
Abstract
The mammalian circadian clock comprises a system of interconnected transcriptional and translational feedback loops. Proper oscillator function requires the precisely timed synthesis and degradation of core clock proteins. Heat shock protein 90 (HSP90), an adenosine triphosphate (ATP)-dependent molecular chaperone, has important functions in many cellular regulatory pathways by controlling the activity and stability of its various client proteins. Despite accumulating evidence for interplay between the heat shock response and the circadian system, the role of HSP90 in the mammalian core clock is not known. The results of this study suggest that inhibition of the ATP-dependent chaperone activity of HSP90 impairs circadian rhythmicity of cultured mouse fibroblasts whereby amplitude and phase of the oscillations are predominantly affected. Inhibition of HSP90 shortened the half-life of BMAL1, which resulted in reduced cellular protein levels and blunted expression of rhythmic BMAL1-CLOCK target genes. Furthermore, the HSP90 isoforms HSP90AA1 and HSP90AB1, and not HSP90B1-GRP94 or TRAP1, are responsible for maintaining proper cellular levels of BMAL1 protein. In summary, these findings provide evidence for a model in which cytoplasmic HSP90 is required for transcriptional activation processes by the positive arm of the mammalian circadian clock.
Keywords
Molecular chaperones are essential for the maintenance of cellular homeostasis in mammals. Initially, they were identified as components of protection systems against proteotoxic stressors such as elevated temperature or heavy-metal exposure. After a proteotoxic event, chaperones bind to denatured proteins, assist in their refolding, and prevent protein aggregation (Gidalevitz et al., 2011). Subsequent studies revealed that also under nonstress conditions, individual chaperones perform a wide range of cellular functions. Heat shock protein 90 (HSP90), for example, regulates the activity, stability, and subcellular localization of a large number of client proteins to which it binds in a selective manner together with associated cofactors (Trepel et al., 2010).
Circadian clocks enable organisms to synchronize behavior and physiology to the light-dark cycle and thereby to anticipate periodically recurring events in their environment. The mammalian molecular transcriptional-translational clock that is ticking autonomously in almost all body cells can be considered to be centered on a negative feedback loop of Period (Per1 and Per2) and Cryptochrome (Cry1 and Cry2) gene transcription. Expression of genes in the negative feedback loop is activated by the essential activator proteins BMAL1 and CLOCK, and in addition retinoic acid–related orphan receptor activators and REV-ERB repressors stabilize the core circadian oscillator by interlocking the expression of the activator component Bmal1 with components of the negative feedback loop (Preitner et al., 2002; Brown et al., 2012). Light serves as a resetting signal for the central oscillator in the suprachiasmatic nucleus (SCN) in the hypothalamus, which synchronizes peripheral oscillators in the rest of the body (Reppert and Weaver, 2002). Body temperature cycles, however, are a strong resetting cue for peripheral clocks (Brown et al., 2002). Accumulating evidence suggests that the temperature-sensitive heat shock response system, the primary driver of inducible chaperone gene expression, is involved in the regulation of the core circadian oscillator. Mice lacking HSF1, the master regulator of the heat shock response, have a longer free-running period than their wild-type littermates (Reinke et al., 2008). In line with this observation, pharmacological inhibition of the heat shock pathway lengthens the circadian period of SCN cells and interferes with phase entrainment and temperature compensation in peripheral clocks of mice (Buhr et al., 2010). Genetic knockdown studies revealed subsequently that phase entrainment of cultured mouse fibroblasts by physiological temperature cycles requires HSF1 but not its close homologue, HSF2 (Saini et al., 2012).
These findings raise the question of how components of the heat shock pathway interact with the molecular oscillator. One possibility is that HSF1 directly binds to regulatory regions of core clock genes and regulates their transcriptional rate. One study reported binding of HSF1 close to E-boxes and physical interaction of HSF1 with BMAL1 and CLOCK after heat shock at the Per2 promoter in mouse (Tamaru et al., 2012). A similar mechanism seems to play a role in the adaptation of mice to cold temperature (Chappuis et al., 2013). Alternatively, HSF1 might affect the circadian oscillator via the expression of designated HSF1 target genes such as Hsp90, which themselves have a wide range of biological functions (Li and Buchner, 2013). Indeed, HSP90 was found to directly regulate the core circadian oscillator in Arabidopsis thaliana by stabilization of the F-box protein ZEITLUPE (Kim et al., 2011). Loss of HSP90 reduced the cellular levels of the ZEITLUPE protein and lengthened the circadian period of the plants. The function of HSP90 in circadian clocks, however, seems to vary widely in different organisms. In contrast to its function in the core clock mechanism in A. thaliana, HSP90 is involved in connecting the circadian clock of Drosophila melanogaster to behavioral output functions but seems to be dispensable in the core oscillator mechanism in the flies (Hung et al., 2009). So far, no function for HSP90 in the mammalian circadian clock has been reported.
Here, we show that inhibition of HSP90 by various small molecules from different substance classes interfered with the circadian clockwork in cultured mouse fibroblasts. Blocking HSP90 function altered predominantly the phase and amplitude of the circadian oscillations, and at the same time, the expression levels of clock genes that are dependent on transcriptional activation by the BMAL1-CLOCK complex were strongly affected. This effect might at least in part be mediated by a shorter half-life and as a result reduced protein levels of BMAL1 that were observed after treatment of cells with HSP90 inhibitors or after small interfering RNA (siRNA)-mediated knockdown of the main cytoplasmic HSP90 isoforms HSP90AA1 and HSP90AB1. This study has therefore identified HSP90 as a regulator of BMAL1 stability and, as a consequence, of BMAL1-dependent transcriptional activation in the mammalian circadian clock.
Materials and Methods
Cell Culture and Real-Time Bioluminescence Monitoring
NIH3T3-Bmal1-luciferase cells stably expressing a Bmal1-luciferase reporter gene (Nagoshi et al., 2004) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 1% penicillin-streptomycin (P/S) (Life Technologies, Darmstadt, Germany). Confluent cells were trypsinized, and 5 × 105 cells were seeded into 35 mm culture dishes 1 day before real-time bioluminescence monitoring was initiated. Cellular clocks were synchronized with either 50% horse serum for 2 h or 100 nM dexamethasone for 30 min. After synchronization, the medium was replaced with phenol red-free DMEM (Life Technologies) that was supplemented with 10% FCS and 100 µM luciferin containing GA (geldanamycin), 17-AAG (17-allylamino-17-demethoxygeldanamycin), 17-AEP-GA (17-[2-(pyrrolidin-1-yl)ethyl]amino-17-demethoxygeldanamycin; InvivoGen, Toulouse, France), or radicicol (Sigma, Taufkirchen, Germany). Real-time bioluminescence was monitored in a light-tight incubator using photomultiplier tube detector assemblies (LumiCycle, Actimetrics, Wilmette, USA). The phase, amplitude, and period length of circadian oscillations were determined with LumiCycle Analysis software.
RNA Analyses
For analyses of clock gene expression, NIH3T3-Bmal1-luciferase cells were treated with 17-AEP-GA for 3 days. After synchronization with 50% horse serum for 2 h, the medium was replaced by 3 mL phenol-red free medium supplemented with 10% FCS, 1% P/S, 100 µM luciferin, and 800 nM 17-AEP-GA. Real-time bioluminescence was monitored, and cells were harvested after 48, 52, 56, 60, 64, or 68 h. Total RNA was isolated by Trizol-chloroform extraction and reverse-transcribed using the Quantitect Reverse Transcription Kit (Qiagen, Hilden, Germany). A quantitative reverse transcription polymerase chain reaction was carried out with the LightCycler480II detection system using the LightCycler480 Probes Master Mix (Roche, Penzberg, Germany) and specific primers and probes (Suppl. Table S1).
Protein Degradation Analyses
To generate cells expressing C-terminally Myc-tagged BMAL1, complementary DNA (cDNA) of Bmal1 (corresponding to NM_007489.3) was cloned into the MluI and ApaI sites of a derivative of the pMC vector (Linka et al., 2007) that was modified to provide C-terminal fusion to a cMyc-epitope tag linked by a Gly-Pro-Pro-Pro-Gly spacer. NIH3T3 cells were transfected with pMC3-Bmal1-cMyc using Effectene Transfection Reagent (Qiagen). Cells were cultured in DMEM supplemented with 10% FCS, 1% P/S, and 100 µg/mL hygromycin, and stable clones were selected. For analysis of protein stability, 3.75 × 105 cells were plated into 60 mm tissue culture dishes. After 24 h, the medium was replaced with medium containing 800 nM 17-AEP-GA. Cells were incubated for 1 day with 800 nM 17-AEP-GA, and then treated with 10 µg/mL cycloheximide (Sigma) for 2, 4, or 8 h. Cells were harvested, and whole-cell protein extracts were prepared by suspending the cells in an equal volume of 2 × NUN buffer (50 mM HEPES NaOH pH 7.6; 600 mM NaCl; 2 M urea; 2% NP-40; 2 mM DTT; protease inhibitor cocktail [Roche]). 50 µg protein lysate was separated by 8% SDS-polyacrylamide gel electrophoresis under reducing conditions and transferred to PVDF membranes (Millipore, Schwalbach, Germany). Membranes were probed with anti-cMyc–horseradish peroxidase (Miltenyi, Bergisch Gladbach, Germany) and anti-U2AF65 (Sigma), and they were developed using the enhanced chemiluminescence detection method (Amersham Biosciences, Frankfurt, Germany).
Quantification of Protein Half-Life
We assumed that protein degradation followed first-order decay kinetics. The measured protein intensity data (denoted by N) was initially log-transformed. The decay rate constant (k) was calculated using the RGP function from Excel (Microsoft). From the decay rate constant, the half-life (T1/2) was calculated: (N = N0e−kt; ln(N) – ln(N0) = –kt; T1/2 = ln(2) / k).
siRNA-Mediated Knockdown
For siRNA-mediated knockdown experiments, 2 × 105 cells were seeded into 35 mm culture dishes. After 24 h incubation, cells were transfected with 10 nM or 25 nM ON-Targetplus smartpool siRNA (Thermo Scientific, Schwerte, Germany) targeting BMAL1 or HSP90 isoforms. ON-TARGETplus non-targeting siRNA was used as a control. Transfection was performed according to the manufacturer’s instructions. Cells were harvested after 96 h, and proteins were analyzed by immunoblotting using anti-BMAL1 (Santa Cruz Biotechnology, Heidelberg, Germany), anti-HSP90 (Abcam, Cambridge, UK), or anti-U2AF65 antibody (Sigma).
Cell Viability Analyses
For analyses of cell viability, trypan blue dye exclusion staining was performed. Cells were harvested by trypsinization. After centrifugation, cells were suspended in 0.5 to 1 mL medium. Cells were mixed with an equal amount of trypan blue stain (0.4%) and analyzed in a Countess cell-counting chamber slide (Invitrogen, Carlsbad, USA). Cell count and viability were determined using the Countess automated cell counter.
Results
Inhibition of HSP90 Alters the Function of the Circadian Clock in Mouse Fibroblasts
The effect of small-molecule-mediated inhibition of HSP90 on the cellular circadian clock was analyzed in NIH3T3 mouse fibroblasts stably expressing the rhythmically transcribed Bmal1-luciferase reporter gene (Nagoshi et al., 2004). Cellular HSP90 activity was inhibited by treating fibroblast cultures with the benzoquinone ansamycin 17-AEP-GA, a low-toxic geldanamycin analog with high stability in aqueous solution, and a Kd of 0.4 µM for binding to HSP90 in vitro (Tian et al., 2004). Incubation of cells at 0.6 µM 17-AEP-GA resulted in a considerable impairment of circadian transcriptional oscillation compared to the solvent-treated sample (Fig. 1A). To quantify the effect of HSP90 inhibition on the cellular circadian clock, eight biological replicates were analyzed for changes in clock parameters on treatment with 17-AEP-GA. Incubation with the HSP90 inhibitor reduced the amplitude of the oscillations on average by about 30% (untreated 921 ± 122, 17-AEP-GA 649 ± 173; Fig. 1B). The phase of the circadian clock was delayed by 2.75 h compared to untreated control samples (untreated 13.1 ± 1.1 h, 17-AEP-GA 15.8 ± 1.1 h; Fig. 1C). The circadian period length did not differ significantly between the 17-AEP-GA treated samples and the untreated controls under these conditions (untreated 24.9 ± 0.7 h, 17-AEP-GA 25.4 ± 0.8 h; Fig. 1D). Importantly, at day 3 after inhibition of HSP90, the expression levels of endogenous Bmal1 messenger RNA (mRNA) were similar to those of the Bmal1-luciferase reporter with regard to the phase shift and amplitude (Fig. 1E), which supports the idea that the reporter faithfully mimicked the transcriptional changes characteristic for the endogenous Bmal1 gene and, by implication, the changes of the entire clock machinery.
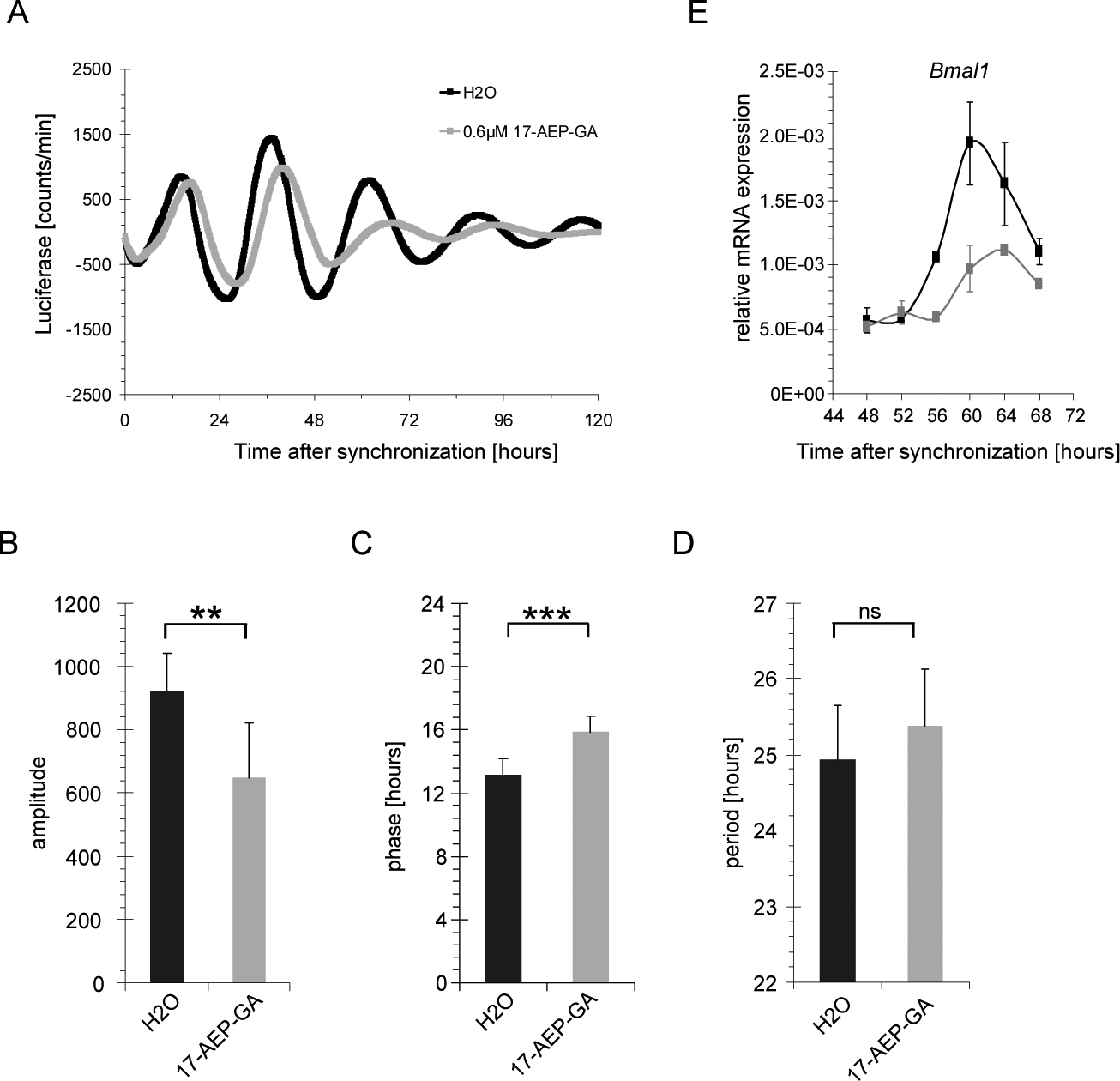
Parameters of the circadian transcriptional oscillator after inhibition of heat shock protein 90 (HSP90). (A) Circadian oscillations in synchronized NIH3T3 mouse fibroblasts stably expressing luciferase (luc) driven by the Bmal1 promoter (NIH3T3-Bmal1-luc) treated with 0.6 µM 17-AEP-GA (17-[2-(pyrrolidin-1-yl)ethyl]amino-17-demethoxygeldanamycin; gray) or solvent (black). Quantification of (B) amplitude, (C) phase, and (D) period length of NIH3T3-Bmal1-luc cells treated with 0.6 µM 17-AEP-GA (gray) or solvent (black) (n = 8). (E) Endogenous Bmal1 messenger RNA (mRNA) levels in NIH3T3-Bmal1-luc cells treated with 0.8 µM 17-AEP-GA (gray) compared to solvent-treated cells (black) (n = 2). Error bars indicate standard deviations. **p < 0.005; ***p < 0.0005 (2-sided t-test).
To exclude that the observed changes in circadian clock parameters resulted from idiosyncratic off-target effects of one particular HSP90 inhibitor, two more benzoquinone ansamycins, geldanamycin and 17-AAG, as well as the structurally distinct HSP90 inhibitor radicicol (Schulte et al., 1998) were compared to 17-AEP-GA in the same assay. Due to the known cytotoxicity of these substances (Guo et al., 2008), cell death was analyzed in all samples by dye exclusion staining. Figure 2 shows that the rhythmic expression of Bmal1-luciferase was impaired by any of the inhibitors in a dose-dependent manner, suggesting that HSP90 as the common molecular target of all substances plays a role in the circadian clock mechanism. Again, changes in amplitude and phase were observed under all conditions, and a complete inhibition of HSP90 might even lead to a lengthening of the circadian period, as was observed with high concentrations of geldanamycin analogs (Fig. 2A-2C). Moreover, while geldanamycin and 17-AAG caused cell death at higher concentrations, effects on the circadian clock were also seen under nontoxic conditions with all substances (Suppl. Fig. S1). Taken together, chemical inhibition of HSP90 by small molecules from different substance classes affects the cellular circadian clock whereby the amplitude and phase of transcriptional oscillations are predominantly affected.
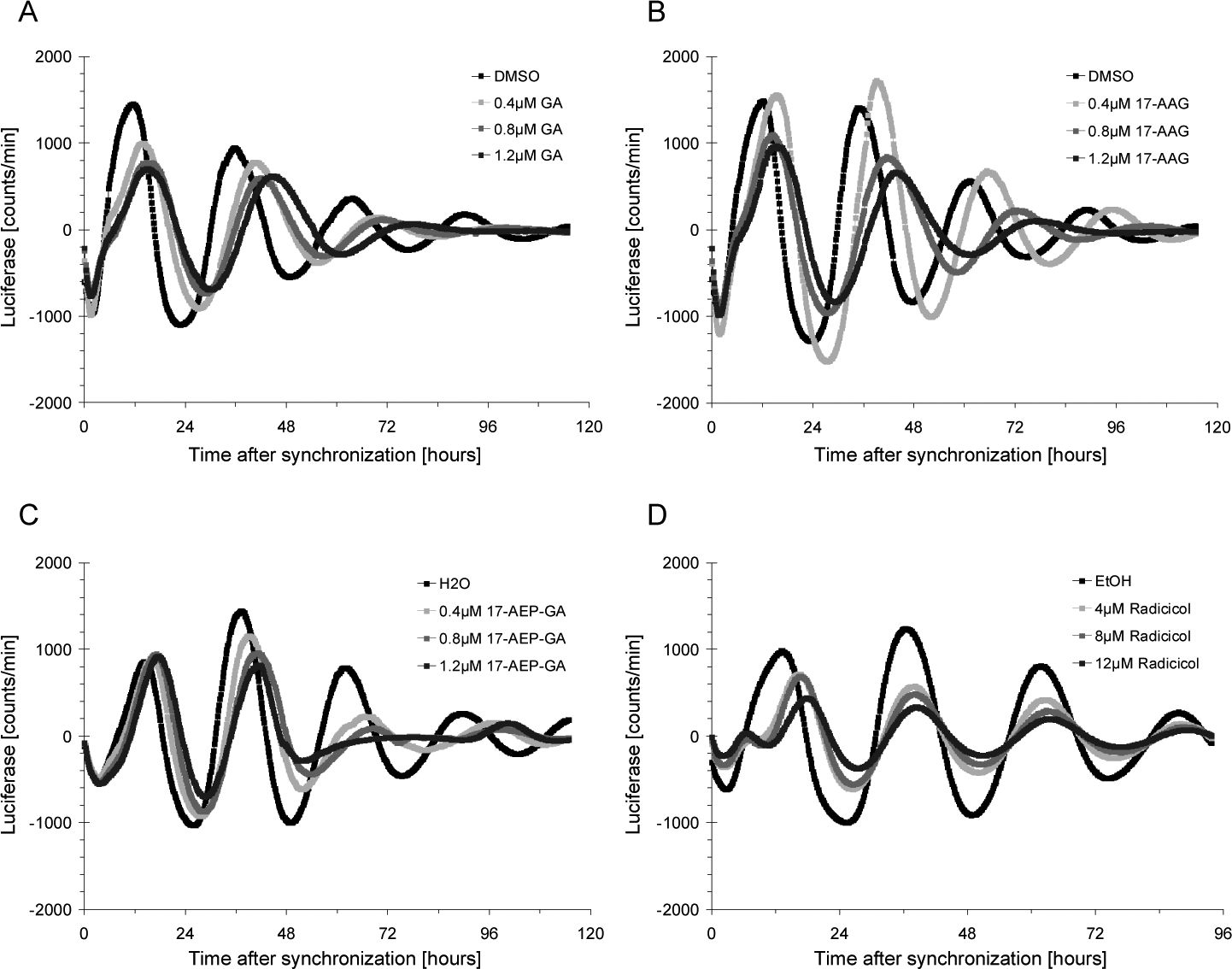
Effects of structurally distinct heat shock protein 90 (HSP90) inhibitors on the cellular circadian clock. Circadian oscillations in synchronized NIH3T3-Bmal1-luciferase cells treated with various concentrations of (A) geldanamycin (GA), (B) 17-allylamino-17-demethoxygeldanamycin (17-AAG), (C) 17-[2-(pyrrolidin-1-yl)ethyl]amino-17-demethoxygeldanamycin (17-AEP-GA), or (D) radicicol.
Loss of HSP90 Activity Shortens the Half-Life of BMAL1
HSP90 controls the levels of many cellular proteins, either of client proteins by direct interaction or indirectly via changing the activity or stability of regulators of protein half-lives (Trepel et al., 2010). Protein degradation is also an important regulatory mechanism in the mammalian circadian clockwork. For example, the stabilities of BMAL1 (Sahar et al., 2010; Zhang et al., 2012b; Ma et al., 2013), Cryptochromes (Godinho et al., 2007; Lamia et al., 2009), and Period proteins (Reischl et al., 2007; Asher et al., 2008) codetermine core clock parameters such as amplitude and period length and are connected to the metabolic control of circadian rhythmicity. BMAL1 and HSP90 have been found to interact as in vitro translated proteins (Hogenesch et al., 1997), and knockdown of BMAL1 disrupts the cellular oscillator (Baggs et al., 2009; Wallach et al., 2013), which prompted us to analyze the effect of HSP90 inhibitors on cellular BMAL1 levels. We found the total levels of BMAL1 protein in cells treated with HSP90 inhibitor to be considerably lower compared to the levels in untreated cells (Fig. 3A). The amount of the slowest migrating protein band, potentially representing a hyperphosphorylated form of BMAL1, was most strongly reduced after inhibition of HSP90, suggesting that dephosphorylation of BMAL1 might precede its degradation. Quantification of four independent experiments revealed a significant reduction of about 40% of BMAL1 protein levels in 17-AEP-GA-treated cells (Fig. 3D). Due to the well-established function of HSP90 in the control of protein stability (Trepel et al., 2010), we supposed that BMAL1 protein is a molecular target of HSP90 and that altered expression of Bmal1 mRNA on inhibition of HSP90 (Fig. 1E) is a consequence rather than the cause for reduced BMAL1 levels. Moreover, Bmal1 is expressed with high circadian amplitude in cells and tissues, whereas BMAL1 protein levels are rather constant throughout the day (Asher et al., 2008), which emphasizes the importance of posttranslational regulation for the control of BMAL1 levels. Therefore, the stability of BMAL1 was analyzed in the presence or absence of HSP90 activity by blocking protein translation and determining the kinetics of BMAL1 protein degradation. Indeed, 17-AEP-GA treatment led to a faster decline of BMAL1 protein levels in the cells compared to the untreated control samples (Fig. 3B and 3E), suggesting that HSP90 controls BMAL1 stability.
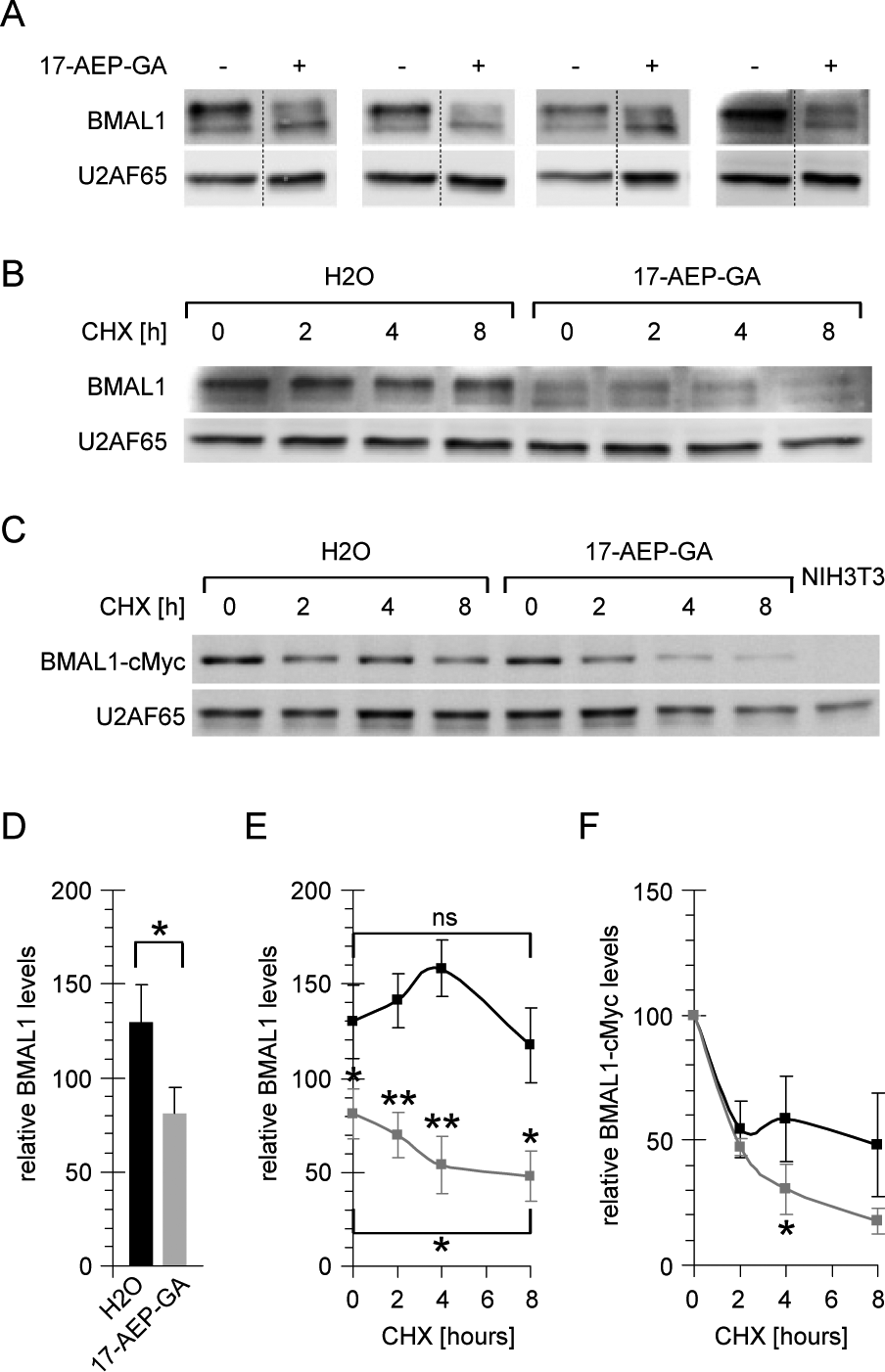
Cellular levels and stability of BMAL1 after inhibition of heat shock protein 90 (HSP90). (A) Cellular levels of BMAL1 protein determined by immunoblotting in four independent experiments in NIH3T3 cells treated with 0.8 µM 17-AEP-GA. U2AF65 protein levels were analyzed as a control for equal sample loading. Gel cuts are indicated by dotted lines. (B) Time course of BMAL1 and U2AF65 protein degradation in NIH3T3 cells treated with 0.8 µM 17-AEP-GA after inhibition of protein synthesis by cycloheximide (CHX) for the indicated duration. The 0-h (CHX) lanes are identical with the last two lanes in (A). (C) Time course of BMAL1 and U2AF65 protein degradation in NIH3T3 cells stably expressing BMAL1-cMyc treated with 0.8 µM 17-AEP-GA after inhibition of protein synthesis by cycloheximide (CHX) for the indicated duration. The most rightward lane shows untransfected NIH3T3 cells that do not express BMAL1-cMyc. (D) Quantification of BMAL1 protein levels from (A). (E) Quantification of BMAL1 protein levels from (B). Protein half-life H2O 42 h, and 17-AEP-GA 16 h. Asterisks above gray data points refer to significance levels of 17-AEP-GA versus H2O. The asterisk below the gray data points refers to the significance level of 17-AEP-GA t = 0 versus t = 8. (F) Quantification of BMAL1-cMyc protein levels from (C) normalized to the respective 0 h (CHX) value. Protein half-life H2O 7 h, and 17-AEP-GA 3 h. (n = 3). Error bars indicate standard deviations. *p < 0.05; **p < 0.005 (2-sided t-test).
To corroborate this result in an independent experimental system, a mouse fibroblast cell line stably expressing cMyc-tagged BMAL1 was created, and the half-life of BMAL1 was determined under the same conditions. Similar to the faster degradation rate that was observed for endogenous BMAL1 in cells treated with HSP90 inhibitor (Fig. 3B and 3E), also BMAL1-cMyc levels expressed from the noncircadian cytomegalovirus promoter (Asher et al., 2008) declined considerably faster in the absence of functional HSP90 compared to the degradation rate in untreated control cells (Fig. 3C and 3F). In summary, these findings provide evidence that HSP90 regulates the cellular amount of BMAL1 and that reduced protein levels can be explained at least in part with a shorter half-life of BMAL1 in the absence of functional HSP90.
The Main Cytoplasmic HSP90 Isoform Is Required for the Stabilization of BMAL1
As mentioned, chemical inhibitors can have side effects on unrelated target proteins. To pinpoint the observed changes in BMAL1 protein stability to the activity of HSP90, single proteins were targeted by siRNA against all HSP90 variants that are known to be inhibited by geldanamycin analogs and radicicol. The main cytoplasmic isoform of HSP90 is expressed from two highly similar genes, Hsp90aa1 and Hsp90ab1. Both genes were knocked down simultaneously, and as a control Bmal1 was targeted directly by a specific siRNA. Figure 4A shows that knockdown of Hsp90aa1 and Hsp90ab1 led to a significant reduction in total cellular BMAL1 levels at the higher siRNA concentration, which was almost as strong as when Bmal1 was targeted directly (Fig. 4B).
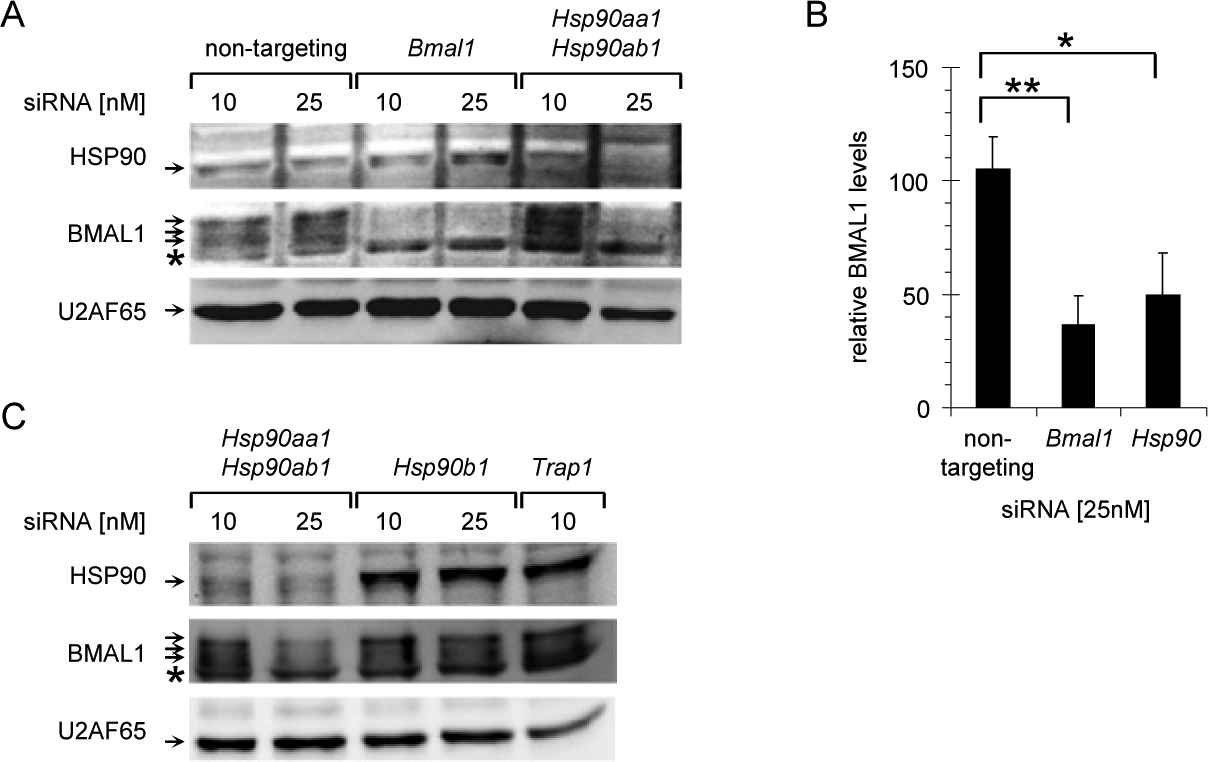
Small interfering RNA (siRNA)-mediated knockdown of heat shock protein (HSP90) isoforms. (A) HSP90, BMAL1, and U2AF65 protein levels in NIH3T3 cells transfected with nontargeting siRNA or siRNAs for Bmal1 or Hsp90aa1-Hsp90ab1. Arrows indicate specific protein bands; the asterisk marks a protein band that is nonspecifically detected by the BMAL1-specific antibody. (B) Quantification of BMAL1 protein levels after knockdown of Bmal1 or Hsp90aa1-Hsp90ab1 from (A) (n = 3). (C) HSP90, BMAL1, and U2AF65 protein levels in NIH3T3 cells transfected with siRNAs for Hsp90aa1-Hsp90ab1, Hsp90b1, or Trap1. Error bars indicate standard deviations. *p < 0.05; **p < 0.005 (2-sided t-test).
HSP90 exists in several isoforms that accumulate in different cellular compartments. The main cytoplasmic HSP90 variants are HSP90AA1 and HSP90AB1, which have been shown to be localized in the nucleus as well (Collier and Schlesinger, 1986). HSP90B1-GRP94 is mainly targeted to the lumen of the endoplasmatic reticulum, where it plays a role in the unfolded protein response (Lee, 1981). The HSP90 isoform TRAP1 is a largely mitochondrial protein with functions in mitochondrial integrity, apoptosis, and oxidative stress (Altieri et al., 2012). Importantly, however, interactions with exclusively cytoplasmic proteins have been described for all HSP90 isoforms, and subpools of all proteins seem to exist in the cytoplasm and/or the nucleus (Welch et al., 1983; Chen et al., 1996; Csermely et al., 1998). Moreover, all HSP90 isoforms are well-established regulators of protein stability (Chavany et al., 1996; Landriscina et al., 2010) and have therefore the potential to control cellular BMAL1 levels. Only downregulation of cytoplasmic HSP90 by simultaneous siRNA-mediated knockdown of the highly similar proteins HSP90AA1 and HSP90AB1, however, lowered the cellular levels of BMAL1 protein (Fig. 4C). The high concentration of Trap1-specific siRNA was highly toxic, and therefore the data were excluded from the analysis. Taken together, these experiments showed that despite the fact that HSP90 is a highly abundant protein, its downregulation reduced the cellular amount of BMAL1 very efficiently (Fig. 4B), suggesting that the stability of BMAL1 is dependent on HSP90.
Inhibition of HSP90 Affects the Expression of BMAL1-CLOCK Target Genes
Finally, we asked if inhibition of HSP90 affects the expression of designated BMAL1-CLOCK target genes and analyzed mRNA levels of several core clock genes in synchronized mouse fibroblasts after inhibition of HSP90. Figure 5 shows that peak mRNA levels of strongly BMAL1-CLOCK-dependent genes such as Nr1d1 (Rev-Erb alpha) (Ripperger, 2006), Dbp (Ripperger and Schibler, 2006), and Per2 (Yoo et al., 2005) were considerably blunted under these conditions (Fig. 5A-5C). In particular, the maximal expression of Nr1d1 68 h after synchronization was reduced more than 8-fold (Fig. 5A). Expression of Clock, however, was unchanged after treatment with 17-AEP-GA (Fig. 5D), which shows that the mechanism by which HSP90 regulates the cellular circadian clock does not affect all components of the oscillator mechanism. Taken together, our findings provide evidence for a model in which HSP90 is required for the stabilization of BMAL1 and thereby guarantees proper transcriptional oscillation of BMAL1-CLOCK-dependent clock genes.
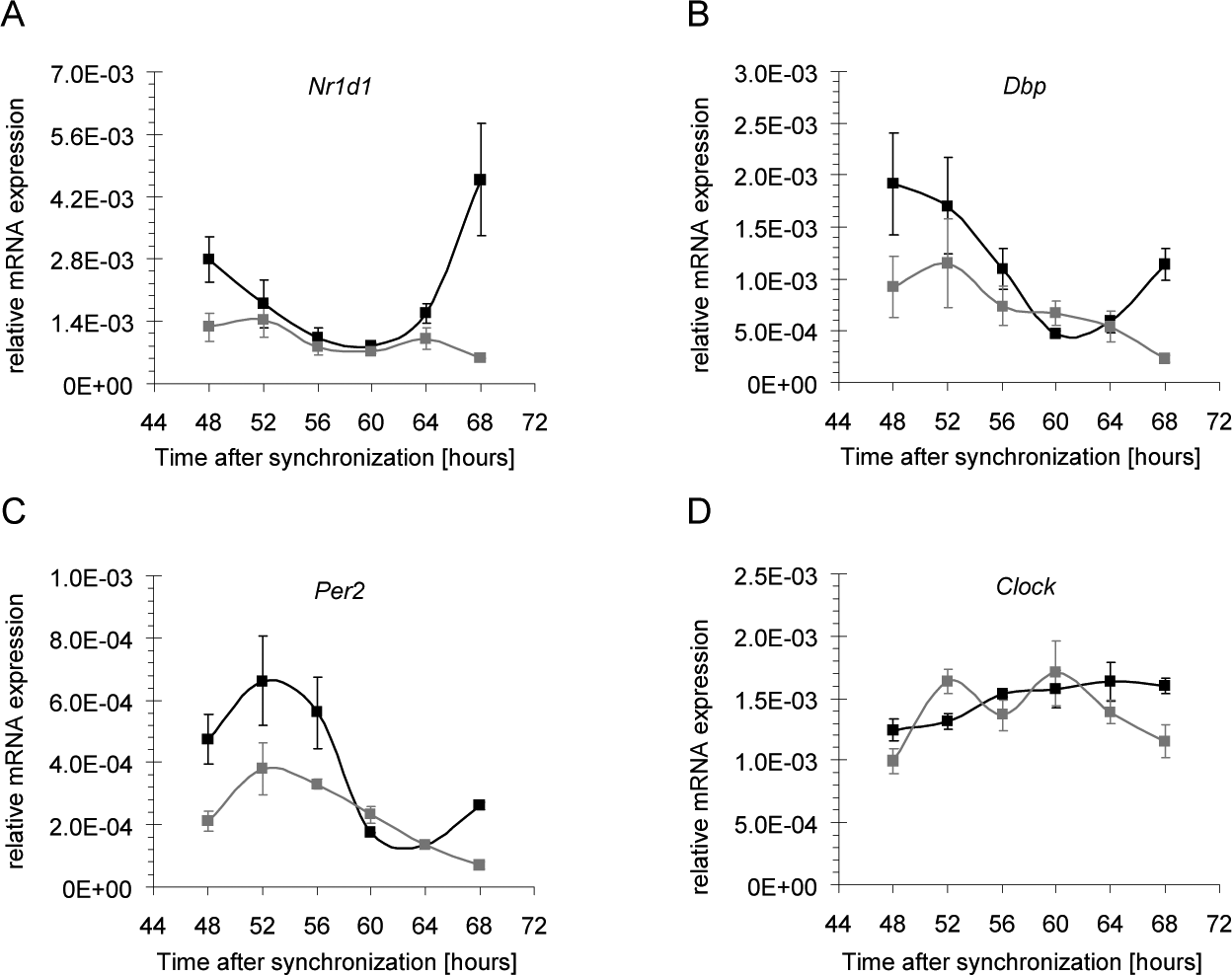
Messenger RNA (mRNA) levels of core clock genes after inhibition of heat shock protein (HSP90). Expression of core clock genes in NIH3T3-Bmal1-luc cells treated with 0.8 µM 17-AEP-GA (gray) or solvent (black). (A) Nr1d1, (B) Dbp, (C) Per2, and (D) Clock (n = 2). Error bars indicate standard deviations.
Discussion
In this study, we set out to analyze the function of HSP90 in the circadian clockwork in cultured mammalian cells. HSP90 seemed to be an obvious candidate for a chaperone regulator of the circadian clock, since it is expressed in a highly circadian manner in mouse (Kornmann et al., 2007), and interplay between HSP90 and circadian clocks has been demonstrated in other model organisms (Hung et al., 2009; Kim et al., 2011). Moreover, HSP90 can be efficiently inhibited by small-molecule inhibitors, most of which compete to its N-terminal ATPase domain, which is essential for all known functions of HSP90 (Nadeau et al., 1993).
The results of this study provide evidence from various experimental approaches that HSP90 plays a role in the BMAL1-CLOCK-containing positive arm of the mammalian circadian clock. First, we found that inhibition of HSP90 interferes with circadian rhythmicity in cultured cells. Treatment of mouse fibroblasts with different geldanamycin derivatives or radicicol altered the phase and amplitude of the circadian clock. Importantly, changes of clock parameters were observed in the absence of cell death in confluent cell cultures excluding cytotoxicity and cell cycle arrest, two common global side effects of HSP90 inhibition (Srethapakdi et al., 2000), as confounding factors. It should be pointed out that the observed effects on transcriptional oscillations do not necessarily reflect the endpoint of HSP90 inhibition. HSP90 activity might not have been fully abolished under any of these conditions, and higher concentrations might lead to a total breakdown of rhythmicity. The observed lengthening of circadian period at higher inhibitor concentrations was indicative of such an effect. Due to the cytotoxicity of HSP90 inhibitors at elevated concentrations, however, it will be difficult to address this point in a comprehensive manner.
Second, in the presence of HSP90 inhibitors, we found considerably reduced mRNA levels, particularly of clock genes that strongly depend on BMAL1-CLOCK for transcriptional activation such as Nr1d1 (Ripperger, 2006). On the level of gene expression, this effect might result from a decrease in transcriptional activation (e.g., due to diminished BMAL1-CLOCK activity; Ripperger and Schibler, 2006) or an increase in transcriptional repression (e.g., due to elevated levels of PER; Asher et al., 2008). In fact, when HSP90 activity was blocked, we observed a shorter protein half-life of the transcriptional activator BMAL1 and, as a consequence, reduced cellular levels of BMAL1. It has been shown before in U2OS cells that diminished levels of BMAL1 lead to lower circadian amplitudes and complete loss of BMAL1 to arrhythmicity (Baggs et al., 2009), supporting the idea that BMAL1 is an important target of HSP90 in the core oscillator mechanism, yet the contribution of other mechanisms cannot be excluded at this point. Finally, gene-specific knockdown revealed that loss of HSP90AA1 and HSP90AB1 has the same effect on cellular BMAL1 levels as chemical HSP90 inhibitors. Cytoplasmic HSP90 has therefore been identified as a regulator of BMAL1 stability.
Future work has to reveal how HSP90 affects BMAL1-CLOCK-dependent processes on the molecular level. A direct interaction between BMAL1 and HSP90 has been reported with in vitro translated proteins (Hogenesch et al., 1997). BMAL1 and CLOCK have been proposed to be kamikaze activators that are primed for degradation when they are bound to chromatin (Stratmann et al., 2012). Inhibition of HSP90 might accelerate this degradation process and thereby lead to reduced mRNA synthesis of BMAL1-CLOCK target genes. In support of such a model would be the reported interaction between BMAL1-CLOCK and HSF1 (Tamaru et al., 2012), since HSF1 interacts with HSP90 and might recruit HSP90 to the BMAL1-CLOCK complex. HSP90 might also associate with BMAL1 in the cytoplasm, alone or in a complex with CLOCK, and protect it from proteasomal degradation or affect nuclear import (Kwon et al., 2006). A potential interaction between BMAL1 and HSP90, however, might be very weak or transient, and we did not succeed in demonstrating such an interaction in mouse liver nuclear extracts, which should contain the majority of BMAL1 (Kwon et al., 2006) and significant amounts of HSP90 (Collier and Schlesinger, 1986), or whole-cell extracts from cultured fibroblasts (data not shown). Therefore, the alternative that HSP90 regulates BMAL1 via an indirect mechanism should also be considered. Several regulatory mechanisms have been described that influence the cellular levels of BMAL1 protein. BMAL1 is rhythmically O-GlcNAcylated,which blocks ubiquitination and thereby degradation (Ma et al., 2013). Inhibition of HSP90 has been shown to destabilize O-linked β-N-acetylglucosamine transferase (OGT) (Zhang et al., 2012a), and the resulting decrease in O-GlcNAcylation levels might promote the degradation of BMAL1. The protein levels of BMAL1 are also controlled via phosphorylation-mediated degradation through the kinase GSK-3β (Spengler et al., 2009; Sahar et al., 2010), and the maturation of GSK-3β is sensitive to geldanamycin-mediated inhibition of HSP90 (Lochhead et al., 2006).
Of further interest is the question of how HSF1 and HSP90 work together in the regulation of the circadian clock. One obvious possibility is that HSF1 regulates the circadian expression of Hsp90. Since HSF1 is required for inducible transcriptional activation of Hsp90 after heat shock (Xiao et al., 1999) and both the circadian activity of HSF1 and rhythmic expression of Hsp90 can be driven by systemic cues (Kornmann et al., 2007; Reinke et al., 2008), HSF1 can be considered a bona fide regulator of circadian Hsp90 expression.
Finally, these findings open the question of what roles the interplay between HSP90 and the circadian clock might play in mammalian homeostasis and disease. HSP90AA1 has recently been identified in silico as a major hub in the human circadian protein-protein interactome (Wallach et al., 2013). Its proposed rhythmic interactions with other cellular proteins inside and outside of the core clock mechanism are supposed to contribute significantly to the temporal organization of cellular physiology. Systemically driven diurnal expression of HSP90 (Kornmann et al., 2007) could be involved in temperature entrainment or amplitude regulation of the circadian clock by timed stabilization of BMAL1 during the circadian cycle. A similar mechanism involving the PKCg-mediated stabilization of BMAL1 has recently been shown to regulate the entrainment of the circadian clock by timely restricted feeding (Zhang et al., 2012b). The requirement for HSP90 in maintaining proper cellular levels of BMAL1, however, adds to the complexity of using HSP90 inhibitors in cancer therapy. Downregulation of BMAL1 accelerates tumor growth (Zeng et al., 2010), and inhibition of HSP90 would further promote this process, providing another example for the often ambivalent role of HSP90 in physiological and pathological processes.
Footnotes
Acknowledgements
The cDNA of Bmal1 was kindly provided by Aziz Sancar. The authors thank Gabriele Schoder for excellent technical assistance and Christian Mielke for valuable comments on the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 728, RE 3046/2-1) and the Human Frontier Science Program (CDA00009/2009-C).
Conflict of Interest Statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.