
Abstract
The circadian clock generates daily cycles of gene expression that regulate physiological processes. The liver plays an important role in xenobiotic metabolism and also has been shown to possess its own cell-based clock. The liver clock is synchronized by the master clock in the brain, and a portion of rhythmic gene expression can be driven by behavior of the organism as a whole even when the hepatic clock is suppressed. So far, however, there is relatively little evidence indicating whether the liver clock is functionally important in modulating xenobiotic metabolism. Thus, mice lacking circadian clock function in the whole body or specifically in liver were challenged with pentobarbital and acetaminophen, and pentobarbital sleep time (PBST) and acetaminophen toxicity, respectively, was assessed at different times of day in mutant and control mice. The results suggest that the liver clock is essential for rhythmic changes in xenobiotic detoxification. Surprisingly, it seems that the way in which the clock is disrupted determines the rate of xenobiotic metabolism in the liver. CLOCK-deficient mice are remarkably resistant to acetaminophen and exhibit a longer PBST, while PERIOD-deficient mice have a short PBST. These results indicate an essential role of the tissue-intrinsic peripheral circadian oscillator in the liver in regulating xenobiotic metabolism.
The efficacy and toxicity of many drugs vary with time of day. These time-dependent effects could be due to differences in pharmacokinetics (absorption, distribution, metabolism, or excretion) or pharmacodynamics (rhythms in cellular sensitivity) (Dallmann et al., 2014).
A network of circadian oscillators exists throughout the body (Mohawk et al., 2012; Weaver and Emery, 2013). The suprachiasmatic nuclei (SCN) of the hypothalamus contain the pacemaker controlling behavioral and physiological rhythms. The SCN receive information about the day/night cycle from the retina and thus are synchronized to the environment. The SCN act as a transducer, generating a variety of behavioral, physiological, and hormonal rhythms that entrain cell-autonomous circadian clocks in peripheral tissues (Mohawk et al., 2012).
The molecular circadian oscillator is a transcriptional-translational feedback loop that oscillates with a cycle length of approximately 24 h. At the core of the oscillator are the bHLH/PAS domain-containing transcription factors, CLOCK and BMAL1. These proteins heterodimerize and enhance transcription through E-box elements in regulatory regions of responsive genes. Among their targets are the Period (Per1-3) and Cryptochrome (Cry1-2) genes, whose protein products feed back to inhibit CLOCK:BMAL1 activity. Numerous transcription factors are rhythmically regulated in parallel, secondarily establishing rhythmicity in their target genes. Assessment of gene expression rhythms in several tissues reveal that ~10% of all genes and proteins are expressed with 24-h rhythmicity, with key, rate-limiting steps often among the rhythmic genes (Koike et al., 2012; Akhtar et al., 2002; Storch et al., 2002; Panda et al., 2002; Mauvoisin et al., 2014).
The 3 PAR-domain–containing, basic leucine zipper (PARbZip) transcription factors, D-site binding protein (DBP), thyrotroph embryonic factor (TEF), and hepatic leukemia factor (HLF), are rhythmically expressed in liver under direct control of the CLOCK:BMAL1 complex (Ripperger and Schibler, 2006). Mice lacking these 3 genes have a greatly reduced capability to metabolize xenobiotics due to loss of their activating function on cytochrome P450 oxireductase (POR) (Gachon et al., 2006). These genes and their rhythmically expressed target genes thus link the loss of the molecular circadian oscillator to time-of-day dependence of xenobiotic metabolism.
The hierarchical nature of the circadian timing system raises the question of the relative importance of SCN-driven, systemic influences versus local, tissue-intrinsic processes in generating functional rhythmicity. Several studies indicate that cell-autonomous circadian oscillations present in peripheral tissues have physiologically important roles (Storch et al., 2007; Sadacca et al., 2011; Marcheva et al., 2010), while other work indicates that SCN-driven rhythmicity can impose molecular rhythmicity on peripheral tissues in which the molecular oscillator is disrupted (Kornmann et al., 2007; DeBruyne et al., 2007b; Hughes et al., 2012).
To assess the functional importance of the hepatic circadian oscillator in xenobiotic metabolism, we investigated the response of wild-type and circadian mutant mice to 2 xenobiotic agents, pentobarbital (PB) and N-acetyl-para-aminophenol (APAP, also known as acetaminophen and paracetamol). Both PB and APAP have time-of-day dependent effects (Scheving et al., 1968; Sato et al., 2005; Xu et al., 2012; Kim and Lee, 1998; Kakan et al., 2011) and are metabolized exclusively by the cytochrome P450 (CYP) system of the liver (Henderson et al., 2006; Henderson et al., 2003). APAP is of significant interest because this widely used analgesic is the leading cause of drug-induced acute liver failure in the United States (Larson et al., 2005). Toxicity results from metabolism of APAP, generating N-acetyl-p-benzoquinone imine (NAPQI). CYP2E1 and CYP1A2 are major cytochromes responsible for APAP metabolism, and disruption of these genes blocks APAP metabolism and results in resistance to APAP toxicity (Zaher et al., 1998). CYP2E1 mRNA and protein are rhythmic in rat liver (Khemawoot et al., 2007), suggesting a molecular mechanism for rhythmicity in APAP toxicity.
Several mouse models are available for investigating the impact of disrupted circadian clock function on physiology. CLOCK-deficient mice have only modest alterations in locomotor activity rhythms, revealing redundancy between CLOCK and NPAS2 in the SCN (DeBruyne et al., 2007a; Dallmann et al., 2011). Peripheral tissues from these mice are arrhythmic when examined ex vivo (DeBruyne et al., 2007b). Mice homozygous for a conditional allele of Clock allow tissue-specific circadian clock disruption. Peripheral tissues of mice deficient in CLOCK or BMAL1 lack positive drive to the circadian oscillator, while mice without PER1, PER2, and PER3 (Per123−/−) lack negative feedback. Both the Bmal1−/− and Per123−/− mice lack central and peripheral circadian rhythmicity.
Using mice with disruption of the circadian clock genes introduced above, we show here that the liver clock can be essential for time-of-day dependent changes in xenobiotic detoxification. Our results further suggest that the manner in which the circadian clock is disrupted determines whether xenobiotic detoxification remains at high or low levels. These results reveal the functional importance of the liver clock.
Materials And Methods
Animals
All experimental animals were generated and maintained in a specific pathogen-free facility at the University of Massachusetts Medical School, Worcester. All experiments involving animals were approved by the Institutional Animal Care and Use Committee at the University of Massachusetts Medical School and were conducted in accordance with international and National Institutes of Health guidelines.
Albumin-Cre mice were purchased from Jackson Labs (Bar Harbor, ME; stock 003574) (Postic et al., 1999). Founder PER2::LUCIFERASE mice (Yoo et al., 2004) were generously provided by Dr. Joseph S. Takahashi (University of Texas Southwestern Medical Center, Dallas). Founder Bmal1+/– mice (Bunger et al., 2000) were generously provided by Dr. Christopher A. Bradfield (University of Wisconsin, Madison). Mice with a null allele of Clock (resulting from deletion of exons 5 and 6) and mice in which these exons were conditional (flanked by loxP) have been previously reported (DeBruyne et al., 2006) and were obtained from our colony. These lines have all been backcrossed to C57BL/6J for at least 9 generations.
Mice with targeted disruption of Per1, Per2, and Per3 on the Sv129 genetic background have been previously described (Bae et al., 2001; Shearman et al., 2000b). Triple-mutant (Per123−/−) mice were obtained from our colony. Congenic wild-type Sv129 mice were used as controls.
Study animals were genotyped by polymerase chain reaction (PCR) amplification of genomic DNA extracted from tail biopsies. Products were separated by agarose gel electrophoresis and visualized with ethidium bromide. Primers and PCR conditions have been previously described for the 3 Per genes (Bae et al., 2001; Shearman et al., 2000a), Clock and Per2::Luc (DeBruyne et al., 2006), Bmal1 (Bunger et al., 2000), and Alb-Cre (Etchegaray et al., 2009).
For all xenobiotic assays, we used adult (3- to 8-month-old) male mice. Bmal1−/− and Clock−/− mice were compared with their wild-type littermate controls. Alb-Cre+;Clockflox/flox mice were compared to Clockflox/flox littermate controls. For PER2::LUCIFERASE recordings, Alb-Cre+;Clockflox/flox;Per2luc/+ mice were compared to littermate controls lacking the Alb-Cre transgene (Clockflox/flox;Per2luc/+).
Unless otherwise noted, animals were housed in cages with food and acidified water available ad libitum and maintained in climate-controlled closets at 21 ± 1 °C and 45% humidity with 12 h of light per day (LD 12:12). During the light phase of the lighting cycle, white light was emitted from fluorescent bulbs (ca. 100 lux). Dim red light (>600 nm) was present at all times.
Pentobarbital Sleep Time Assay
For the pentobarbital sleep time (PBST) assay, pentobarbital sodium salt (Sigma-Aldrich, St. Louis, MO) dissolved in sterile saline was injected intraperitoneally (i.p.) at a dose of 40 or 50 mg/kg body weight as indicated for each experiment. Unless otherwise noted, animals were fasted 12 h before injection by placing them in a fresh cage with an empty food hopper but with water available at all times. Immediately after injection, animals were returned to their home cage until they lost consciousness. Then, they were gently placed on their back. “Sleep time” was calculated as time from injection to regaining of the righting reflex (i.e., the time at which the animal was able to right itself 3 times within 1 min; modified after Dandiya and Cullumbine, 1959). All injections were made on the first day of constant darkness (DD) at indicated times with the exception of the experiment to examine the influence of previous food availability, in which mice were tested on the first and second day in DD. Food removal occurred during darkness, less than 24 h after the last light-to-dark transition. Mice were fasted for 12 or 24 h before testing at CT2 or were fasted for 24 or 36 h before testing at CT14 (see Suppl. Fig. S1A,B).
Pentobarbital Measurement
Twelve hours after food removal, PB (50 mg/kg) was injected into wild-type mice on the first day of DD (at CT14 or at CT2). Animals were randomly split into 2 groups. In 1 group of mice, blood samples were obtained 30 min after injection. In the second group, blood samples were obtained at the time the animals regained the righting reflex. Blood samples were collected by decapitation and allowed to clot on ice for >20 min. Samples were centrifuged, and serum was obtained and frozen at −20 °C. PB levels in serum were assayed by high-performance liquid chromatography (HPLC) at the Core Laboratory of the University of Massachusetts Memorial Hospital (Worcester, MA).
Acetaminophen (APAP) Toxicity
APAP toxicity was assessed by measuring alanine aminotransferase (ALT) activity in serum 24 h after APAP injection on the first day in constant darkness at the indicated times (see paradigm in Suppl. Fig. S1C). For these experiments, APAP (Sigma-Aldrich) was dissolved in prewarmed sterile saline and injected i.p. at a dose of 250 mg/kg unless otherwise noted. In each experiment, 2 animals were injected with sterile saline only as controls. In all cases, these control values were comparable to ALT levels in saline-injected mice from the literature (<100 IU/L) and thus are reported only once in the Results section. To assess the impact of different doses of APAP, mice (n = 3 per group) were injected i.p. with saline or with APAP at 100, 250, 400, or 600 mg/kg.
After injection with APAP, animals were returned to their home cage for 24 h. Then, animals were decapitated and blood was collected from the trunk. After clotting, serum was collected and ALT levels analyzed in duplicate with a colorimetric end-point measurement kit scaled from the manufacturer’s protocol (Teco Diagnostics, Anaheim, CA).
Data Analysis
All data are given as mean ± standard error of the mean (SEM). Data were compared using 1- or 2-way analysis of variance (ANOVA) with factors CT (time) and genotype and Bonferroni’s post-hoc test if appropriate. All statistical tests were 2-tailed, and the level of significance was set to α = 0.05.
Results
Circadian Rhythmicity in PBST Is Due to Hepatic PB Metabolism
Previous studies reported diurnal changes in PBST, measured with a lighting cycle present (Davis, 1962; Sato et al., 2005; Scheving et al., 1968). Because the ambient lighting condition could influence PBST (“masking”), we first assessed whether PBST is rhythmic in constant darkness (i.e., whether PBST is under circadian control). Groups of wild-type mice injected with PB differed significantly depending on circadian time (1-way ANOVA, p < 0.001). We observed a strong, nearly 2-fold circadian variation in PBST with peak sleep times at CT2 (125.8 ± 7.1 min) and shortest sleep times at CT14 (68 ± 6.0 min). Intermediate values were observed at CT8 (88.0 ± 2.1 min) and CT20 (88.6 ± 5.3 min) (Fig. 1).
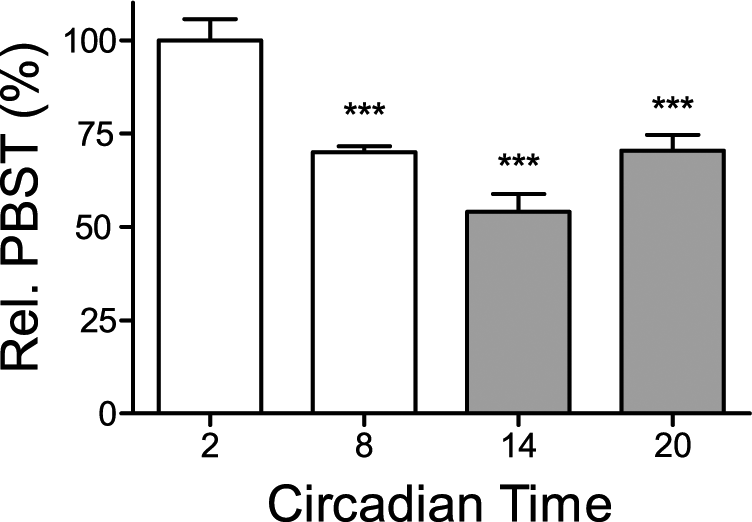
PBST is time-of-day dependent in wild-type mice. Relative pentobarbital sleep time (PBST) of wild-type mice injected intraperitoneally (i.p.) with 50 mg/kg pentobarbital (PB) at indicated circadian times (n = 4-5 per group) as percent of PBST at CT2 (100%
Prior food intake is known to influence PBST in mice (Lovell, 1986), which is why we removed food from all animals 12 h before assessing PBST. Due to rhythmicity in food intake, however, these groups likely differed in the food consumed prior to removing their food. To assess whether this could contribute to the rhythm in PBST, animals were fasted for 12, 24, or 36 h before administration of PB. PBST differed between CT2 and CT14 irrespective of the duration of prior fasting. PBST was only dependent on the circadian time of PB injection (1-way ANOVA, p = 0.001, Fig. 2).
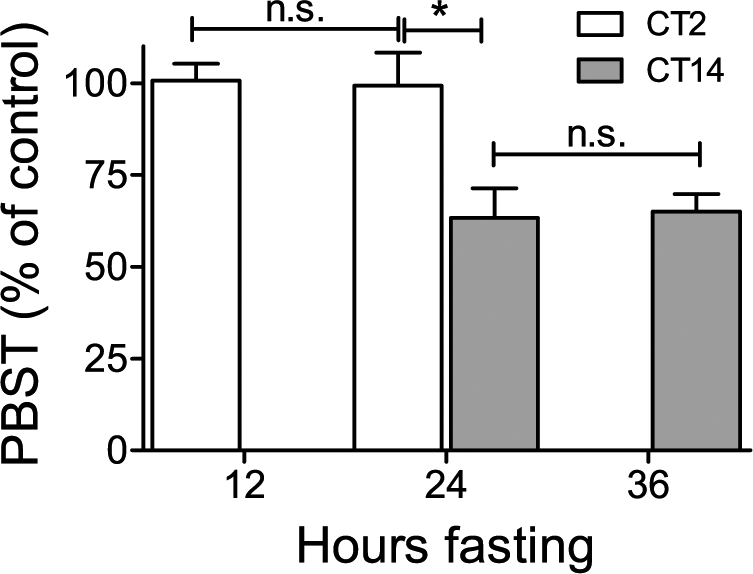
Pentobarbital sleep time (PBST) is not dependent on duration of prior fasting. The influence of fasting on PBST in wild-type mice injected i.p. with 40 mg/kg pentobarbital (PB) at CT2 or CT14 with prior fasting of 12, 24, or 36 h (see Suppl. Fig. S1B). PBST was determined for n = 5 animals per group. Values are given as relative PBST (percent of average PBST after 12 h of fasting at CT2) (control, 100 %
Pentobarbital acts mainly through GABAA receptors in the brain (Rudolph and Antkowiak, 2004), which show daily variation in their abundance (Chassard and Bruguerolle, 2004). Thus, rhythmicity in PBST could be caused by rhythmicity in the rate of hepatic PB metabolism, by rhythmicity in the hypnotic effects of a given concentration of PB, or by a combination of these mechanisms. To address this, we assessed PB levels in serum in one group of mice 30 min after injection (before the animals regained the righting reflex) and assessed PB levels in another group of mice upon regaining the righting reflex (“awakening”). At 30 min after injection, levels of PB in serum were significantly higher in mice injected at CT2 than in those injected at CT14 (p < 0.05; Fig. 3), suggesting a more rapid clearance of PB at CT14. When assayed at the time the animals regained the righting reflex, serum PB levels did not differ between mice injected at CT2 and CT14 (p = 0.49). Thus, rhythmicity in PBST appears to be primarily due to rhythmicity in hepatic metabolism rather than to rhythmicity in the hypnotic effects of PB. Although pharmacodynamic effects based on, for example, receptor availability cannot be excluded by our data, this interpretation is further substantiated by the results obtained in the liver-specific CLOCK-deficient mice (see below).
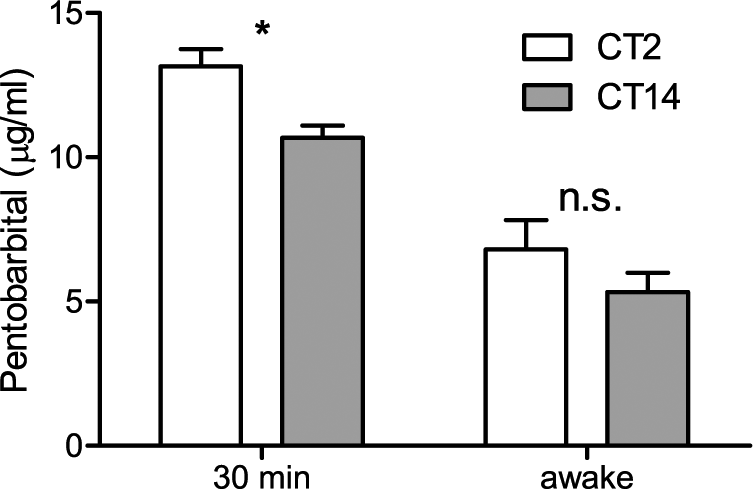
Pentobarbital (PB) clearance from serum is dependent on time of day. PB (40 mg/kg, i.p.) was injected at CT2 or CT14 in wild-type mice (n = 5-7 per group), and PB concentration in serum was measured 30 min after injection (left) or (in a second independent group of animals) after the mice regained their righting reflex (“awake”; right). In post-hoc testing of 2-way analysis of variance, PB levels at 30 min after injection differed significantly between CTs (*p = 0.01, Bonferroni’s post-hoc test) but not at the time of recovery from anesthesia (n.s., p = 0.49).
Circadian Rhythmicity in APAP Toxicity
Previous publications indicate that peak APAP toxicity in rodents occurs at the time of the maximal rate of metabolism of APAP, early in the nighttime (Kim and Lee, 1998; Matsunaga et al., 2004). Our results with PBST indicate that the highest rate of PB metabolism occurs early in the subjective night (CT14). Thus, for examining APAP toxicity, we focused our efforts on the expected peak and trough times, CT14 and CT2, respectively. In wild-type mice, injection of APAP (250 mg/kg, i.p.) at CT2 was well tolerated and led to only slight elevation in ALT levels compared to saline-injected control mice. In contrast, ALT levels were greatly elevated following injection of the same APAP dose at CT14 (Fig. 4).
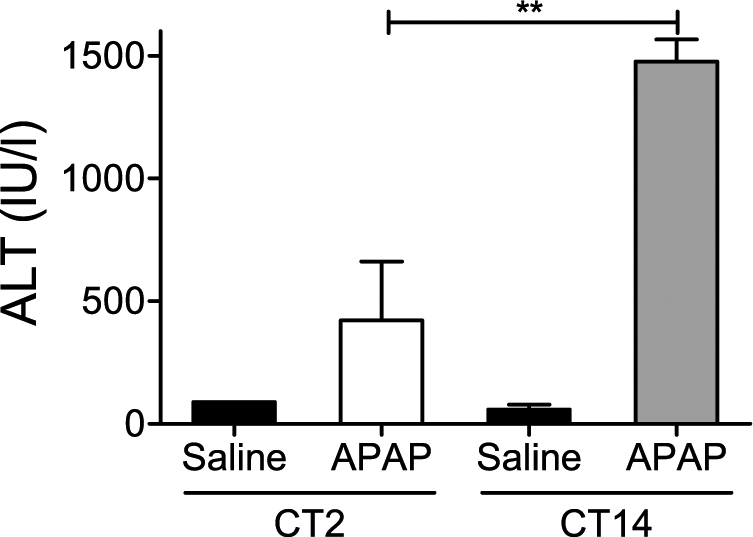
Effect of intraperitoneal (i.p.) saline (Saline, n = 2 per group) or 250 mg/kg acetaminophen (APAP, n = 6 per group) injection at 2 circadian times in wild-type animals. Statistically significant differences versus CT2 in Bonferroni’s post-hoc test are given as **p < 0.01. Black bars = saline controls; open bars = subjective day (CT2); gray bars = subjective night (CT14).
We attempted to rule out the impact of food intake preceding the APAP injection, because shifting food intake into the day has been shown to affect APAP toxicity (Matsunaga et al., 2004), by removing food 12 h prior to APAP injection (as in the PBST experiments). Fasting for 12 h prior to APAP injection at CT2 induced high levels of toxicity, which were not reversed by a 1-h refeeding opportunity (Suppl. Fig. S2). The increased toxicity following fasting could be due to induction of CYP2E1 by fasting (Leclercq et al., 2000). Irrespective of the mechanism, this experiment indicates that fasting has pronounced effects on APAP toxicity, and thus fasting cannot be used to probe the potential impact of prior food intake on APAP toxicity.
Rhythms in PBST Are Absent in Mice With Disrupted Circadian Clocks
In contrast to wild-type controls with an intact circadian clockwork, disruption of the molecular clock led to a loss of the rhythm in PBST (Fig. 5). CLOCK-deficient mice maintain locomotor activity rhythms in constant darkness (DeBruyne et al., 2006). Despite disruption of their molecular clockwork in peripheral tissues (as revealed by loss of rhythmicity in liver and lung explants recorded ex vivo) (DeBruyne et al., 2007b), there is moderate imposed rhythmicity on liver proteins and gene expression in vivo, with no apparent change in phase (DeBruyne et al., 2006). We thus expected that Clock−/− mice would show rhythmic PBST. Surprisingly, PBST in Clock−/− mice did not differ between CT2 and CT14 (108 ± 8 min vs. 98 ± 5 min, respectively, p > 0.05). The CLOCK-deficient mice had sleep times at both CT2 and CT14 that were comparable to that of wild-type controls at CT2 (108 ± 7 min) but were more than twice that of wild-type mice at CT14 (45 ± 4 min; Fig. 5).
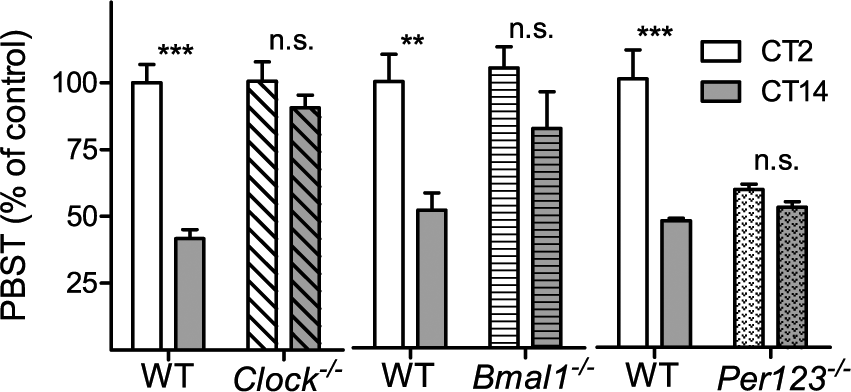
Rhythm in pentobarbital sleep time (PBST) is disrupted in mice with whole-body disruption of circadian clock genes. PBST in Clock−/− (n = 6 per group, left, diagonally hatched), Bmal1−/− (n = 4-5 per group, middle, horizontally hatched), and Per123−/− (n = 6 per group, right, dotted) compared to their respective control wild-type mice after injection of 50 mg/kg pentobarbital (PB). To facilitate easy comparison between experiments, PBST is normalized to CT2 of the wild-type control value for each experiment (100% values of controls at CT2: Clock-WT 107.6 ± 7.3 min, Bmal1-WT 62.8 ± 6.4 min, Per123-WT 50.2 ± 5.4 min). Statistical differences in Bonferroni’s post-hoc test between CTs are indicated as n.s. (not significant), **p < 0.01, or ***p < 0.001. Open bars = subjective day (CT2); filled bars = subjective night (CT14).
Similarly, disruption of the positive drive within the molecular clock in a different mouse line (i.e., the arrhythmic Bmal1−/− mouse) disrupted the PBST rhythm in the same way: there was no difference in PBST between Bmal1−/− mice injected at CT2 (66 ± 5 min) and CT14 (52 ± 8 min). As with CLOCK-deficient mice, Bmal1−/− mice had long sleep times at both time tested comparable to control mice injected with PB at CT2 (Fig. 5).
In contrast, ablation of the negative limb of the feedback loop (i.e., in mice with disruption of all 3 Period genes) “clamped” the PBST at low values similar to those of strain-matched wild-type mice at CT14 (Fig. 5). PBST of Per123−/− mice was not different between CT2 (30 ± 1 min) and CT14 (26 ± 1 min). Thus, Clock−/−, Bmal1−/−, and Per123−/− lack rhythmicity in PBST, but they differ in whether they resembled controls injected at CT2 (Clock−/−, Bmal1−/−) or at CT14 (Per123−/−).
APAP Toxicity Is Reduced and Is Not Rhythmic in CLOCK-Deficient Mice
Clock−/− mice tolerated APAP treatment remarkably well at both times of day examined (Fig. 6A). Even more unexpectedly, Clock−/− mice were relatively resistant to APAP across a range of doses up to 600 mg/kg, even when studied at CT14 (the time at which wild-type controls were maximally sensitive) (Fig. 6B).
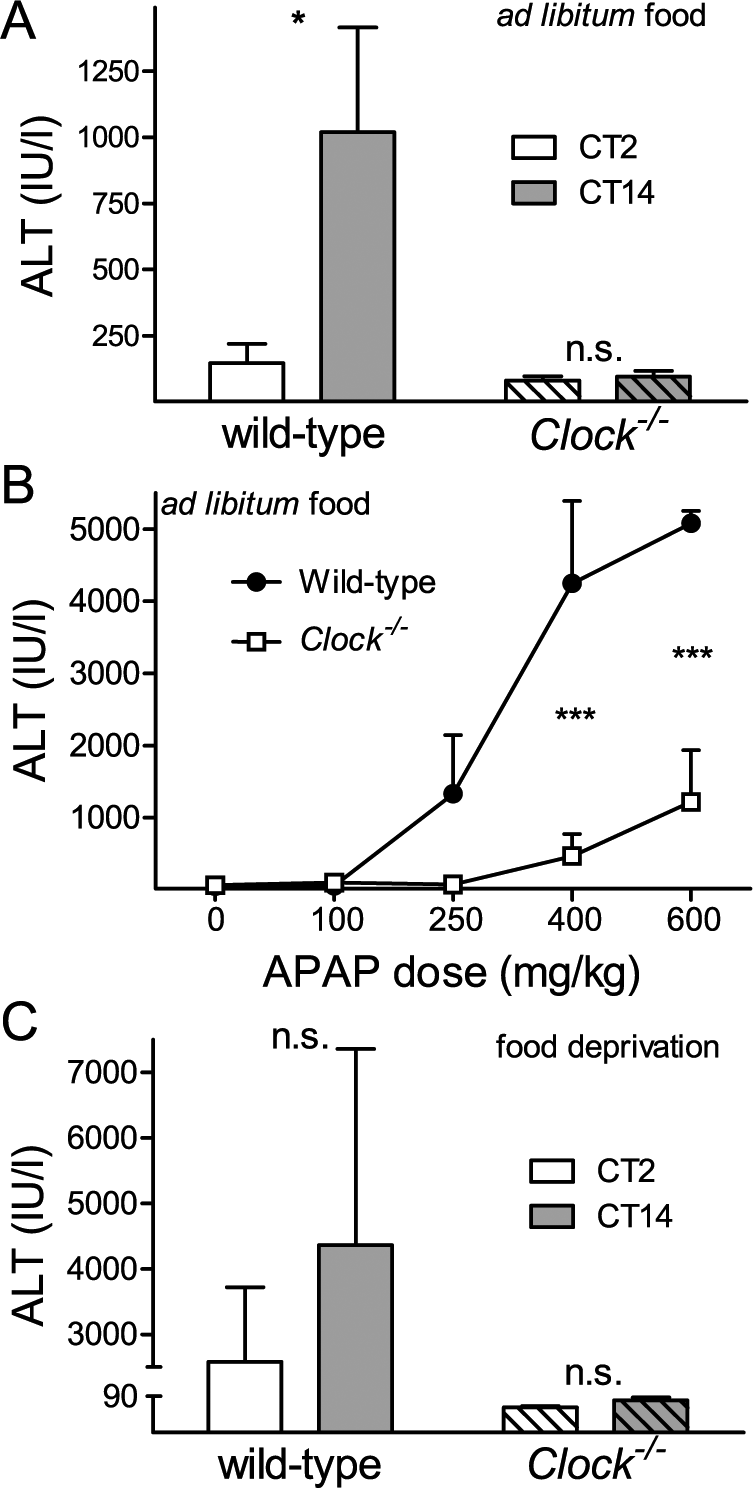
Clock−/− mice are resistant to N-acetyl-para-aminophenol (APAP) toxicity. (A) Alanine aminotransferase (ALT) levels in wild-type and Clock−/− (hatched) mice following APAP (250 mg/kg) administration at CT2 or CT14 (n = 5 per group). (B) Dose range experiment with indicated doses of APAP in wild-type (closed circles) and Clock−/− (open squares) mice at CT 14 (n = 3 per group). (C) Twelve hours of food deprivation before APAP (250 mg/kg) injection increases APAP toxicity in wild-type but not in Clock−/− (hatched) mice at CT2 and CT14 (note the difference in values between panel A and panel C). Statistically significant differences between genotypes in Bonferroni’s post-hoc test after 2-way analysis of variance are indicated as n.s. (not significant), *p < 0.05, or ***p < 0.001.
To further examine the resistance of Clock−/− mice to APAP toxicity, we examined their response to APAP after fasting for 12 h. As noted above, fasting increased APAP toxicity in wild-type mice even at the low-toxicity time (CT2) (Fig. 6C, Suppl. Fig. S2). In contrast, APAP toxicity remained low in fasted Clock−/− mice at both times examined (Fig. 6C).
Rhythms in PBST and APAP Toxicity Are Absent in Mice Lacking CLOCK Only in the Liver
We generated mice in which CLOCK expression was disrupted specifically in the liver by intercrossing mice with the widely used Albumin-Cre driver with mice bearing conditional alleles of Clock. As expected, mice with liver-specific disruption of Clock (Alb-Cre+; Clockflox/flox) lacked CLOCK protein in the liver and lacked circadian reporter gene oscillations in explants of liver, while normal rhythms were present in lung explants (Suppl. Fig. S3).
In their response to both PBST and APAP, the Alb-Cre+;Clockflox/flox mice were similar to mice lacking CLOCK throughout the body. PBST of Alb-Cre+;Clockflox/flox mice at CT2 (129 ± 8 min) and at CT14 (119 ± 8 min) did not differ (Bonferroni’s post- hoc test, p > 0.05), and both were comparable to sleep times of Clockflox/flox control mice at CT2 (129 ± 11 min), which was significantly longer than control Clockflox/flox mice at CT14 (62 ± 2 min; Bonferroni’s post-hoc test, p < 0.001; Fig. 7A). Similarly, liver toxicity following APAP treatment of Alb-Cre+;Clockflox/flox mice was low at both CT2 and CT14 (Fig. 7B), as in Clock−/− mice. Thus, in both assays, mice lacking Clock (either throughout the body or specifically in the liver), responded to these 2 xenobiotics administered at either CT2 or CT14 in a manner similar in magnitude to wild-type mice receiving comparable treatments at CT2. Thus, the increase in hepatic xenobiotic metabolism that normally occurs at CT14 in wild-type mice is absent in mice lacking hepatic CLOCK.
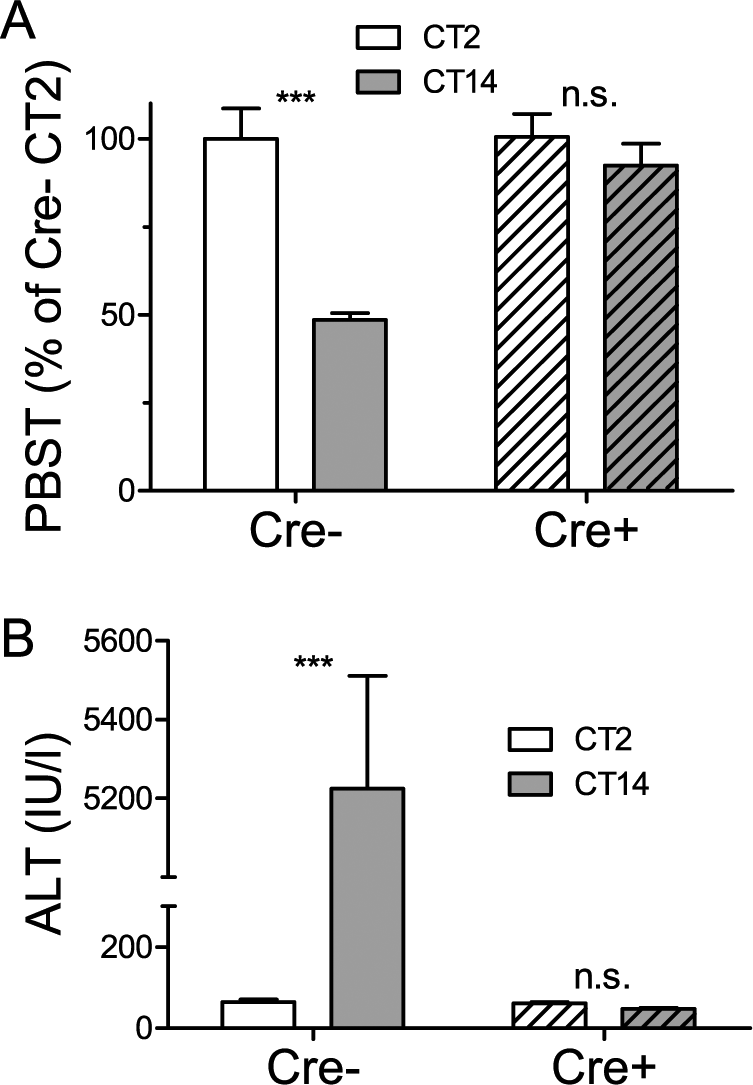
Rhythms in pentobarbital sleep time (PBST) and N-acetyl-para-aminophenol (APAP) toxicity are absent in liver-specific CLOCK-deficient mice. (A) PBST in Alb-Cre–;Clockflox/flox mice (Cre–) and Alb-Cre+;Clockflox/flox mice (Cre+, hatched) (n = 6 per group) after 50 mg/kg pentobarbital (PB) injection. PBST is normalized to CT2 of the wild-type control value (100%
Discussion And Conclusions
The circadian system is an important modulator of metabolism in general (Dallmann et al., 2012; Eckel-Mahan et al., 2012) and xenobiotic metabolism in particular (Gachon et al., 2006; Dallmann et al., 2014). How the circadian clock controls metabolism is less clear. For example, systemic cues can generate rhythms in expression of a subset of hepatic genes even when the hepatic circadian clock is crippled (Kornmann et al., 2007; DeBruyne et al., 2006). Conversely, disruption of food intake greatly reduced rhythmic gene expression even in a genetically intact liver clock (Vollmers et al., 2009). This poses the question of whether the cell-autonomous clock in the liver is essential to generate rhythmicity in xenobiotic metabolism.
Here, we used 2 xenobiotic agents metabolized by the liver to show that the liver clock is essential for rhythmic xenobiotic metabolism of the whole organism. The lines of evidence indicating the importance of the tissue-autonomous hepatic clock in rhythmic xenobiotic metabolism are (1) hepatic metabolism (rather than neuronal sensitivity) is responsible for circadian differences in PBST (Fig. 3), and (2) rhythmicity in PBST and APAP toxicity is lost in mice in which the circadian clock was selectively disrupted in liver, using Alb-Cre+;Clockflox/flox mice (Fig. 7). These findings extend previous work indicating rhythmicity in barbiturate sleep time and APAP toxicity (Xu et al., 2012; Dai et al., 2006; Matsunaga et al., 2004; Kim and Lee, 1998; Arthur et al., 2003; Scheving et al., 1968; Sato et al., 2005; Gachon et al., 2006). While others have speculated “that dosing time dependent hypnotic durations might be explained by differing sensitivity of the CNS (pharmacodynamics) rather than pharmacokinetics” (Sato et al., 2005), our work demonstrates that the circadian clock within the liver is necessary for this rhythmicity, strongly arguing that this effect is predominantly pharmacokinetic in nature.
In wild-type mice, PBST was shorter and APAP-induced toxicity was greater at CT14 compared to CT2. It is important to keep in mind that APAP toxicity is due to its conversion to the toxic metabolite, NAPQI, and more rapid metabolism of APAP to NAPQI is associated with more severe hepatotoxicity (Kim and Lee, 1998; Zaher et al., 1998). Our results are thus consistent in showing increased xenobiotic metabolism in the liver of wild-type mice at CT14.
Mice lacking CLOCK or BMAL1 lack rhythmicity in PBST and APAP toxicity (Figs. 5-7) and are similar to wild-type mice at CT2 in both assays, apparently being unable to increase xenobiotic metabolism above this basal level. Conversely, mice lacking all 3 Per gene products behave like wild-type mice at CT14 in the PBST assay, whether injected with PB at CT2 or CT14 (Fig. 5). Our APAP studies were limited to 2 time points, so we cannot completely exclude the possibility that rhythms in APAP toxicity persist with altered phase in the mutant mice, but this seems very unlikely based on the comparable phase in peak clock gene and protein expression previously reported in CLOCK-deficient mice (DeBruyne et al., 2006) and the absence of rhythmicity in mice lacking the 3 Per genes or BMAL1.
The pattern of results indicates that the differences in xenobiotic metabolism among these mutant lines are direct effects of a malfunctioning circadian clock, as opposed to representing pleiotropic, clock-independent effects. Breaking the clock by disabling the CLOCK:BMAL1 activator complex stops the transcriptional feedback loop in the opposite phase compared with breaking the clock by disabling the PER/CRY repressor complex. In our studies, CLOCK or BMAL1 deficiency has the opposite effect compared to disruption of the Per genes, consistent with a role for these complexes and the circadian oscillator as a whole in activating and repressing xenobiotic metabolism, respectively. In this respect, our data are in line with greatly reduced and arrhythmic CYP-dependent detoxification capacity in mice lacking Dbp/Hlf/Tef genes due to low levels of hepatic POR expression (Gachon et al., 2006). CLOCK:BMAL1 complexes act as direct transcriptional activators of the Dbp/Hlf/Tef genes. While other rhythmically regulated genes also likely contribute to rhythmic metabolism of xenobiotics, the remarkable resistance of CLOCK-deficient mice to high APAP doses (Fig. 6B) and their resistance to fasting-induced enhancement of APAP toxicity (Fig. 6C) suggest that the circadian transcriptional activator complex is a very important “modulator” of xenobiotic metabolism. This is further substantiated by published data for hepatic POR expression in mice lacking BMAL1 specifically in liver, in which POR expression is clamped at minimal levels throughout the circadian cycle (Lamia et al., 2008). To underscore the magnitude of this modulation, note that Clock−/− mice are as well protected from APAP toxicity as CYP2E1-deficient mice (Lee et al., 1996) and mice with liver-specific POR deletion (Henderson et al., 2003); CYP2E1 and POR are the major enzymes responsible for APAP’s hepatotoxicity.
As noted above, it is useful to compare the effect of breaking the circadian clock by disabling the transcriptional activator complex with breaking the clock by disabling the repressor complex. Our results are consistent with a role for these complexes in activating and repressing xenobiotic metabolism, respectively. This pattern of modulation implicates the circadian clock rather than pleiotropic functions of the affected genes. A similar pattern of results is seen for the contribution of circadian clock genes to wound healing (Kowalska et al., 2013) and regulation of NAD+ and glucose (Ramsey and Bass, 2011). Circadian clock function has also been implicated by studies indicating that disruption of clock function by any methods leads to a similar phenotype or pathology (Cheng et al., 2011; reviewed in Yu and Weaver, 2011), although in these cases, the mechanisms are less clear. In other studies, disrupting the circadian clock in “opposite” ways leads to phenotypes that differ and are not obviously opposite (e.g., in neurodegeneration) (Musiek et al., 2013), making it more difficult to identify the role of the circadian clock separately from other actions of circadian clock proteins. In contrast, in our study, the clock-controlled Dbp/Hlf/Tef genes likely provide a direct link between the circadian clock and xenobiotic metabolism.
Small-molecule modulators have been described that act on peripheral and central clocks through various molecular targets (Chen et al., 2012; Solt et al., 2012; Yamaguchi et al., 2013; Hirota et al., 2012). It seems likely that development of “chronobiotic” compounds with biological activity in vivo may provide a novel mechanism to rapidly adjust the hepatic circadian clock to a favorable molecular time, leading to a favorable profile of genes involved in xenobiotic metabolism. This may afford a new approach to reducing APAP-induced toxicity. As noted above, disruption of CLOCK:BMAL1 activity is one approach. Another approach is suggested by our finding that disruption of the 3 Per genes leads to short PBST, suggestive of rapid xenobiotic metabolism. Conversely, enhancing activity within the negative limb of the feedback loop may reduce xenobiotic metabolism. PER and CRYPTOCHROME (CRY) proteins work together to produce negative feedback. Small-molecule compounds that can stabilize CRY proteins have been described recently (Hirota et al., 2012). Compounds with similar activity and biological activity in vivo could augment the negative feedback effect within the circadian clockwork, altering output gene expression rhythms to reduce xenobiotic metabolism. CRY1 is an especially interesting target because it also appears to be involved in the rhythmic modulation of CYP2E1 expression through inhibition of hepatic nuclear factor-1α (HNF-1α) (Matsunaga et al., 2008). In addition, this work suggests the possibility that single-nucleotide polymorphisms and other genetic variations in circadian clock genes may also contribute to interindividual differences in xenobiotic metabolism, as has been shown for other metabolic functions (Garcia-Rios et al., 2012).
Recently, it has been suggested that one possible mechanism of action of APAP is dependent on electrophilic metabolites, generated by CYPs expressed in the spinal cord, acting on TRPA1 (Andersson et al., 2011). Our data indicating circadian rhythmicity of APAP metabolism (as assessed by hepatotoxicity) suggest that the rate of production of these electrophilic metabolites may also be time-of-day dependent. If this is the case, both therapeutic (analgesic) and side effects (hepatotoxicity) of APAP may be influenced by time of day.
In summary, the hepatic circadian clock plays an important role in modulating xenobiotic metabolism. The hepatic circadian clock contributes to enhancing metabolism of PB and APAP early in the nighttime. Disruption of the liver clock by disrupting genes involved in the circadian transcriptional activator complex (CLOCK or BMAL1) leads to consistently low xenobiotic metabolism, while disruption of the feedback repressive complex (e.g., Per genes) leads to persistence of elevated xenobiotic metabolism. These findings suggest novel mechanisms by which xenobiotic detoxification can be manipulated, possibly preventing liver damage by APAP, chemotherapeutics, or other xenobiotics.
Footnotes
Acknowledgements
We thank Christopher Lambert for technical assistance, Drs. J. S. Takahashi and C. A. Bradfield for providing mouse lines, S. M. Reppert for encouragement during early phases of this project, and Michael Arand for comments on a previous version of the manuscript. This work was supported by the National Institutes of Health (R21 NS051458 and R01 NS056125 to DRW and NRSA F32 GM074277 to JPD) and the Deutsche Forschungsgemeinschaft (DA525/2-1 to RD) and Alexander von Humboldt Foundation (3.3-DEU/1131488 FLF to RD). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the sponsoring agencies.
Author Contributions
Designed and conducted experiments: JPD, DRW & RD. Analyzed data: JPD & RD. Wrote manuscript: JPD, DRW & RD.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.