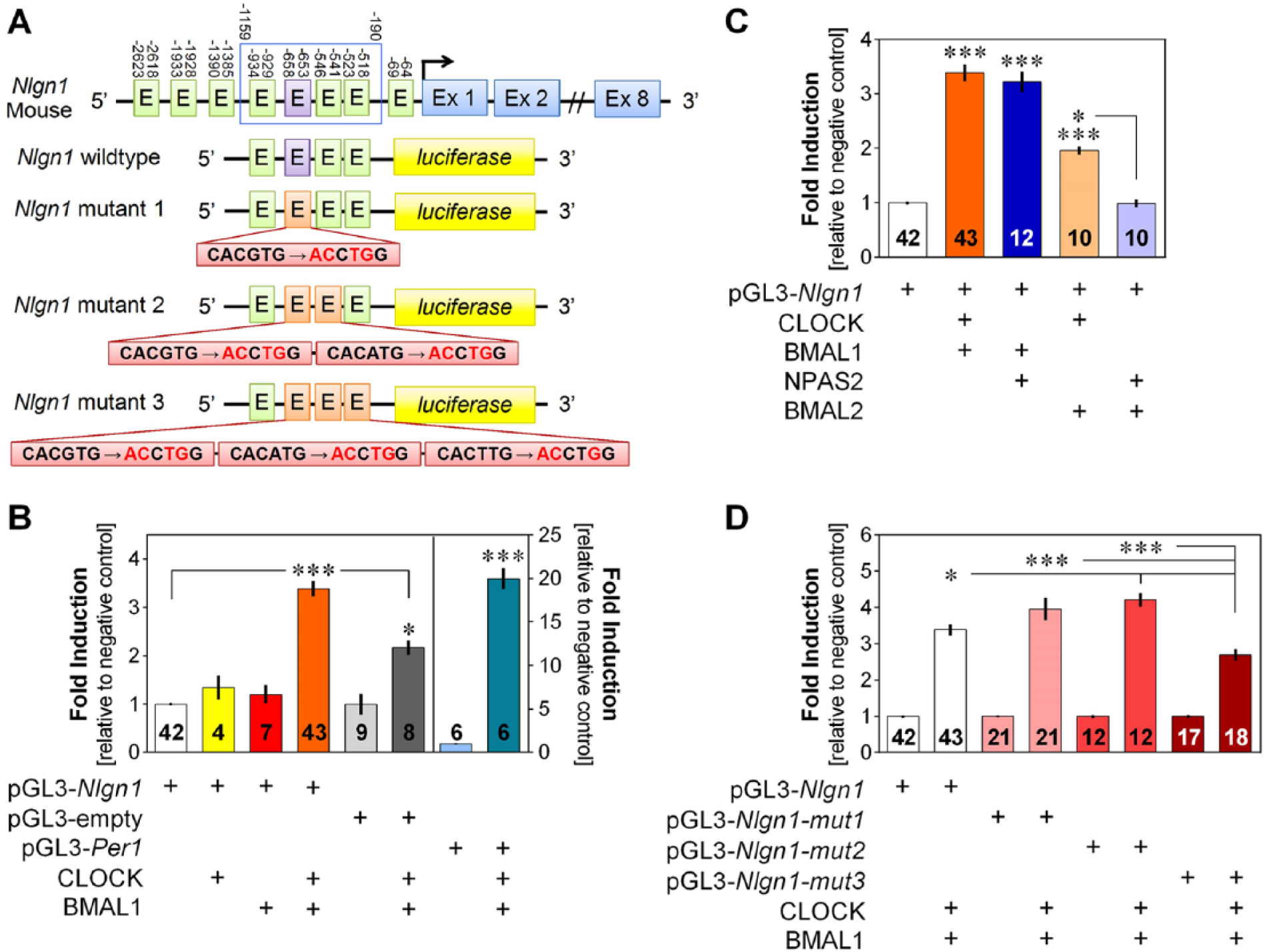
Transcriptional activation of Nlgn1 and Per1 by core clock components. (A) Scheme of the region of the Nlgn1 gene selected for luciferase assay (-1159 to -190 bp before transcription start site indicated by an arrow) containing 4 E-boxes including 1 canonical E-box CACGTG (purple) and 3 noncanonical E-boxes CANNTG (green, where N corresponds to A, T, C, or G), as well as mutations of 4 nucleotides in 3 E-boxes. (B) Transcriptional activation by the heterodimer CLOCK/BMAL1 via the Nlgn1 and Per1 gene fragments, and the empty pGL3 vector. The left and right y-axes show the fold induction relative to the negative control without CLOCK and BMAL1 for Nlgn1 (and empty vector) and Per1, respectively. *p < 0.05 and ***p < 0.001 between indicated points or compared with the negative control (same in C and D). Numbers on bars represent n values (same for all figures). (C) Transcriptional activation by CLOCK, BMAL1, and their homologs NPAS2 and BMAL2 via the Nlgn1 gene fragment. Fold inductions are relative to the negative control without CLOCK and BMAL1 (same in D). CLOCK/BMAL1 and NPAS2/BMAL1 conditions significantly differ from the 3 other conditions (p < 0.001). (D) Fold induction of the transcriptional activation of Nlgn1 with and without mutated E-boxes (mut1 refers to mutant 1 depicted in panel A, mut2 to mutant 2, and mut3 to mutant 3). All CLOCK/BMAL1 conditions significantly differ from their respective negative control (p < 0.001).
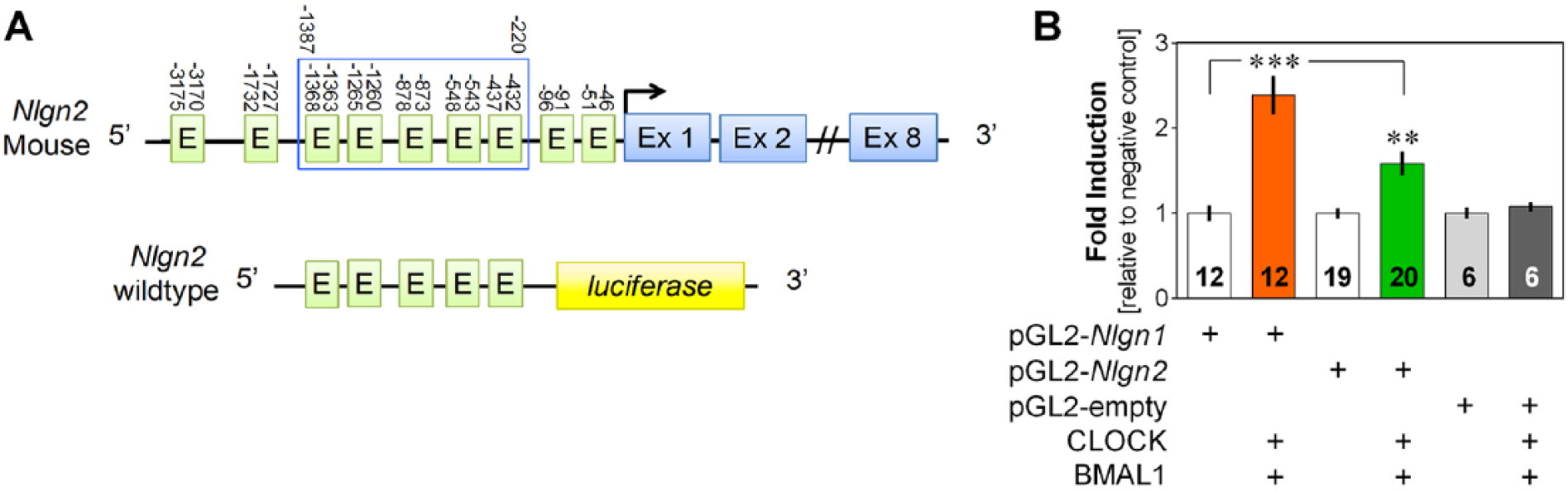
Transcriptional activation of Nlgn1 and Nlgn2 by core clock components. (A) Scheme of the region of the Nlgn2 gene cloned for luciferase assay (-1387 to -220 bp before transcription start site indicated by an arrow) containing five non-canonical E-boxes labeled E (CANNTG, where N corresponds to A, T, C, or G). (B) Transcriptional activation by the heterodimer CLOCK/BMAL1 via the Nlgn1 and Nlgn2 gene fragments, and the empty pGL2 vector. The left y-axis shows the fold induction relative to the negative control without CLOCK and BMAL1. **p < 0.01 compared to the negative control; ***p < 0.001 between indicated points.
Cell Culture and Transfection
COS-7 cells were grown in Dulbecco’s modified Eagle medium (DMEM; HyClone, Logan, UT) supplemented with 10% fetal bovine serum and 1% L-glutamine (Thermo Fisher Scientific, Waltham, MA) in a humidified atmosphere containing 5% CO2 at 37 °C. COS-7 cells were transfected in 24-well plates (seeded at 1 × 105 cells/well) using 2 µL of Lipofectamine LTX (Invitrogen, Carlsbad, CA) with a total of 700 ng of expression plasmids. The plasmid mix included 50 ng of luciferase reporter plasmid (pGL3-Basic or pGL2-Basic) containing an insert or not (insert: nonmutated or mutated Nlgn1 or Nlgn2 sequence), 200 ng of pSG5-CLOCK (Travnickova-Bendova et al., 2002) or pSG5-NPAS2 (Dardente et al., 2007) or pSG5, 200 ng of pCS2-MTK-BMAL1 (Travnickova-Bendova et al., 2002) or pCS2-MTK-BMAL2 (Dardente et al., 2007) or pCS2-MTK, 25 ng of pCR3-LacZ (Invitrogen), and 225 ng of pBluescript (Stratagene, San Diego, CA) as a carrier. As a positive control, the assay was also performed in parallel with 25 ng of a pGL3-Basic plasmid containing an ~1.8-kb portion of the Per1 promoter (Travnickova-Bendova et al., 2002).
For the experiments aimed to examine whether GSK3β can inhibit the CLOCK/BMAL1-driven activation of the Nlgn1 selected region, 50 ng of pcDNA3-HA-GSK3β WT (Addgene, Cambridge, MA; Jim Woodgett, Mont Sinai Hospital, Toronto, ON; He et al., 1995) or pcDNA3-GSK3β-S9A (Stambolic and Woodgett, 1994) or pcDNA3.1(+) (Part No. V790-20, Invitrogen) was added to the plasmid mix described above. To maintain a total of 700 ng plasmid mix, 175 ng of pBluescript vector was included as a carrier. Five to 6 hours following transfection, 500 µL of COS-7 medium was added to each well, and cells were grown overnight at 37 °C. For experiments aimed to examine whether 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD-8; Sigma-Aldrich, St. Louis, MO) can counteract the inhibitory effect of GSK3β on the selected Nlgn1 region, 50 and 100 µM of TDZD-8 was added to conditions of interest described above and incubated for 1 h at 37 °C before luciferase assay.
Luciferase Assay
Luciferase assays were performed as described previously (Dardente et al., 2007; Mongrain et al., 2008). Cells were rinsed with PBS 1× and lysed by shaking 10 min in 150 µL of lysis buffer (25 mM Tris, 2 mM EDTA, 1 mM DTT [dithiothreitol], 10% [v/v] glycerol, 1% Triton X-100). Lysate (12 µL) was transferred in a 96-well luminometer microplate, and 50 µL of luciferase buffer (20 mM Tris/phosphate pH 7.8, 1 mM MgCl2, 2.7 mM MgSO4, 0.1 mM EDTA, 33.3 mM DTT, 530 µM ATP, 270 µM Coenzyme-A, 470 µM D-Luciferin) was injected automatically and luciferase counts measured using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA).
A β-galactosidase assay was then performed by mixing 30 µL of lysate (or lysis buffer as a blank) with 750 µL of a solution containing 0.27% β-mercaptoethanol (Bio-Rad, Hercules, CA) in buffer Z (40 mM NaH2PO4.H2O, 60 mM Na2HPO4.7H2O, 1 mM MgSO4.7H2O, 10 mM KCl), and incubated 5 min at 37 °C. Then, 150 µL of a solution containing 4 mg/mL of 2-nitrophenyl β-D-galactopyranoside (ONPG; Sigma-Aldrich) and buffer Z was added to the mix and incubated for 5 to 20 min at 37 °C, until a yellow coloration appeared (except for the blank). To stop the reaction, 375 µL of 1 M Na2CO3 (Sigma-Aldrich) was added, and 200 µL of the mix was transferred in a 96-well microplate. The absorbance was measured at 420 nm.
Finally, a DC protein assay (Bio-Rad) was performed by transferring 20 µL of lysate (or lysis buffer as a blank) in a 96-well microplate and adding 25 µL of a solution composed of 2% of reagent S and 98% of reagent A. Then, 200 µL of reagent B was added, and the mix was incubated 15 min at room temperature. The absorbance was measured at 750 nm. Relative luciferase activity was calculated by dividing luciferase counts by β-galactosidase and DC assay absorbances. Data were then expressed in fold induction over the negative control (empty vectors). Experiments were carried out at least in triplicate, and the number of replicates is indicated on each bar of the associated figures.
Animals and Protocol
ClockΔ19/Δ19 homozygous mice and their WT littermates were the same animals as those used in previous studies (Mongrain et al., 2008; Freyburger et al., 2016). Briefly, ClockΔ19 heterozygous mutant mice described by Vitaterna et al. (1994) were obtained from Jackson Laboratory (Bar Harbor, ME) and backcrossed with BALB/c mice (Charles River Laboratories, Wilmington, DE) to increase breeding efficiency (50:50 C57Bl/6J and BALB/c background). Clock homozygous mice (ClockΔ19/Δ19) and WT littermates were entrained to a 12-h light, 12-h dark cycle (LD 12:12) for 2 weeks (with zeitgeber time 0 [ZT0] = lights-on) and then sacrificed every 6 h under LD12:12 (ZT2, ZT8, ZT14, ZT20; n = 4 or 5 per genotype per time). All mice were sacrificed by cervical dislocation, and brains were immediately frozen on dry ice. Right forebrains were subsequently dissected to perform RNA extraction, while left forebrains were used for protein extraction (see below). Experiments were approved by the Animal Care and Use Committee of the Douglas Mental Health University Institute and carried out in accordance with guidelines of the Canadian Council on Animal Care.
RNA Extraction and Quantitative PCR
RNA extraction, reverse transcription, and quantitative PCR (qPCR) for detection of Nlgn1 transcript variants were performed as outlined previously (El Helou et al., 2013; Massart et al., 2014). Quantification of Per2 steady-state mRNA level was also performed as a positive control. In brief, RNA was extracted from right forebrains using the RNeasy Lipid Tissue Midi kit and was DNase treated (Qiagen, Hilden, Germany). RNA amount and quality were verified with a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific) and agarose gel electrophoresis. Reverse transcription was performed with 0.5 µg of RNA using random hexamers and Superscript II reverse transcriptase (Life Technologies, Carlsbad, CA), according to the manufacturer’s instructions. qPCR was performed using a ViiA 7 real-time cycler (Life Technologies). Individual mouse cDNA was diluted and used in a 10-µL reaction with Fast TaqMan Master Mix reagent (Life Technologies) under fast cycling conditions: 50 °C for 2 min and 95 °C for 20 sec, followed by 40 cycles of 95 °C for 1 sec and 60 °C for 20 sec. PCR for each sample was performed in triplicate. Normalization was performed against 3 endogenous controls: beta-glucuronidase (GusB), ribosomal protein S9 (Rps9), and TATA-box binding protein (Tbp) using Expression Suite Software v1.1 (Life Technologies). The primer and probe sequences used for qPCR are shown in Table 1. Relative quantification was calculated using a modified ΔΔCt method (Expression Suite Software v1.1).
Protein Extraction and Western Blot
Total protein was extracted from left forebrains of ClockΔ19/Δ19 mutant mice and WT littermates as follows (procedure adapted from Giannone et al., 2013): tissues were rinsed in ice-cold PBS 1× before being placed in ice-cold modified RIPA buffer (50 mM HEPES, 10 mM EDTA, 0.1% SDS, 1% IGEPAL, 0.5% sodium deoxycholate, protease and phosphatase inhibitors [Sigma-Aldrich]) and homogenized on ice (PowerGen 125 homogenizer, Thermo Fisher Scientific) until translucid (2-3 min). Homogenates were centrifuged at 8000 g for 40 min at 4 °C, and the supernatant was kept at –80 °C for subsequent analysis.
Next, 40 µg of protein of each sample (4) was loaded on gel for each time point and genotype. SDS-PAGE was carried out using a 10% precast gel (Bio-Rad) at 125 V for 65 min. Proteins were then transferred on a nitrocellulose membrane (Bio-Rad) at 100 V for 40 min. The membrane was blocked with blocking buffer (5% nonfat dry milk diluted in PBS-T) for 1 h at room temperature. Primary anti-NLGN1 antibody (1:1000, No. 129111; Synaptic System, Göttingen, Germany) diluted in blocking buffer was incubated overnight at 4 °C. After washes in PBS-T, mouse anti-Actin antibody (1:5000, No. A5441; Sigma-Aldrich) was added for 1 h at room temperature. Membrane was washed again and a fluorescent anti-mouse secondary antibody (1:15,000, IRDye 680RD; LI-COR, Lincoln, NE) diluted in blocking buffer was incubated for 1 h at room temperature. Membrane was revealed using Odyssey CLx imaging system (LI-COR) and analyzed using ImageStudio 3.1 software (LI-COR). Quantification of NLGN1 was normalized relative to Actin.
Statistical Analyses
Fold inductions of relative luciferase activity were compared between conditions using 1-way analyses of variance (ANOVAs) for WT and mutated Nlgn1 and Nlgn2 sequences and using unpaired t tests for Per1. Gene expression of Per2 and Nlgn1 transcript variants and NLGN1 protein level were analyzed using 2-way ANOVAs with factors Genotype and Time. Significant effects were decomposed using post hoc Tukey’s multiple comparisons, simple effect analysis, or unpaired t tests. The threshold for statistical significance was set to 0.05, and results are reported as mean and standard error of the mean.
Results
Transcriptional Activation of
Nlgn1
by CLOCK/BMAL1
To assess whether CLOCK and BMAL1 can directly activate the transcription of the Nlgn1 gene, a 969 bp fragment from the putative promoter of Nlgn1 that contained 4 E-boxes was cloned in front of a luciferase gene in pGL3 vector to perform luciferase assays in COS-7 cells (Figure 1A). The condition with both CLOCK and BMAL1 showed a 3.4-fold induction compared with the negative control without CLOCK and BMAL1, and no significant induction was observed for conditions with only CLOCK or BMAL1 (Figure 1B; F5,107 = 56.6, p < 0.0001). This induction was significantly higher than that observed with CLOCK and BMAL1 using the empty pGL3 vector (p < 0.001).
To compare this transcriptional activation with that of a known clock gene and to confirm validity of the luciferase assay, transcriptional activation via the Per1 promoter was quantified in parallel. Compared with the negative control condition with empty plasmids, Per1 promoter had a 20-fold induction with CLOCK- and BMAL1-expressing plasmids (Figure 1B; t = 15.4, p < 0.0001). Given that the empty pGL3 vector contains a series of 3 E-boxes in the 225 bp just upstream the luciferase gene in the absence of insert, and that CLOCK and BMAL1 increased luciferase counts using this vector (Figure 1B), we further validated transcriptional activation by CLOCK and BMAL1 via the Nlgn1 gene fragment using a second expression vector, pGL2. In addition, we simultaneously assessed whether transcriptional activation by CLOCK and BMAL1 could be observed for another gene of the same adhesion system by performing the assay using a fragment of the Nlgn2 putative promoter, which contained 5 E-boxes (Figure 2A). Transcriptional activation by CLOCK and BMAL1 via the Nlgn2 gene showed a 1.6-fold induction compared with the negative control, which was significantly lower than the 2.4-fold induction observed with Nlgn1 (Figure 2B; F5,69 = 15.3, p < 0.0001). CLOCK/BMAL1 did not induce transcriptional activation of the empty pGL2 vector. These results suggest that CLOCK and BMAL1 can activate transcription of the Nlgn1 gene and to a lesser extent that of the Nlgn2 gene.
To investigate whether homologs of CLOCK and BMAL1, respectively NPAS2 and BMAL2, can also drive transcription via the promoter of Nlgn1, luciferase assays were performed with different combinations of plasmids expressing CLOCK, NPAS2, BMAL1, and BMAL2. With NPAS2 and BMAL1, a 3.2-fold induction was observed compared with the negative control, which is similar to the 3.4-fold induction seen with CLOCK and BMAL1, whereas a 2-fold induction was observed with CLOCK and BMAL2 (Figure 1C; F4,112 = 80.2, p < 0.0001). In contrast, the condition with NPAS2 and BMAL2 did not result in a significant induction compared with the control. This indicates that Nlgn1 transcription can also be controlled by homologs of CLOCK and BMAL1.
To identify which specific nucleotide sequences are responsible for transcriptional activation of the Nlgn1 gene by CLOCK and BMAL1, mutations were introduced in 3 E-boxes of the cloned sequence of Nlgn1 putative promoter (Figure 1A) and used for luciferase assays. The first mutation was targeted to the canonical E-box, and the second and third mutations targeted the 2 noncanonical E-boxes just downstream of the canonical E-box. Contrary to expectations, the first 2 mutations did not decrease transcriptional activation driven by CLOCK and BMAL1. Indeed, compared with the 3.4-fold induction obtained with the WT sequence, the induction was around 4 with the mutated canonical E-box and around 4.2-fold with the addition of the following noncanonical E-box mutation, which was significantly higher than the WT sequence (Figure 1D; F7,178 = 79.8, p < 0.0001). However, the addition of a third mutated E-box led to a 2.7-fold induction, which was significantly lower than that observed for the WT Nlgn1 gene fragment as well as that of the first 2 mutant constructs.
GSK3β Represses CLOCK/BMAL1 Transcriptional Activation of
Nlgn1
GSK3β is known to phosphorylate BMAL1, which leads to destabilization of the heterodimer with CLOCK (Sahar et al., 2010). We thus tested the modulatory role of GSK3β on the transcriptional activity of CLOCK and BMAL1 upon Nlgn1. To do so, luciferase assays were performed with conditions including CLOCK, BMAL1, and a WT GSK3β or a mutated form of GSK3β where serine 9 is replaced by an alanine (GSK3β S9A). This mutation leads to a constitutively active form of GSK3β, as serine 9 phosphorylation inhibits GSK3β kinase activity (Stambolic and Woodgett, 1994; Beaulieu, 2012). The addition of WT GSK3β to the condition with CLOCK and BMAL1 significantly decreased transcriptional activation via the cloned Nlgn1 fragment, whereas the addition of GSK3β S9A blocked it (Figure 3A; F3,54 = 49.8, p < 0.0001).
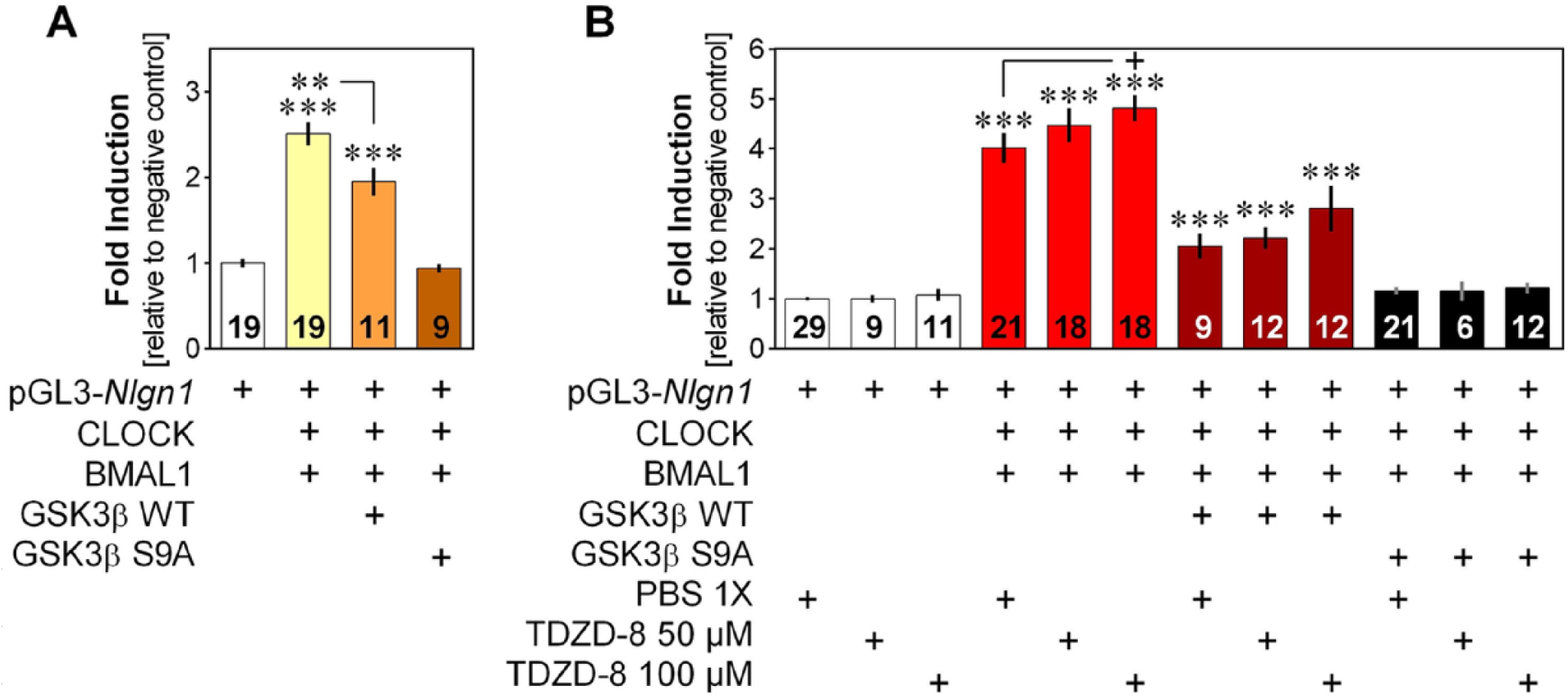
Modulatory role of GSK3β on transcriptional activation of Nlgn1. (A) Fold induction of the transcriptional activation of the Nlgn1 gene fragment by CLOCK and BMAL1, with and without wild-type GSK3β (GSK3β WT) or constitutively active GSK3β (GSK3β S9A), expressed relative to the negative control without CLOCK and BMAL1. **p < 0.01 between indicated points; ***p < 0.001 compared with the negative control (same in B). (B) Fold induction of the transcriptional activation of Nlgn1 gene fragment with CLOCK, BMAL1, GSK3β WT, and GSK3β S9A exposed to the vehicle PBS or 2 concentrations of the GSK3β inhibitor TDZD-8, expressed relative to the negative control without CLOCK and BMAL1 and exposed to PBS. +p = 0.056 between indicated points.
To test the effect of a GSK3β inhibitor on the transcriptional activation of Nlgn1, TDZD-8 has been used in luciferase assays since it selectively inhibits GSK3β by acting as a noncompetitive inhibitor of substrate binding (Martinez et al., 2002). TDZD-8 was also shown to increase phosphorylation of GSK3β serine 9 (Collino et al., 2008). To investigate the potential to inhibit endogenous GSK3β, conditions with only CLOCK and BMAL1 were tested. For these conditions, the fold induction was around 4 after 1 hour of incubation with the vehicle (PBS 1×). An induction of 4.5-fold was seen with a 1-h incubation with 50 µM TDZD-8, and a 4.8-fold induction was observed with a 1-h incubation with 100 µM TDZD-8 (Figure 3B; F11,166 = 42.1, p < 0.0001), which showed a tendency for a higher induction compared with the vehicle condition (p = 0.056). TDZD-8 at the 2 tested doses did not significantly prevent the inhibition of CLOCK/BMAL1 transcriptional activation of Nlgn1 by WT GSK3β and did not affect that of the constitutively active GSK3β S9A (Figure 3B). These results may suggest that the inhibitory effect of TDZD-8 requires physiological GSK3β level (i.e., no overexpression) and point to a modulatory role of GSK3β on CLOCK/BMAL1-driven transcriptional regulation of Nlgn1.
Altered
Nlgn1
mRNA in
Clock
Mutant Mice
We then aimed at verifying the specific implication of CLOCK in the regulation of Nlgn1 expression in vivo. To do so, the mRNA steady-state level of 4 different transcripts of Nlgn1 was measured in the right forebrain of ClockΔ19/Δ19 mutant mice and WT littermates at 4 different times of the day (Figure 4). We used probes specifically targeting the presence of an insert in splice site A, the absence of insert in splice site A, the presence of an insert in splice site B, and the absence of insert in splice site B (Figure 4A) as done previously (El Helou et al., 2013). The expression of Nlgn1 with the presence or the absence of insert A was significantly decreased in ClockΔ19/Δ19 mutant mice compared with WT mice (Figure 4B; Genotype effect: F1,3 > 5, p < 0.05). There was no significant difference between genotypes or interaction with time-of-day concerning the expression of Nlgn1 with an insert B (Genotype effect: F1,3 = 0.9, p = 0.3; Genotype by Time interaction: F3,29 = 2.2, p = 0.1). However, the expression quantified during the early dark phase (ZT14), which corresponds to the lowest expression in WT mice, was higher in ClockΔ19/Δ19 mutant than in WT mice (p < 0.02). Similarly, the expression of Nlgn1 without insert B was significantly changed in ClockΔ19/Δ19 mutant mice in comparison to WT mice (Figure 4B; Genotype by Time interaction: F3,27 = 3.7, p = 0.02), with expression during the light period being significantly lower in mutant than in WT mice. In parallel, we validated that the Clock∆19/∆19 mutation abolished the rhythmic expression of the clock gene Per2 in the mouse forebrain, which peaks at ZT14 (Figure 4C; Genotype by Time interaction: F3,32 = 5.2, p < 0.01).
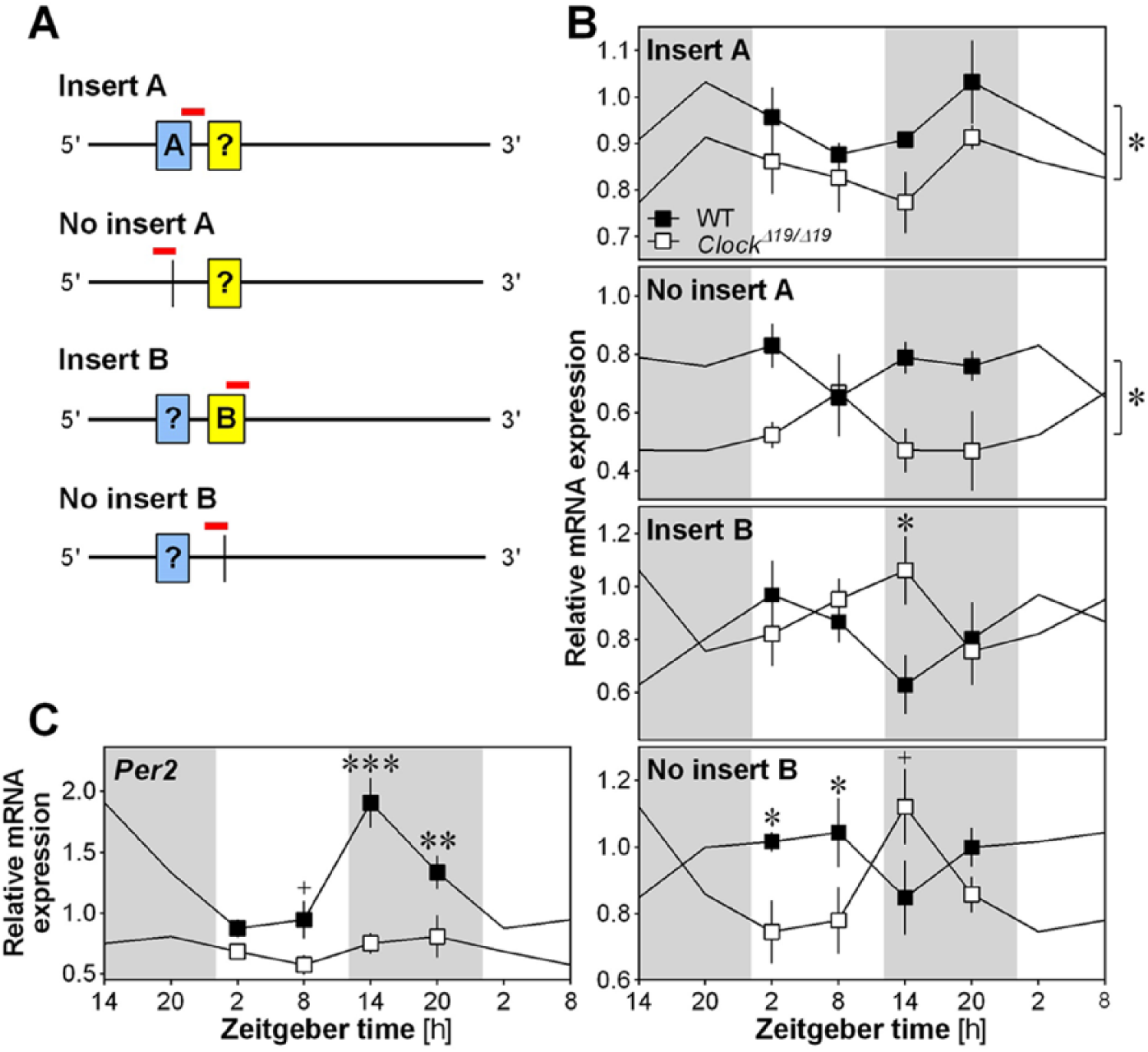
Nlgn1 mRNA in Clock mutant mice. (A) Schematic view of the position of qPCR amplicons (denoted by red lines in the online version) on the Nlgn1 mRNA for targeting different transcript variants (Insert A, No insert A, Insert B, No insert B). Insert A is depicted in blue and insert B in yellow online. The question mark inside the boxes indicates that the presence and absence of the other insert could not be simultaneously assessed by qPCR because the inserts are separated by more than 300 nucleotides. (B) Relative expression of Nlgn1 transcripts in the right forebrain of ClockΔ19/Δ19 mutant mice (n = 3 to 5 per time point) and WT littermates (n = 4 to 5 per time point) at ZT2, ZT8, ZT14 and ZT20. Data are shown once but curves have been double-plotted. Mutant mice are shown in white and WT in black, and white backgrounds indicate the 12-h light periods and grey backgrounds the 12-h dark (same in C). +p < 0.06 and *p < 0.05 between WT and mutant mice. The stars on the right of the top two panels are indicative of a significant genotype effect. (C) Relative expression of Per2 in the right forebrain of ClockΔ19/Δ19 mutant mice and WT littermates at ZT2, ZT8, ZT14 and ZT20 (n = 5 per genotype per time point). **p < 0.01 and ***p < 0.001 between WT and mutant mice.
NLGN1 Protein in
Clock
Mutant Mice
Since a change in mRNA levels does not necessarily reflect a change at the protein level, total NLGN1 protein was quantified at the same 4 times of the day in the left forebrain of ClockΔ19/Δ19 mutant mice and WT mice (Figure 5). Global NLGN1 level was not significantly changed in Clock mutant mice in comparison to WT mice (Genotype effect: F1,3 = 0.2, p = 0.6), and there was also no significant impact of the mutation on rhythmic NLGN1 protein level (Genotype by Time interaction: F3,24 = 0.8, p = 0.5). Together, mRNA and protein measurements in Clock mutant mice may suggest a complex regulation of Nlgn1 by clock elements in vivo that does not necessarily affect total protein level.
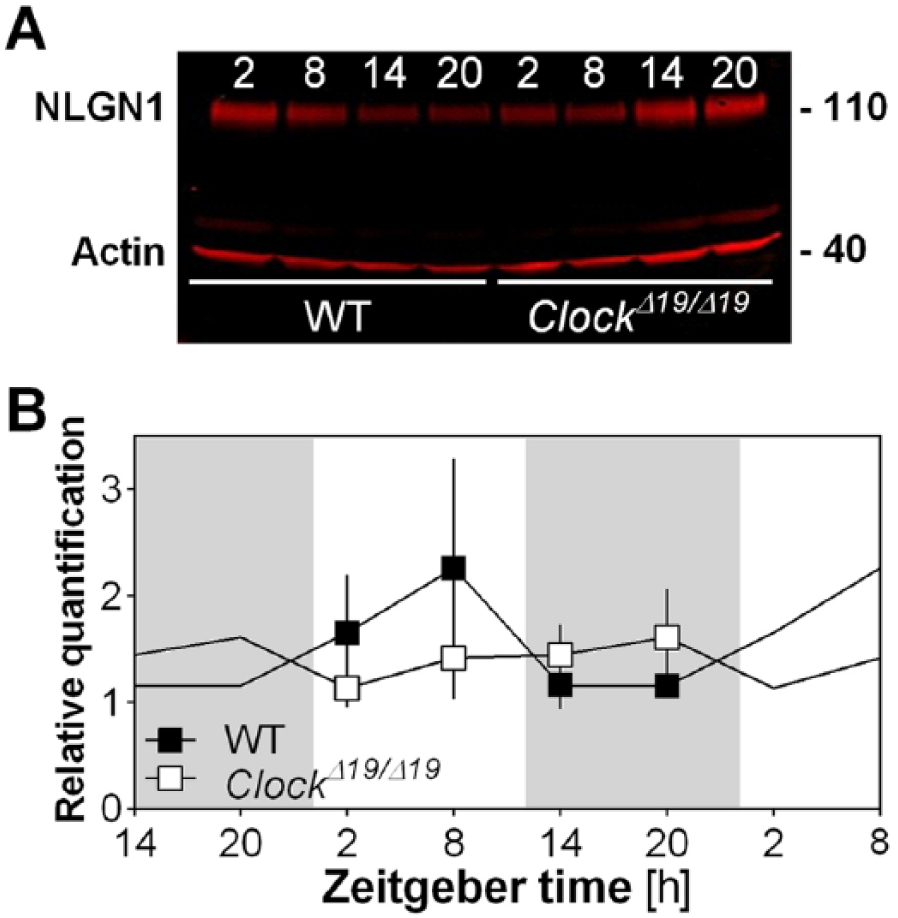
NLGN1 protein level in Clock mutant mice. (A) Representative Western blot showing NLGN1 in the left forebrain of ClockΔ19/Δ19 mutant mice and WT littermates at ZT2, ZT8, ZT14, and ZT20. The loading control protein actin is also shown. Note that total NLGN1 level is detected, not the level of the different NLGN1 isoforms (i.e., insert A, no insert A, insert B, no insert B). (B) Quantification of Western blots for NLGN1 in forebrain of ClockΔ19/Δ19 mutant mice and WT littermates (n = 4 per genotype per time point). Data are shown once but curves have been double-plotted. Mutant mice are shown in white and WT in black, and white backgrounds indicate the 12-h light periods and gray backgrounds the 12-h dark periods.
Discussion
Our previous work showed that the core clock transcription factors, CLOCK and BMAL1, bind to the Nlgn1 gene in the mouse cerebral cortex in a time-of-day dependent manner (El Helou et al., 2013). Here, we showed that CLOCK and BMAL1 can together induce transcriptional activation via a sequence of Nlgn1 putative promoter region, an activation that is repressed by the clock regulator GSK3β. We also observed that NPAS2 and BMAL2 can substitute for CLOCK and BMAL1, respectively, to induce expression of the Nlgn1 gene. Moreover, gene expression data in Clock mutant mice further support a transcriptional regulation of Nlgn1 by the molecular clock, an effect that appears to be transcript variant-specific. Overall, our present findings point to a contribution of core clock proteins in the transcriptional regulation of the synaptic component NLGN1.
Our in vitro data indicate that the dimers CLOCK:BMAL1 and NPAS2:BMAL1 can equally activate transcription via a fragment of the Nlgn1 gene. The activation by CLOCK:BMAL2 was lower, whereas no activation was observed in the case of the dimer NPAS2:BMAL2. Our findings of lower transcriptional activation with BMAL2 are similar to those made previously for the Per1 gene (Dardente et al., 2007). NPAS2 is expressed in the mouse forebrain and was reported to form a heterodimer with BMAL1 that is transcriptionally active (Garcia et al., 2000), which is in line with our findings for Nlgn1. Also, NPAS2 was shown to compensate for the absence of CLOCK when considering circadian functions (DeBruyne et al., 2006, 2007). Concerning BMAL2, it was earlier shown not to compensate for deficits in circadian features of the locomotor activity rhythm in Bmal1 knockout mice (Bunger et al., 2000), but this was found to result from a Bmal2 downregulation in Bmal1 knockout (Shi et al., 2000). However, BMAL2 cannot rescue circadian rhythm of gene expression in Bmal1 knockout fibroblasts, which was proposed by the authors to originate from the lack of a nuclear localization signal in BMAL2 following assays with BMAL1-BMAL2 chimeric constructs (Xu et al., 2015). Contrary to our findings, this study nevertheless reported a similar efficiency of BMAL1 and BMAL2 in steady-state transcriptional activation (Xu et al., 2015). In our study, only steady-state transcriptional activation was measured, and whether core clock transcription factors can drive a transcriptional rhythm of Nlgn1 expression in cells remains unknown. Future work should thus investigate whether CLOCK, BMAL1, NPAS2, and BMAL2 can drive a rhythm of Nlgn1 expression in vitro using continuous bioluminescence records.
CLOCK and BMAL1 induced transcriptional activation via the Nlgn1 promoter sequence that contained 4 E-boxes including a consensus CACGTG, and to a lesser extent via the Nlgn2 promoter sequence that contained 5 E-boxes. This may suggest a particular role of the consensus E-box present in the Nlgn1 fragment but absent from the Nlgn2 targeted sequence. We indeed reported previously that CLOCK and BMAL1 were binding to a 77 bp DNA fragment containing this consensus E-box in a time-of-day dependent manner in the mouse cerebral cortex (El Helou et al., 2013). Nonetheless, we observed no decrease in transcriptional activation via the Nlgn1 promoter when mutating the CACGTG into ACCTGG. This exact same mutation was previously shown to reduce by more than half the transcriptional activity of CLOCK and BMAL1 upon a portion of the Dbp gene (Rey et al., 2011). Our previous work showed that mutating only 2 nucleotides of 2 E-boxes of Rorγ reduced CLOCK/BMAL1 transcriptional activation also by more than 50% (Mongrain et al., 2008), while the work of others revealed that mutating 5 or 6 nucleotides from 1 E-box-like element of Per2 almost completely abolished CLOCK/BMAL1 steady-state or rhythmic transcriptional activation, respectively (Yoo et al., 2005; Nakahata et al., 2008). The lack of decrease in transcriptional activation of the Nlgn1 gene after mutating the consensus E-box as well as in combination with the following E-box indicates that other nucleotides of the cloned Nlgn1 sequence are mostly mediating CLOCK/BMAL1-dependent transcription. This is indeed supported by our observation of significantly decreased transcriptional activation in the triple mutant. Intriguingly, the finding that mutating 2 E-boxes significantly increased CLOCK/BMAL1-mediated transcriptional activation is puzzling and may indicate that these specific sites play a repressor role in the control of Nlgn1 expression by core clock transcription factors, which has been proposed previously for other targets (Kondratov et al., 2006; Nguyen et al., 2013).
The repression of Nlgn1 transcriptional activation by CLOCK/BMAL1 when adding GSK3β could provide further support for a direct role of the heterodimer in the regulation of Nlgn1 transcription. Indeed, GSK3β-dependent BMAL1 phosphorylation drives BMAL1 degradation (Sahar et al., 2010), which would inhibit CLOCK/BMAL1 activity in vitro. In addition, a role for GSK3β in the regulation of Nlgn1 expression could reveal an intracellular signaling pathway by which NLGN1 amount can be adjusted as a function of synaptic activity since GSK3β responds to, notably, glutamatergic and dopaminergic transmission (Nishimoto et al., 2008; Beaulieu, 2012). When inhibiting endogenous GSK3β in COS-7 cells using TDZD-8, we observed an increasing trend for the CLOCK/BMAL1-dependent transcriptional activation of Nlgn1. Higher doses of TDZD-8 could not be used, as these decreased cell viability. The lack of strong effect of the inhibitor could reside in low basal GSK3β activity in COS-7 cells or in too high CLOCK/BMAL1-mediated activation, preventing inhibition by endogenous GSK3β. Similarly, the lack of significant effect of the inhibitor on overexpressed WT GSK3β activity may come from saturation due to overexpression. In our study, the transfection of the constitutively active GSK3β S9A (Stambolic and Woodgett, 1994) ensured maximal inhibition of the CLOCK:BMAL1 dimer but rendered inhibition with TDZD-8 impossible. Therefore, our findings may support that this inhibitor affects serine 9 to modulate GSK3β activity, as proposed by others (Collino et al., 2008), in parallel to supporting a role for molecular clock components in the regulation of Nlgn1 gene expression.
We further characterized the role of clock components in the regulation of Nlgn1 expression using around-the-clock quantification of Nlgn1 transcripts in the forebrain of Clock mutant mice. The expression of specific Nlgn1 variants was decreased in Clock mutant mice, while others appeared to show an inverted rhythm in their pattern of expression. This observation is reminiscent of, among others, that of different splice variants of the Presenilin-2 gene reported to show different rhythms of expression in the liver (Bélanger et al., 2006), an effect that may depend on core clock transcription factors or other circadian regulators. In fact, the expression of specific splicing factors has been shown to be clock-controlled (McGlincy et al., 2012). In addition, the temperature rhythm was recently shown to importantly influence alternative splicing in the mouse (Preußner et al., 2017), and alterations of the body temperature rhythm have been reported in mice with the Clock mutation (Ochi et al., 2003). Alternatively, Nlgn1 expression could also be regulated by negative regulators of the clock such as REV-ERBα, whose gene expression is decreased in Clock mutant mice (Oishi et al., 2002). Even if we did not observe retinoic acid-related orphan receptor response elements (RORE: binding sites for REV-ERBα) in the promoter of Nlgn1, at least when considering 3000 bp preceding the transcription start site, assessment of Nlgn1 gene expression in Rev-Erbα knockout mice would help to understand the role of clock proteins in the transcriptional regulation of Nlgns. In sum, our findings suggest a complex, transcript variant-specific, regulation of Nlgn1 transcription by CLOCK in vivo.
Several circadian studies have shown that rhythmicity in mRNA expression is not always reflected at the protein level and vice versa (Chiang et al., 2014; Mauvoisin et al., 2014; Reddy et al., 2006), which is in line with our observations of changes in the expression of Nlgn1 transcripts in Clock mutant mice that are not paralleled with a change in NLGN1 protein level. Absence of effect at the protein level could reside in low statistical power. Otherwise, given that we made different observations for the expression of the different Nlgn1 transcript variants, the absence of difference at the protein level likely resides in the incapacity to detect distinct protein isoforms (no isoform-specific antibodies available). In addition, both mRNA expression and protein levels were examined in half brain samples, which may mask effects that would be specific for brain regions or even cell types. Future studies should thus assess the influence of CLOCK, BMAL1, and their homologs on the expression of different Nlgn1 variants and isoforms in precise brain areas and cell populations.
In conclusion, our findings support a role for core clock transcription factors in regulating the gene expression of Nlgn1 in the mouse. Importantly, CLOCK and BMAL1 may not be main transcriptional regulators of Nlgn1 as implied by moderate transcriptional activation in vitro and variant-specific gene expression changes in Clock mutant mice. Nevertheless, our results suggest that Nlgn1 could represent an additional “clock-controlled gene” that would link molecular clock mechanisms to synaptic functioning and plasticity. In fact, NLGN1 has been implicated in synaptogenesis, presynaptic glutamate release, and postsynaptic glutamate responses as well as disorders of the central nervous system such as Alzheimer’s disease (Jung et al., 2010; Kwon et al., 2012; Suzuki et al., 2012; Bie et al., 2014). Therefore, the control of Nlgn1 expression by clock proteins would provide a route by which molecular clock components can influence these cellular and neuronal functions and dysfunctions.
Footnotes
Acknowledgements
We thank Xuan Ruan, Gaétan Tremblay, and Mélanie Welman for technical help with experiments and equipment and Aude Villemain for mouse colony management. This research was funded by the Canada Research Chair in Sleep Molecular Physiology (V.M.), the Canada Research Chair in Molecular Psychiatry (J.M.B.), salary awards from the Fonds de recherche du Québec–Santé (FRQS) and the Canadian Institutes of Health Research (CIHR) to V.M. and N.C., a grant from CIHR to V.M., a grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) to N.C., and fellowships from FRQS and NSERC to V.M., from NSERC to P.G.R., from the Université de Montréal to L.H., and from the Hôpital du Sacré-Coeur de Montréal to E.K.O.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.