
Abstract
In plants, the circadian clock regulates the expression of one-third of all transcripts and is crucial to virtually every aspect of metabolism and growth. We now establish sumoylation, a posttranslational protein modification, as a novel regulator of the key clock protein CCA1 in the model plant Arabidopsis. Dynamic sumoylation of CCA1 is observed in planta and confirmed in a heterologous expression system. To characterize how sumoylation might affect the activity of CCA1, we investigated the properties of CCA1 in a wild-type plant background in comparison with ots1 ots2, a mutant background showing increased overall levels of sumoylation. Neither the localization nor the stability of CCA1 was significantly affected. However, binding of CCA1 to a target promoter was significantly reduced in chromatin-immunoprecipitation experiments. In vitro experiments using recombinant protein revealed that reduced affinity to the cognate promoter element is a direct consequence of sumoylation of CCA1 that does not require any other factors. Combined, these results suggest sumoylation as a mechanism that tunes the DNA binding activity of the central plant clock transcription factor CCA1.
Introduction
Circadian rhythms are driven by an intrinsic biological timekeeping system, the circadian clock, which exists in animals, plants, fungi, and even certain prokaryotes (Zhang and Kay, 2010). In plants, clock defects can decrease both carbon fixation and biomass production by about half (Dodd et al., 2005), highlighting the importance of circadian timekeeping for food crop productivity. The circadian clock drives the rhythmic expression of about 30% of all genes in the model plant Arabidopsis thaliana (Covington et al., 2008). This daily transcriptional reprogramming is achieved through a set of clock genes that indirectly regulate their own expression via a complex circuitry of transcriptional-translational feedback loops (TTFLs). The TTFL system in Arabidopsis contains a crucial morning-phased transcription factor complex of CIRCADIAN CLOCK ASSOCIATED (CCA1) and LATE ELONGATED HYPOCOTYL (LHY). The CCA1/LHY complex represses expression of the evening-phased gene TIMING OF CAB2 EXPRESSION 1 (TOC1) and is thought to activate the expression of other clock genes such as PSEUDO-RESPONSE REGULATOR 9 (PRR9) (Farre et al., 2005). However, recent studies have found that CCA1/LHY also repress the PRR genes (Kamioka et al., 2016). TOC1 and PRR9 proteins feed back to repress CCA1 and LHY expression. CCA1/LHY expression is indirectly activated by a so-called Evening Complex (EC), which represses PRR9 expression (Hsu and Harmer, 2013).
In all rhythmic organisms, timekeeping depends on posttranslational modification of TTFL proteins, to tune the expression and localization dynamics of clock proteins and turn the clock system into a 24-h oscillator. Phosphorylation is the most characterized posttranslational modification on circadian clock proteins in any organism, but it is evident that other modifications dynamically regulate clock proteins and might be equally important for clock function. For example, the cellular machinery that modifies proteins by the covalent attachment of the Small Ubiquitin-related MOdifier (SUMO) is necessary for clock function in mammals (Cardone et al., 2005; Lee et al., 2008), is fully conserved across eukaryotes (Johnson, 2004), and has now been shown in this issue of the Journal of Biological Rhythms also to influence timekeeping in the model plant Arabidopsis. In this study, we identified the crucial clock transcription factor CCA1 as a target of sumoylation. Although sumoylation is an abundant modification, very few examples exist (especially in plants) of functional consequences on specific targets. Our results show that reduced DNA binding affinity is a direct effect of CCA1 sumoylation, providing a striking new example of the effect of sumoylation on a specific target protein.
Materials and Methods
All chemicals were obtained from Sigma Aldrich unless otherwise stated. Restriction enzymes were from New England Biolabs. H2O means double-distilled water. All oligonucleotides used in this study are listed in Supplementary Table S1, and all novel DNA constructs reported here were verified by sequencing. All results presented are representative of at least 2 independent experiments.
Plant Lines, Genotyping, and Growth Conditions
The Arabidopsis Columbia-0 plant lines ots1 ots2 (Conti et al., 2008), CCA1pro:CCA1-3xHA-YFP cca1 (Yakir et al., 2009), here referred to as CCA1-3HY, were described previously. CCA1-3HY was crossed with ots1 ots2 to give CCA1-3HY ots1 ots2. F1-F3 progeny were selected for T-DNA insertions and transgenes by polymerase chain reaction (PCR)–based genotyping. Soil-grown plants were grown under long day conditions (16 h light/8 h dark) at 22 °C unless otherwise stated. Seedlings were grown on half-strength Murashige and Skoog media without vitamins (0.5 MS) at 21 °C in 100 µmol × m–2 × s–1 white light.
Hemagglutinin-Immunoprecipitation
For hemagglutinin (HA)–pulldowns of sumoylated CCA1, leaf material was homogenized in liquid nitrogen and ground in extraction buffer (300 mM NaCl, 50 mM Tris-HCl pH 7.4, 5 mM EDTA, 1 mM DTT, 10% glycerol, 0.2% NP-40, 0.1% Triton X-100, 20 mM N-ethylmaleimide (NEM), 50 µM MG132, and 50 µM PR-619, Complete protease inhibitor tablet (Roche, Basel, Switzerland), PhosStop cocktail (Roche), 1% PVPP). Cell debris was removed by centrifugation and filtration through a 0.22-µm filter. The crude extract was incubated with anti-HA antibody-conjugated agarose beads (Roche No. 11815016001) for 4 h rotating at 4 °C. Beads were pelleted and washed 4 times in extraction buffer. Bound protein was eluted from the beads in lithium dodecyl sulfate (LDS) protein loading buffer (Invitrogen, Carlsbad, CA) at 70 °C. Proteins were separated on 4% to 12% polyacrylamide gels (Invitrogen Novex) and transferred to nitrocellulose membranes using the iBlot system (Invitrogen). Primary antibodies were anti-HA-tag (1:5000, No. ab20084, abcam, Cambridge, UK), anti-SUMO1/2 (1:2000-5000, abcam No. ab5316), and anti-Ubiquitin (1:1000, No. 04-263, Millipore, Burlington, MA). An important note is that the results in Figure 1 are consistently observed with batch numbers GR143233-1, GR38587-1, and GR272641-1 of the SUMO antibody but not with batches GR94030-1 and GR244508-1. SuperSignal West Pico or Dura Chemiluminescent Substrate (Thermo Scientific, Waltham, MA) was used to visualize hybridized bands according to the manufacturer’s instructions.
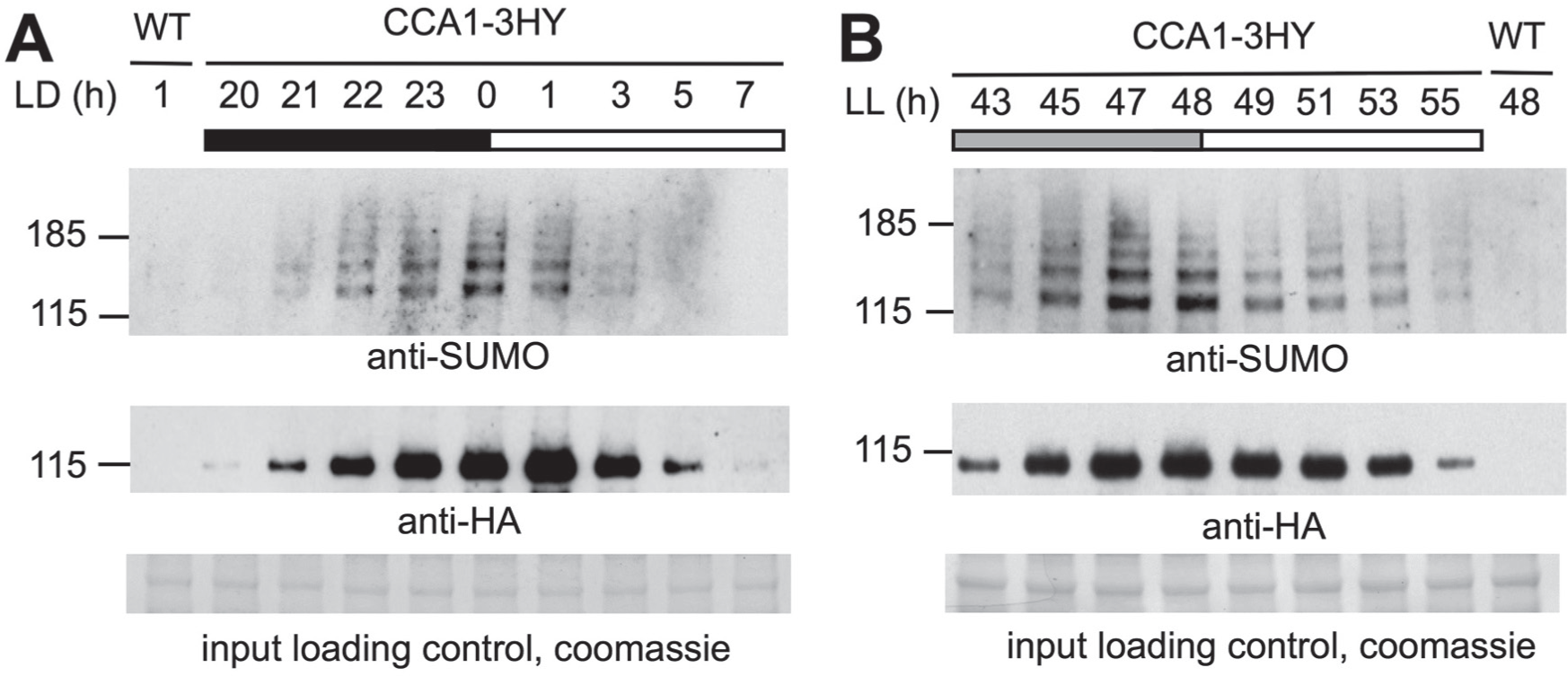
The clock transcription factor CCA1 is sumoylated in planta. Plants were sampled at the indicated times under light:dark cycles (LD; A) or upon transfer to constant light (LL; B). In both, time is hours since the last dawn. CCA1-3HY was immunoprecipitated and detected on immunoblot using anti-HA antibody (mid panel). Sumoylation of CCA1 was detected with anti-SUMO (upper panel). Input protein loading control in lower panel.
Heterologous Sumoylation of CCA1
An Escherichia coli sumoylation system (Okada et al., 2009) was used for heterologous sumoylation of CCA1 with a His-tag or an MBP-tag. A culture of BL21 (DE3) cells expressing pCDFDuet-AtSUMO1-(GG or AA)-AtSCE1, pACYCDuet-AtSAE1a-AtSAE2, and pET28a-CCA1-His/MBP was grown ~3 h to OD600 0.8 at 37 °C. Overnight expression at 17 °C was induced by adding 200 µM IPTG.
For MBP-tag purification, cells were pelleted and resuspended in 60 mL ice-cold wash buffer (1x phosphate-buffered saline [PBS], 1x Complete protease inhibitor [Roche], 1 mM DTT, 20 mM MgSO4), aliquoted into 10-mL tubes, and flash frozen after the addition of 0.1% Triton. Per experiment, 2 tubes were thawed by rotation at room temperature in the presence of lysozyme (1 mg/mL) and benzonase nuclease (0.5 µL/ml, Merck Millipore). When lysed, the cell extract was cleared by centrifugation for 60 min at 50228 g. To the supernatant, 0.4 mL amylose beads was added and incubated rotating in cold room for ~1 h. Beads were pelleted and washed in wash buffer 2 (HBS-EP+ buffer, 1 mM TCEP, 1x Roche Complete protease inhibitors). Washed beads were pelleted and eluted in 5 mL elution buffer (wash buffer, 20 mM maltose) for 10 min. Beads were removed by centrifugation, and the supernatant was dialyzed overnight to remove the maltose.
For His-tag purification, the samples were pelleted and snap frozen. Samples were thawed on ice and resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 1% Triton X100, 5% glycerol, 1 mM DTT, 1x protease inhibitor without EDTA [Roche], 1 mM PMSF, pH 8.0) with 1 mg/mL lysozyme. The cells were further lysed by sonication. RNase A and DNase I were added to reduce the viscosity. Cell debris was pelleted by centrifugation at 20,000 g for 20 to 30 min or 4000 g for 60 min at 4 °C. The supernatant was collected for His-purification. Ni-NTA agarose beads were equilibrated in lysis buffer. The protein extract was applied to the beads, and the mix was incubated 1 to 2 h rotating at 4 °C. The beads were pelleted by centrifugation at 100 g at 4 °C for 1 min, and the supernatant was removed. The beads were washed 3 to 5 times in wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole 1x protease inhibitor without EDTA [Roche], 1 mM PMSF, pH 8.0) before eluting the proteins from the beads with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0). A second aliquot of elution buffer was added to the beads, they were heated to 96 °C for 10 min, and the eluate was collected.
For blotting (MBP- and His-purified), protein samples were separated on 4% to 12% polyacrylamide gels (Invitrogen Novex) and transferred to nitrocellulose membranes using the iBlot system (Invitrogen). The primary antibody was anti-T7-tag (1:15,000, Novagen).
Protein Identification by Mass Spectroscopy
The His-purified protein samples were concentrated by TCA precipitation. One volume ice-cold 20% TCA was added to the sample and incubated on ice for 30 min. Protein was pelleted by centrifugation at 17,000 g at 4 °C for 15 min. The pellet was washed in ice-cold acetone and centrifuged for 5 min at 17,000 g at 4 °C twice, then air dried. The pellet was resuspended in 1x LDS sample buffer (Invitrogen) with 100 mM DTT and separated on a 4% to 12% polyacrylamide gel (Novex, Winston-Salem, NC) and stained with SimplyBlue Safestain (Invitrogen). The bands of interest were excised and prepared for mass spectrometric analysis. Gel slices were shrunk by twice adding 200 µL methanol for 10 min and replacing it with 200 µL 50 mM ammonium bicarbonate (ABC) for 10 min. Two hundred microliters of methanol was added a third time, replaced by 100 µL 20 mM DTT, and the samples were incubated at 60 °C for 30 min. After shrinking the gel pieces in methanol again, 100 µL 55 mM NEM was added and incubated at room temperature for 60 min to block cysteine residues. After incubating the gel pieces twice in methanol, they were digested overnight with 20 µL trypsin in 80 µL 50 mM ABC. The supernatant was saved, and additional peptides were extracted by incubating the gel pieces for 15 min in 80 µL 1% FA, followed by 80 µL 1% FA in 50% methanol, and 80 µL methanol twice. The solutions were combined, dried in a SpeedVac, and resuspended in methanol for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis. LC-MS/MS data were analyzed with MASCOT. The SUMO Interaction Motif at CCA1 amino acids 598-593 was identified using the online GPS-SUMO tools available at http://sumosp.biocuckoo.org/online.php.
Site-directed Mutagenesis and Cloning of Hepta-mutant
The putative sumoylation sites (K117, K124, K393, K483, K559, K595, K598) identified by mass spectrometry were mutated from lysine to arginine by site-directed mutagenesis. The primers, containing a single nucleotide mutation, were designed using Quickchange software (Agilent Technologies, Santa Clara, CA). Template DNA was removed by DpnI digestion from the PCR product, which was transfected to OneShot E. coli cells (Invitrogen). The N-terminal triple mutant of CCA1 was generated by successive site-directed mutagenesis reactions of K117, K124, and K393 in one pET28a-AtCCA1 plasmid (pET28a-CCA1-Nterm) and the quadruple C-terminal mutant containing substitutions of K483, K559, K595, and K598 in another pET28a-AtCCA1 plasmid (pET28a-CCA1-Cterm). The 2 parts of CCA1 were combined through restriction digest at the AgeI and XhoI sites and subsequent T4 DNA ligation to generate the hepta-mutant.
Protoplast Assays
For confocal imaging vectors, the CCA1pro:CCA1-YFP DNA sequence was amplified from a vector obtained from Laszlo Bodnar (Hungarian Academy of Sciences), adding the restriction sites NotI and StuI, and this product was ligated into pOmega+ to give CCA1pro:CCA1-YFP. For localization studies of SUMO fusions, the coding sequences of SUMO1 (At4g26840), SUMO2 (At5g55160), and SUMO3 (At5g55170) were amplified to add BamHI sites at the 5′ and 3′ ends, and the fragment was introduced at the BamHI site, yielding an N-terminal fusion protein of SUMO to CCA1 in the pOmega CCA1pro:CCA1-YFP plasmid. Overexpresser constructs were created by replacing the CCA1 promoter sequence with the 35S promoter sequence amplified from pCambia 35Spro:NanoLUC kindly provided by Andrew Millar (University of Edinburgh), using PCR primers to introduce NotI and BamHI sites that allowed cloning into the pOmega vector set. The YFP controls CCA1pro:YFP and 35Spro:YFP were generated by excising the coding sequences of SUMO and CCA1 with BamHI and HindIII, blunting the linearized vectors (NEB E1201S), and religation with T4 DNA ligase. Protoplasts for confocal imaging were generated as described by Hansen and van Ooijen (2016), transfected with 30 to 40 µg plasmid, and resuspended in 1 mL W5 solution (Hansen and van Ooijen, 2016) before being transferred to a 6-well plate pretreated with 1% bovine serum albumin in W5 solution. The protoplasts were kept in 30 µmol m–2 s–1 white light overnight at 21 °C. A 35 µL aliquot of protoplasts was imaged on a Leica SP5 in photon counting mode with a 20× objective. The excitation wavelength was 514 nm for detection of YFP signal and 594 nm for chloroplast autofluorescence detection. The images were analyzed in Fiji (Schindelin et al., 2012).
For circadian gene expression assays in protoplasts, techniques were modified from Kim and Somers (2010) identically to what is detailed in the accompanying article.
Cell-free Protein Degradation Assay
The cell-free protein degradation assay was adapted from Más et al. (2003). Col-0, ots1 ots2, CCA1-3HY, and CCA1-3HY ots1 ots2 adult plants grown at 12 h light/12 h dark were sampled at dawn. Plant material was homogenized in liquid nitrogen and protein extracted in extraction buffer (50 mM Tris-HCl pH 7.4, 300 mM NaCl, 10 mM MgCl2, 5 mM DTT, 5 mM ATP, 0.2% NP-40, 0.1% Triton X-100). Cell debris was removed by centrifugation and filtration through a 0.22-µm filter. Protein content was quantified by Bradford assays. The samples were transferred to a room temperature water bath, and at the indicated times, aliquots were transferred to fresh tubes containing 4xLDS containing DTT and stored at −20 °C immediately. Samples were separated on 4% to 12% polyacrylamide gels (Invitrogen Novex), transferred to nitrocellulose membranes using the iBlot system (Invitrogen), and detected using an anti-HA antibody (1:5000, abcam No. ab20084).
ChIP Analyses
Chromatin immunoprecipitation (ChIP) was based on procedures described by Nagel et al. (2015). Briefly, 0.5 g 3-week-old seedlings were submerged in 1% formaldehyde, and a vacuum (–70 kPa) was drawn for 2 × 15 min. Glycine was added to a final concentration of 0.127 M to quench the cross-linking, and a vacuum (–70 kPa) was drawn for a further 5 min. Seedlings were washed once in ice-cold PBS and snap frozen in liquid N2. Tissue was ground in nuclei extraction buffer (100 mM MOPS pH 7.6, 10 mM MgCl2, 250 mM sucrose, 5% Dextran T-40, 2.5% Ficoll 400, 40 mM β-mercaptoethanol, 20 mM NEM, 50 µM MG132, 50 µM PR-619, 1x Complete protease inhibitor [Roche], 1x PhosStop [Roche]) and filtered through nylon mesh, and the nuclei were pelleted by centrifugation at 10,000 g at 4 °C. The nuclei were lysed in nuclei lysis buffer (50 mM Tris-HCl pH 8, 10 mM EDTA pH 8, 1% sodium dodecyl sulfate [SDS]) and diluted in ChIP dilution buffer (16.7 mM Tris-HCl pH 8, 167 mM NaCl, 1.2 mM EDTA pH 8, 0.01% SDS, 20 mM NEM, 50 µM MG132, 50 µM PR-619, 1x Complete protease inhibitor [Roche], 1x PhosStop [Roche]) before the DNA was sheared by sonication (Diagenode BioRuptor Plus). The samples were further diluted, and Triton X-100 was added to give a concentration of 1.1%. Cellular debris was removed by centrifugation, and samples were precleared with Protein G agarose beads (Invitrogen). Of the samples, 1/50 was kept as input before the remainder was split in two. Twenty micrograms of GFP antibody (abcam, ab290) was added to one aliquot, the other was used as mock. Samples were incubated for 5 h rotating at 4 °C. Protein G beads were added, and the samples were incubated for 2 h. The beads were collected by centrifugation and washed once in a series of ice-cold wash buffers: low-salt buffer (0.1% SDS, 1% Triton X-100, 20 mM Tris-HCl pH 8, 2 mM EDTA pH 8, 150 mM NaCl), high-salt buffer (0.1% SDS, 1% Triton X-100, 20 mM Tris-HCl pH 8, 2 mM EDTA pH 8, 500 mM NaCl), LiCl buffer (250 mM LiCl, 1% NP-40, 1% sodium deoxycholate, 10 mM Tris-HCl pH 8, 1 mM EDTA pH 8), and 0.5x TBE. DNA was precipitated from the input samples by ethanol precipitation. Ten percent Chelex resin was added to the input, and IP samples and the samples were incubated at 95 °C for 10 min, followed by Proteinase K treatment for 30 min at 55 °C. DNA was purified from the supernatant using a PCR purification Kit (Qiagen, Hilden, Germany). DNA was quantified by quantitative reverse transcription polymerase chain reactions (qRT-PCR) with primer pairs listed in Supplementary Table S1. Abundance of DNA in the IP samples was calculated as % of Input = (2–ΔCt) × 100%.
Electrophoretic Mobility Shift Assay
Biotin-labeled DNA duplexes PRR9 or mutPRR9 (
Surface Plasmon Resonance
Surface plasmon resonance (SPR) was performed as published previously (O’Neill et al., 2011) with minor modifications to reflect the higher sensitivity of the Biacore T200 (GE Healthcare, Chicago, IL) used in this study. Biotinylated oligonucleotides were immobilized on a pretreated streptavidin chip to 3 response units by injection of 1 nM for at 5 µL/min. Reverse oligonucleotides were bound by injecting 10 µM at 5 µL/min until saturation. One flow cell was always kept blank for reference subtractions. Protein and buffers were filter-sterilized before use; dilutions of protein were made in HBS-EP+ buffer (GE Healthcare), injected at 75 µL/min for 90 sec, and dissociated for 600 sec. Surfaces were regenerated with 3 cycles of 60 sec of regeneration buffer (0.1% SDS, 3 mM EDTA) in between each protein run. Steady-state affinity analyses were performed to deduce KD using BiaEvaluation software (GE Healthcare) using the standard 1-site binding model. Chi-squared values are reported for all fits (any value below 2 indicates a reliable fit). Curves were exported and plotted in GraphPad Prism.
Results
CCA1 Is Sumoylated In Vivo
Targets of sumoylation are predominantly nuclear localized and often function in DNA transcription, chromatin modification, and other nuclear-related processes (Miller et al., 2010). TTFL timekeeping in plants involves numerous proteins that fit that description. Dynamic posttranslational regulation of CCA1 is known to affect its function (Yakir et al., 2009), and it belongs to the MYB-like superfamily of transcription factors that are particularly overrepresented among sumoylated proteins (Miller et al., 2010). To investigate whether CCA1 is modified by SUMO in vivo, we employed Arabidopsis plants that express a fusion protein of CCA1 with a triple HA-tag and the YFP epitope from the native CCA1 promoter (Yakir et al., 2009; referred to in this article as CCA1-3HY). This transgene rescues the period effect of the cca1 mutation (Yakir et al., 2009). Plants were grown in light-dark cycles, harvested over a time series spanning the CCA1 expression window, and subjected to immunoprecipitation in an extraction buffer inactivating SUMO protease activity. The circadian accumulation of CCA1 was confirmed using immunoblotting with an anti-HA antibody (Fig. 1A, middle panel), and dynamic sumoylation of CCA1 was observed over this time series using an antibody against SUMO isoforms 1 and 2 (anti-SUMO; upper panel). To test whether sumoylation of CCA1 relies on the light-dark cycle, this experiment was repeated under circadian conditions of constant light and temperature. Under these conditions, both rhythmic accumulation of CCA1 as well as rhythmic sumoylation of CCA1 persisted (Fig. 1B), even though total sumoylation remained stable over this experiment (
In either condition, multiple distinct bands are visible using a SUMO antibody on CCA1 pulldowns (Fig. 1), at molecular weights that are consistent with either SUMO modification at multiple sites, SUMO chain formation, or SUMO modification(s) combined with additional modifications.
Identifying Sumoylation Sites in CCA1
To identify potential sumoylation sites in CCA1, a recombinant expression system was employed to allow easy production of larger quantities of CCA1. Previously, a system was reported to reconstitute part of the Arabidopsis sumoylation pathway in E. coli, which has no endogenous sumoylation pathway (Okada et al., 2009). Recombinant T7- and His-tagged CCA1 protein was expressed in this system to verify that the protein contains sumoylation sites and to subsequently identify these by mass spectrometry. As a positive control, the Arabidopsis transcription factor MYB30 was included, a known sumoylation target (Zheng et al., 2012). Target proteins were expressed along with 2 variants of SUMO; an intact conjugatable SUMO (+) or, as a negative control, an unconjugatable SUMO (–) lacking the double glycine conjugation motif (Okada et al., 2009). Upon immunoblotting using anti-T7 antibody, CCA1 and MYB30 were detected in His-tag affinity purified protein extracts. Clear sumoylation of the positive control MYB30 was observed (Fig. 2A) using intact SUMO (+) and not in negative controls (–), indicating that the heterologous coexpression system worked as expected. Sumoylation of CCA1 was evident in this assay, verifying that CCA1 can indeed be sumoylated.
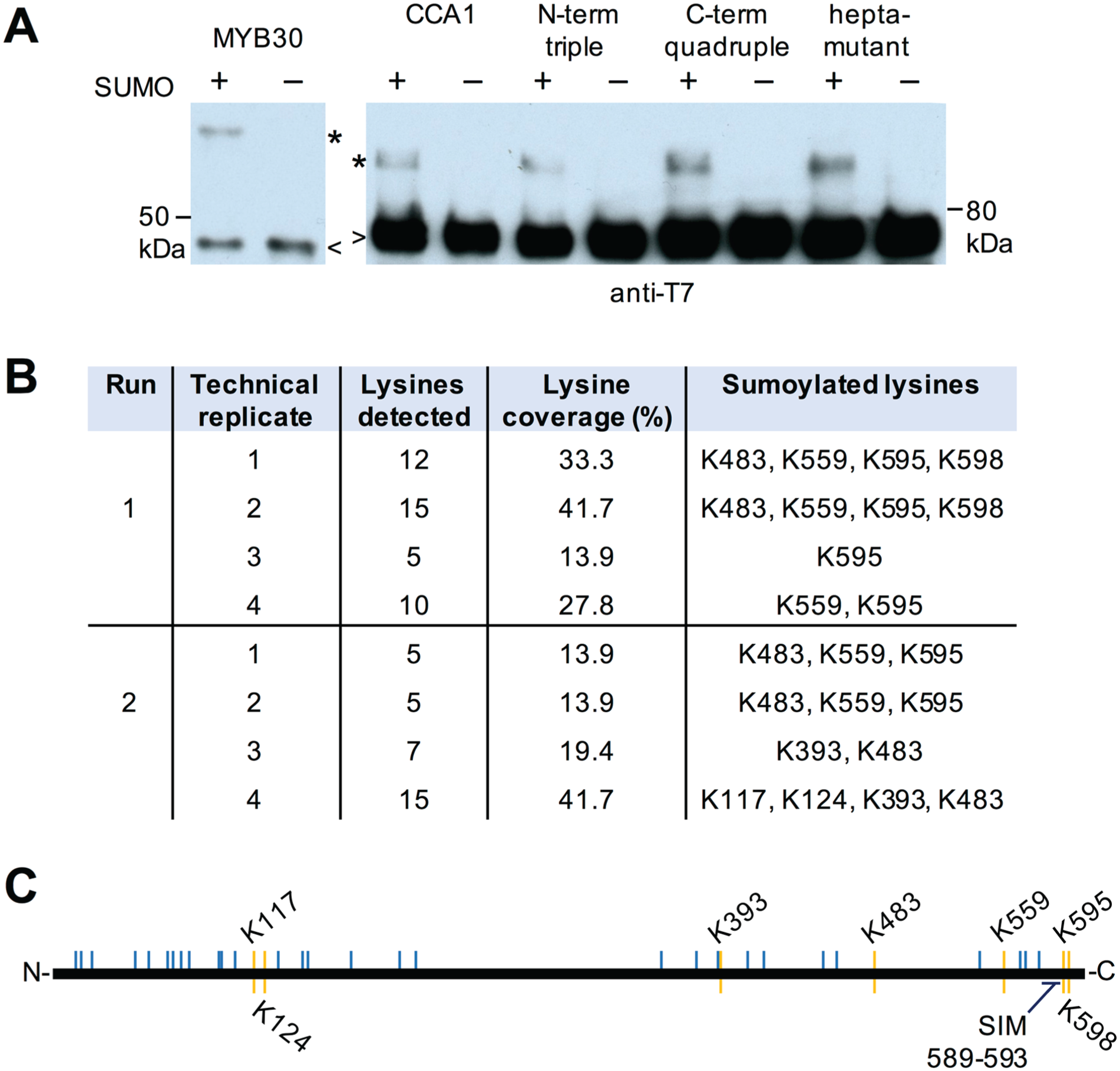
CCA1 is sumoylated in E. coli on several lysine residues. (A) CCA1 and positive control MYB30 protein was coexpressed with the Arabidopsis sumoylation machinery and wild-type (+) SUMO or nonconjugateable SUMO (–) in E. coli, and His-tag affinity purified protein was detected on immunoblots using an anti-T7 tag antibody. Asterisks indicate the expected molecular weight band of sumoylated target protein; arrowheads indicate the nonsumoylated bulk of the protein. (B) Sumoylation sites identified by mass spectrometry in 2 independent experiments each with 4 replicates. (C) Schematic of CCA1 protein with lysines indicated by blue lines and observed sumoylated lysines by yellow lines. A predicted SUMO interaction motif (SIM) is also indicated. The amino acid number is indicated above and below the schematic.
Identification of sumoylation sites was attempted by mass spectrometric analysis. CCA1 was sumoylated in E. coli using a variant of SUMO suitable for mass spectrometric detection. In this variant, the threonine immediately upstream of the double glycine motif has been substituted with an arginine to introduce a trypsin cleavage site. In LC-MS/MS, lysines modified with this SUMO variant are distinguishable by the double glycine modification. The CCA1 peptides detected in 2 independent experiments covered between 13.9% and 41.7% of the lysines in the full amino acid sequence (Fig. 2B). In total, 7 sumoylation sites were detected (Fig. 2B, C). To validate whether any of these lysines were the predominant sumoylation sites, point mutations from lysine to arginine were introduced. Neither single mutant showed a decrease in sumoylation in E. coli (data not shown), and therefore, several sites were mutated in a single construct: either the 3 most N-terminal, the 4 most C-terminal, or all 7 lysines. No obvious differences in sumoylation were detected in any of these protein variants, suggesting that these sites do not account for the bulk of sumoylation detected on CCA1 and that additional sumoylated lysines have remained undetected by our mass spectrometric analyses.
Functional Relevance of CCA1 Sumoylation
The most unequivocal way of testing the contribution of differential sumoylation of CCA1 to the circadian clock system would be to complement the cca1 lhy double mutant with wild-type CCA1 versus a nonsumoylatable version of CCA1. Since it was not possible to generate a nonsumoylatable version of CCA1 (Fig. 2B), we tested instead the characteristics of the CCA1 protein in planta in the wild-type background compared with a SUMO protease double mutant (ots1 ots2). In this background, increased global sumoylation has been reported (Conti et al., 2008; Suppl. Fig. S1B) as well as a long-period circadian phenotype (see accompanying article). To test the expression profile of CCA1 target promoters in this background, a protoplast assay was used (Kim and Somers, 2010). While the long period phenotype was consistent across reporters, the altered expression levels suggest that increased global sumoylation affects more than 1 point in the complex clock network (Suppl. Fig. S1C, D), at least as reported in this protoplast assay.
Sumoylation can regulate the localization, stability, or activity of a protein, as well as its intermolecular interactions (Wilkinson and Henley, 2010; Mazur and van den Burg, 2012). CCA1 accumulates in the nucleus at subjective dawn (Yakir et al., 2011), and we first tested the effects of sumoylation on this nuclear localization. YFP-tagged CCA1 was expressed in protoplasts of wild-type plants (Col-0) versus ots1 ots2 using either the native, circadian CCA1 promoter or the constitutive 35S promoter. No difference in CCA1 localization was observed between the genotypes with either promoter (Fig. 3A), suggesting that changes to overall sumoylation levels do not influence localization of CCA1. To verify that result, the effects of N-terminal fusion of SUMO isoforms to the CCA1 protein were studied, as expression of a fusion protein of SUMO with a target protein can mimic constitutive sumoylation of that target (Georges et al., 2011). No difference in localization was observed between CCA1 and SUMO-CCA1 (Fig. 3B). In conclusion, neither the predominant nuclear localization nor the detectable amount of protein that remains in the cytoplasm were differential between genotypes or CCA1 constructs, supporting the notion that either CCA1 sumoylation is not differential in this ots1 ots2 background or sumoylation does not have a major influence on CCA1 protein localization.
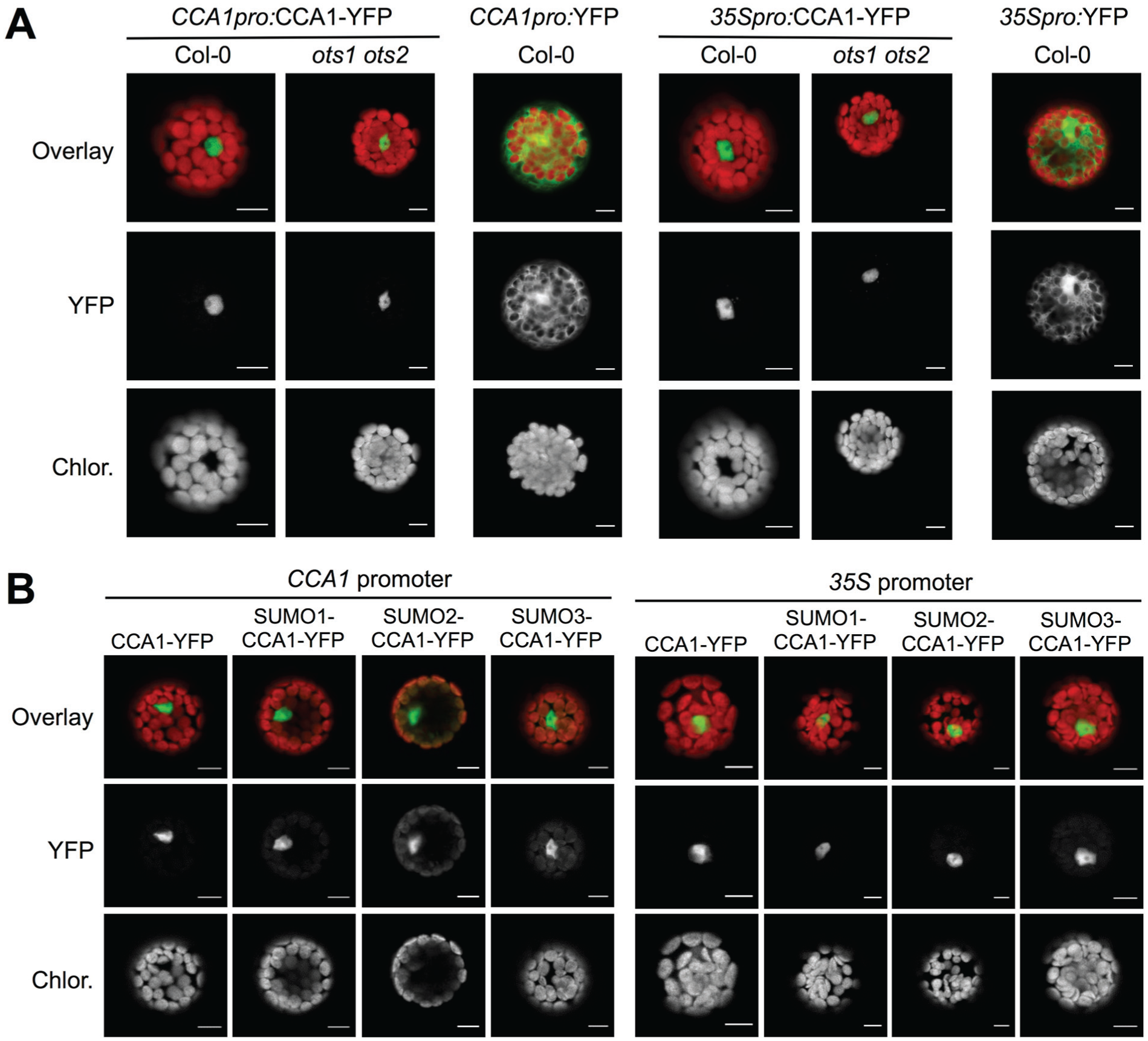
CCA1 localization is not altered in the ots1 ots2 mutant background. (A) CCA1-YFP fusion protein or control YFP protein were expressed from the indicated promoters in the Col-0 and ots1 ots2 backgrounds and imaged by confocal microscopy. (B) N-terminal fusion to SUMO isoforms does not change localization of CCA1. Expression of CCA1 in Col-0 protoplasts as fusion proteins with C-terminal YFP tag and N-terminal SUMO isoforms as indicated, expressed from the CCA1 promoter (left) or 35S promoter (right), and imaged by confocal microscopy. Individual YFP (middle panel) and chlorophyll (lower panel) channels are provided in gray scale and as an overlay in color (upper panel). Scale bar = 10 µm.
Second, sumoylation can affect target protein stability (Miura et al., 2009; Zheng et al., 2012), and the dynamics of several clock components were previously demonstrated to be fine-tuned through regulation of their stability (Más et al., 2003; Song and Carré, 2005; Kim et al., 2007; Para et al., 2007; Baudry et al., 2010; van Ooijen et al., 2011). To test whether changes in global sumoylation affect CCA1 stability, the CCA1-3HY line was crossed to the ots1 ots2 background to allow comparative stability assays. Total protein lysates from both lines were generated at the peak of CCA1 accumulation, and CCA1 protein stability was analyzed by immunoblotting (Fig. 4A, B). No significant changes to protein stability were observed between the 2 genetic backgrounds, indicating either that CCA1 sumoylation levels are not altered in the ots1 ots1 background or that sumoylation has no major influence on CCA1 stability.
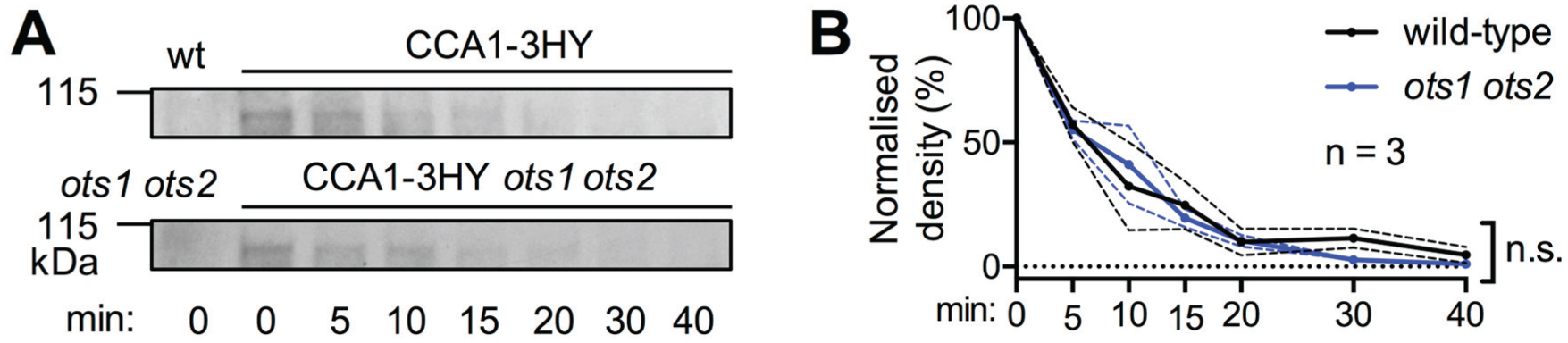
CCA1 protein stability is not altered in the ots1 ots2 mutant background. (A) Stability of CCA1-3HY protein was analyzed in ots1 ots2 mutant and control background plants. Aliquots of total protein extracts were sampled at the indicated time points and immunoblotted. Representative blots of 3 biological replicates. (B) Densitometry data of blots in (A) and 2 additional biological replicates (mean value ± SEM). Paired t test; p = 0.5.
Third, sumoylation can regulate the specific activity of target proteins involved in transcription (Lee et al., 2008; Miura et al., 2009; Zheng et al., 2012; Li et al., 2013). The main activity of CCA1 in the clock is to inhibit or activate the expression of genes through binding to a conserved promoter element called the “evening element,” found in CCA1-controlled gene promoters such as the PRR9 promoter (Hsu and Harmer, 2013). ChIP experiments were carried out to elucidate whether binding activity of CCA1 to the PRR9 promoter sequence was differential between wild-type and ots1 ots2 backgrounds. Seedlings were harvested at dawn, the samples were cross-linked, and the abundance of the PRR9 promoter sequence in the immunoprecipitated CCA1 complexes was analyzed using qRT-PCR. A greater enrichment was consistently observed in the wild-type compared with ots1 ots2 background (Fig. 5A; Suppl. Fig. S1E). Nonspecific binding was not observed in negative control Col-0 plants lacking the CCA1-3HY transgene nor in no-antibody controls, and no signal was detected on control DNA fragments (actin). Combined, this verifies that reduced binding of CCA1 to the PRR9 evening element is observed in the SUMO protease-defective line ots1 ots2 compared with control plants.
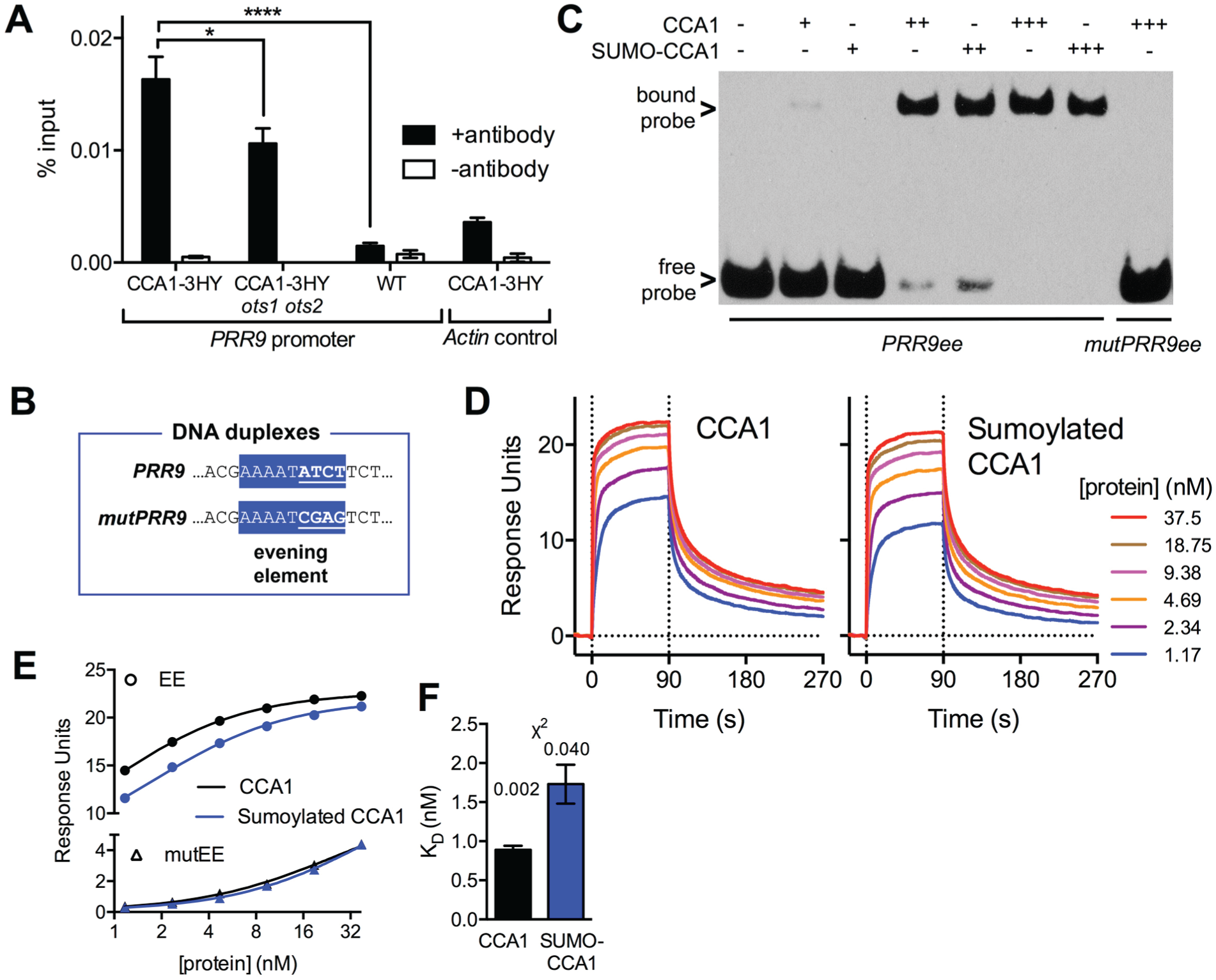
DNA binding of CCA1 is reduced in the ots1 ots2 mutant background in vivo and directly by sumoylation in vitro. (A) Indicated plant lines were harvested at dawn and subjected to chromatin immunoprecipitation of CCA1-3HY along with negative controls without antibody. PRR9 promoter sequences were detected using qRT-PCR along with negative controls of the actin sequence (mean ± SEM of 4 technical replicates, ordinary 1-way analysis of variance with Dunnett’s multiple comparisons test; p-value: <0.0001 [****]; <0.05 [*]). (B) The evening element in the PRR9 promoter sequence (PRR9ee) is highlighted in blue. Four nucleotides were mutated (mutPRR9ee) to act as a negative control for CCA1 binding (underlined; mutPRR9ee). (C) Electrophoretic mobility shift assay (EMSA) of biotinylated DNA duplexes of the PRR9 evening element (PRRee) or a mutated negative control (mutPRRee) with 3 concentrations of sumoylated or nonsumoylated CCA1 protein (1 nM, 10 nM, and 45 nM). Arrows indicate free, unbound probe and probe bound in protein-DNA complexes. (D) Surface plasmon resonance (SPR) experiments detecting the binding of sumoylated or nonsumoylated CCA1 protein to the PRR9 evening element sequence during a 90-sec interaction (0-90 sec) at the indicated protein concentrations, along with the following dissociation (90-270 sec). (E) SPR analysis of recombinant sumoylated (blue) and nonsumoylated CCA1 (black) protein, binding to the wild-type evening element (EE; circles) or mutated sequence (triangles, mutEE). The lines indicate the steady-state affinity fits to the data points. (F) Binding constant KD as determined by steady-state affinity analyses of the data in (E). Chi-squared values are provided, indicating goodness of fits.
Sumoylation of CCA1 Directly Suppresses DNA Binding
A limitation of the approach taken above is that it remains unclear whether (part of) the changes to overall clock dynamics or even to CCA1 promoter binding in the ots1 ots2 background are due to potentially differential sumoylation of CCA1 directly or from altered sumoylation of additional proteins involved in the intricate clock feedback system. The only unequivocal way to establish whether direct effects exist for CCA1 sumoylation is to use purified recombinant CCA1 protein in binding assays without any other proteins present. Sumoylated and nonsumoylated CCA1 protein (as in Fig. 2) was purified from E. coli and compared in in vitro DNA binding assays. To test whether sumoylation has a direct effect on DNA binding, biotin-labeled oligonucleotide duplexes containing the evening element of the PRR9 promoter (PRR9ee, Fig. 5B) or a mutated negative control probe (mutPRR9) were added to multiple concentrations of sumoylated or control CCA1 protein. DNA duplexes and protein-DNA complexes were resolved in an electrophoretic mobility shift assay (EMSA). At a highest protein concentration (45 nM, Fig. 5C), all of the PRR9ee probe was bound to either sumoylated or nonsumoylated CCA1, indicating that both can bind DNA. However, at intermediate CCA1 protein concentrations (10 nM), more free probe remained in the reactions with sumoylated compared with nonsumoylated CCA1. At low protein concentrations (1 nM), bound probe was detected only in reactions containing nonsumoylated CCA1. This demonstrates that the binding affinity of CCA1 to the evening element in the PRR9 promoter is reduced by sumoylation of CCA1. The shift indicative for a protein-DNA complex was not observed with negative control probe, verifying the binding of CCA1 to the evening element is specific in this assay.
To confirm this result and to quantify the difference in DNA binding affinity, SPR analyses were conducted. Biotinylated DNA duplexes of PRR9ee and mutPRR9ee were immobilized on streptavidin-coated SPR chip surfaces, and binding of a wide concentration range of recombinant protein was analyzed. Consistent across a protein concentration range, reduced binding of sumoylated CCA1 was observed versus control CCA1 (Fig. 5D). To quantify the difference in affinity to the 2 heterologous proteins, the steady-state affinity was fitted to the binding curves (Fig. 5E). Nonspecific binding to the mutPRR9ee probe was low and identical between the 2 protein samples. The binding constant KD in the control protein was lower (0.9 nM) than in the sumoylated CCA1 protein (1.7 nM; Fig. 5F), indicating that sumoylation of CCA1 directly reduces DNA binding.
Discussion
Phosphorylation of CCA1 by CK2 affects circadian period length, and this phosphorylation is necessary to form the dimers required for DNA binding (Daniel et al., 2004). Here we demonstrated that CCA1 is additionally modified by sumoylation (Fig. 1). It is important to note that in vivo and in vitro (Fig. 5), only a small amount of the total CCA1 pool is sumoylated, yet the function of the total pool is significantly affected. In fact, this is perfectly consistent with all previous observations. Without exceptions, the effects of sumoylation on target proteins are mediated by only a small percentage of the total pool of that particular target protein in vivo. This phenomenon is referred to as the “SUMO enigma” (Lee et al., 2008; Wilkinson and Henley, 2010), first coined in 2005 (Hay, 2005). Especially enigmatic, this phenomenon occurs not only in vivo but also in vitro: sumoylation of a small percentage of the mammalian RNA binding protein La produced in E. coli results in a striking change to the binding activity, as detected by electrophoretic mobility shift (Kota et al., 2016). In this respect, CCA1 appears to be a typical sumoylation target both in vivo and in vitro. Like CCA1, La is additionally controlled by CK2 phosphorylation. Interplay between phosphorylation and sumoylation is a widespread mechanism for modulating the activity or function of a protein (Gareau and Lima, 2010; Nukarinen et al., 2017).
In plants, phosphorylation-dependent sumoylation was demonstrated on the brassinosteroid-responsive transcription factor CESTA (Khan et al., 2014) and on the master regulator of plant immunity, NPR1 (Saleh et al., 2015). Sumoylation is predominantly a nuclear modification (Miller et al., 2010), and within the nucleus, sumoylation of CESTA induces the assembly of nuclear bodies that are proposed to promote protein-protein interactions required for the brassinosteroid response (Khan et al., 2014). Although sumoylation does not affect the predominantly nuclear localization of CCA1 (Fig. 3), it could still have effects on suborganellar localization. Confocal imaging of the mammalian CLOCK/BMAL1 transcription factor complex revealed that SUMO is required for localization to nuclear bodies within the nucleus (Lee et al., 2008). Further investigation into the associations between phosphorylation and sumoylation of CCA1 could reveal whether these modifications are codependent and act as a regulatory mechanism of transcriptional regulation by CCA1 within the nucleus.
CCA1 binds specifically to the highly conserved Evening Element promoter sequence, but the resultant phase of expression is dependent on flanking DNA sequences (Harmer and Kay, 2005). Furthermore, recent ChIP-Seq studies identified additional binding motifs including morning-phase specific sequences (Nagel et al., 2015). How CCA1 is directed toward one or the other motif is still poorly understood but could result from differential protein-protein interactions that might be affected by sumoylation (Ouyang et al., 2009; Wilkinson and Henley, 2010; Saleh et al., 2015). Although our studies were limited to the effects of sumoylation on the binding of CCA1 to the Evening Element (Fig. 5), it will now be useful to identify differential effects on binding to other sites (either directly or through differential interactions with co-regulators). Clearly, given the complexity of (1) the feedback loop structure, (2) posttranslational regulation of clock proteins, and (3) the number of CCA1 binding elements, a complicated pattern is bound to emerge. Elucidating the full role of sumoylation on these aspects would ultimately require detailed mathematical modeling to predict the poorly understood difference between resultant expression phase as well as repression or induction.
Sumoylation regulates timekeeping in plants (see accompanying article), and we have now established that the crucial plant transcription factor CCA1 is a sumoylation target. Although it is possible that part of the overall in planta effect of sumoylation mutants on the clock is via CCA1, circumstantial evidence exists that sumoylation modulates additional clock proteins. First, the evening-phased clock components EARLY FLOWERING 3 (ELF3) and GIGANTEA (GI) were identified as SUMO targets (Miller et al., 2010; López-Torrejón et al., 2013). However, both results were obtained upon exposure to nonphysiological heat stress, and a functional role has not been demonstrated in either case. Sumoylation does functionally influence red light signaling via PhyB and photomorphogenesis via COP1, but whether these events influence timekeeping is not evident (Sadanandom et al., 2015; Lin et al., 2016). Based on these studies and the results presented here, it remains unclear whether any percentage of the overall clock defect in the ots1 ots2 background is mediated via CCA1. What we have unequivocally shown, however, is that the central plant clock transcription factor CCA1 undergoes dynamic sumoylation, which directly alters the binding affinity to the evening element. The CCA1 protein binds to the promoter of >1500 genes in Arabidopsis (Nagel et al., 2015; Kamioka et al., 2016), indicating that these observations could have wide consequences for the growth and health of plants.
Footnotes
Acknowledgements
The authors would like to thank Rachel Green for the CCA1-3HY plant line, Steven Spoel is acknowledged for help with the EMSA and Michael Skelly for discussing ChIP experiments. This research was supported by the Royal Society Research grants RS120372 and RS140275 and by the Royal Society University Research Fellowship UF110173 (all awarded to G.v.O.).
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.