
Abstract
Circadian misalignment occurs with age, jet lag, and shift work, leading to maladaptive health outcomes including cardiovascular diseases. Despite the strong link between circadian disruption and heart disease, the cardiac circadian clock is poorly understood, prohibiting identification of therapies to restore the broken clock. Exercise is the most cardioprotective intervention identified to date and has been suggested to reset the circadian clock in other peripheral tissues. Here, we tested the hypothesis that conditional deletion of core circadian gene Bmal1 would disrupt cardiac circadian rhythm and function and that this disruption would be ameliorated by exercise. To test this hypothesis, we generated a transgenic mouse with spatial and temporal deletion of Bmal1 only in adult cardiac myocytes (Bmal1 cardiac knockout [cKO]). Bmal1 cKO mice demonstrated cardiac hypertrophy and fibrosis concomitant with impaired systolic function. This pathological cardiac remodeling was not rescued by wheel running. While the molecular mechanisms responsible for the profound cardiac remodeling are unclear, it does not appear to involve activation of the mammalian target of rapamycin (mTOR) signaling or changes in metabolic gene expression. Interestingly, cardiac deletion of Bmal1 disrupted systemic rhythms as evidenced by changes in the onset and phasing of activity in relationship to the light/dark cycle and by decreased periodogram power as measured by core temperature, suggesting cardiac clocks can regulate systemic circadian output. Together, we suggest a critical role for cardiac Bmal1 in regulating both cardiac and systemic circadian rhythm and function. Ongoing experiments will determine how disruption of the circadian clock causes cardiac remodeling in an effort to identify therapeutics to attenuate the maladaptive outcomes of a broken cardiac circadian clock.
Circadian rhythms define physiology and behaviors ranging from sleep-wake cycle, hormonal levels, body temperature, and many other processes that occur throughout a 24-h period (Aguilar-Arnal et al., 2013). In the cardiovascular system, blood pressure, heart rate (Degaute et al., 1991), and cardiac output oscillate by time of day (Delp et al., 1991). Disruption of the circadian rhythm is a feature of several cardiovascular diseases (Rudic and Fulton, 2009; Young and Bray, 2007). In the last decade, the central and peripheral molecular circadian clocks have been actively investigated to understand the composition and function of the circadian clock at the organism and tissue levels (Bell-Pedersen et al., 2005). Both the central and peripheral clocks are endogenously generated and self-sustained by a well-defined set of genes (Lowrey and Takahashi, 2011). BMAL1 binds to CLOCK which together initiate the rhythmic expression of the core clock genes Periods (Per1 and Per2), Cryptochromes (Cry1 and Cry2), and Rev-erb (Rev-erbα and Rev-erbβ). PERs and CRYs form a repressive complex that rhythmically inhibits BMAL1-CLOCK-mediated transcription for feedback inhibition (Koike et al., 2012). Thus, in this regulatory feedback loop, robustness of circadian oscillation is achieved by rhythmic regulation of BMAL1 and concomitant control of transcription of other clock genes (Preitner et al., 2002).
Transgenic experiments in mice have attempted to elucidate the impact of circadian clock disruption on cardiac function and disease. The first transgenic mouse designed to answer these questions was the cardiomyocyte-specific Clock mutant (CCM) mouse that developed a hypertrophic phenotype with impaired contractile function (Bray et al., 2008). Follow-up studies in cardiomyocyte-specific Bmal1 knockout (CBK) mice also demonstrated a hypertrophic phenotype that was evident by 12-16 weeks of age (Durgan et al., 2011). However, subsequent work with this mouse did not report cardiac hypertrophy despite elevated myocyte cross-sectional area compared with wild-type (WT) controls. The authors did note that the CBK mouse developed diastolic dysfunction likely due to cardiac fibrosis (Ingle et al., 2015). More recently, inducible models of Bmal1 deletion in the postnatal heart have started to emerge. Interestingly, although these mice demonstrate impaired rhythmicity of heart rate and elevated arrhythmia susceptibility, they were not reported to undergo hypertrophic or fibrotic remodeling (Schroder et al., 2013), Together, although it is clear that deletion of cardiac clock genes is detrimental to cardiac function, the resultant phenotype lacks consensus. In part, this may be due to differences in the timing of clock deletion—that is, whether constitutive or later in life with inducible approaches (Schroder et al., 2015). Perhaps further contributing to the lack of consensus of the impact of deletion of clock proteins in the heart is the exclusion of female mice from previous studies. Thus, while the cardiac field is growing in its understanding of the significance of the molecular clock, the impact of clock manipulation on heart function is not yet clear.
Exercise reduces cardiovascular disease risk factors and is potently cardioprotective through functional and anatomical changes referred to as exercise-induced cardiac remodeling (Weiner and Baggish, 2012). These changes render the heart with improved function and improve resistance to stressful stimuli. Recently, exercise has also emerged as a stimulus capable of shifting the molecular clock in non-cardiac tissue (Schroeder et al., 2012; Gannon and Rea, 1995). However, the molecular framework of exercise and clock is not well defined, nor has exercise been tested as a timekeeper in the heart. Therefore, we set out to determine the impact of cardiac Bmal1 deletion on cardiac function and to test whether exercise could ameliorate the negative effects of clock disruption.
Materials and Methods
Animals and Experimental Design
All experiments and methods were conducted in accordance with institutional guidelines and were approved by the Institutional Animal Care Users Committee at the University of Wyoming. All mice were on the C57Bl6 background and were bred in-house. Experiments were conducted in male and female mice at ~8 weeks of age. Mice were fed standard laboratory chow (Lab Diet 5001) with water and food provided ad libitum. Mice were maintained on a 12-h light/dark cycle, with lights on at zeitgeber 0 (ZT0; 0700 h) and lights off at ZT12 (1900 h).
To test the role of cardiac-specific BMAL1 in regulating circadian rhythm and cardiac function, we generated a model of inducible myocyte-specific deletion of Bmal1 using Cre-Lox recombinase. We crossed mice homozygous for the Bmal1-loxP-targeted allele with transgenic mice expressing tissue-specific Cre from the α-MHC promoter (α-MHC-MerCreMer; cardiomyocyte-specific). Both strains were purchased from Jackson Laboratories (Stock Nos 007668 and 0056587). Genotyping was used to confirm generation of α-MHC-Cre (forward: 5′-TGAGCATTCTCCTGCTGTTTC-3′, reverse: 5′-CAGCATTGTGAGAACAAGG-3′) and Bmal1 (forward: 5′-ACTGGAAGTAACTTTATCAAACTG-3′; reverse: 5′-TGACCAACTTGCTAACAATTA-3′). To delete Bmal1 in Cre-positive tissues, tamoxifen (30 mg/kg) was administered by intraperitoneal injection for 5 consecutive days at ZT2 followed by a 1-week washout period. At this dose, tamoxifen achieves maximum recombination with minimal cardiac toxicity (Rouhi et al., 2022). Consistent with previous reports of this model and Cre-Lox publications, Bmal1 fl/fl mice served as the genetic WT control (Liang et al., 2022; Hou et al., 2018). However, we do acknowledge that the lack of tamoxifen-treated α-MHC MerCreMer controls is a limitation.
Wheel Running Engagement and Exercise Capacity
Mice assigned to the exercise group were singly housed with a running wheel (Columbus Instruments) to be used voluntarily. Each wheel had a magnetic indicator and Hall effect sensor to record wheel revolutions (converted to kilometers). Wheel running data were collected daily, and mice were checked to ensure the wheel was still functioning properly. A 5-day introductory period preceded 4 days of hourly recorded data, ending with 5 additional days of daily running wheel data for a total of 14 days (Bruns et al., 2020). The average daily running distance was quantified on the last 5 days of running (days 9-14). Sedentary mice were also individually housed. Wheels were pulled the night before sacrifice to permit capture of the 2-week training period, rather than the acute exercise bout. An exhaustive exercise test was conducted on a subset of mice following the 2-week training period at ZT2 to determine differences in exercise capacity. Briefly, the test was performed on a treadmill with a fixed incline of 25°. The initial (warm-up) speed was set to 6 m/min for 6 min, with an increase in speed by 3 m/min every 3 min until the animal could no longer keep pace and ceased to run (Brown et al., 2020). Twenty-four hours following the exhaustive exercise test, echocardiography was performed and animals were humanely euthanized (FatalPlus; pentobarbital). The hearts were dissected and weighed, and the left ventricle (LV) and septum were dissected from the right ventricle. Tissues were flash-frozen in liquid nitrogen for subsequent assessments. The experimental design is summarized in Figure 1.
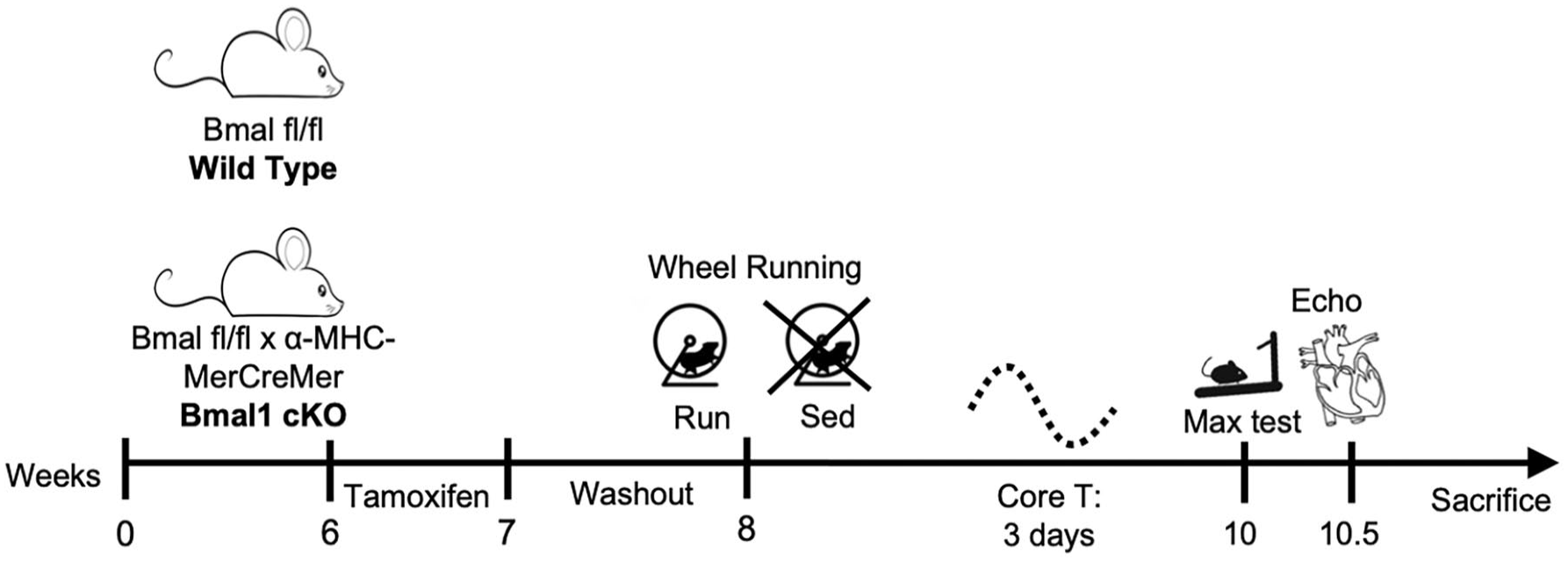
Experimental design. Generation of cardiomyocyte-specific deletion of Bmal1 by breeding of α-MHC-MerCreMer and Bmal1 fl/fl mice. Deletion of Bmal1 in Cre-positive tissues was achieved by daily intraperitoneal injections of tamoxifen (30 mg/kg) for 5 consecutive days at ZT2 followed by a 1-week washout period. Mice were then placed in experimental groups for assessment of voluntary wheel running activity and circadian rhythm. Maximum exercise capacity by treadmill time to exhaustion was performed at least 24 h before echocardiography and sacrifice.
Global Assessment of Circadian Rhythm
To assess circadian temperature profiles, we implanted pre-programmed SubCue Data Loggers to track core body temperature. Mice were anesthetized with isoflurane (3% for induction, 1.5% for maintenance) and dataloggers were inserted into the abdominal cavity. For analgesia, animals received buprenorphine post-surgery (0.1 mg/kg, subcutaneously) for 12 h for 3 days. After recovery, we recorded 3 days of unstimulated core temperature with animals housed on a normal 12-h light/dark cycle. Hour-by-hour values were averaged in Excel over 5-7 days for body temperature and 3-5 days for wheel running. Bathyphase (daily trough, for body temperature averaged over 5 days) and acrophase (daily peak, for wheel running averaged over 3-5 days) were determined in ClockLab Analysis 6 (Actimetrics). Actograms for body temperature and wheel running were also generated in ClockLab Analysis.
Echocardiography
Cardiac function was assessed by transthoracic echocardiography using Visual Sonics Vevo 2100 with a 40-mHz probe (FujiFilm). Mice were induced with 2% isoflurane in 1 L/min compressed air and maintained on an operative circuit nose cone (Harvard Apparatus) where isoflurane was titrated in 1 L/min compressed air to maintain a heart rate >400 bpm. Core temperature was maintained at 37 °C with a heating platform (FujiFilm). Respiratory rate and heart rate were monitored with 4-lead limb electrocardiogram (ECG) (FujiFilm). Two-dimensional parasternal long-axis (LA) B-mode views and parasternal short-axis (SA) B-mode and M-mode views of the LV were captured. Using VevoLab software, cine loop analysis was conducted on cardiac cycles between respirations to minimize respiratory artifact. Endocardial borders were traced in LA B-mode up to the aortic valve to assess total areas, volumes, ejection fraction (EF) [EF = (LVEDv − LVESv)/LVEDv × 100], fractional shortening (FS) [FS = (LVIDd − LVIDs)/LVIDd × 100], and cardiac output (CO). SA M-mode tracings measured parallel to the axis of the papillary muscles were used to assess anterior and posterior LV wall thickness during diastole and systole. All echocardiography was performed at ZT2.
Immunoblotting
LV lysates were homogenized in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCL, 150 mM NaCl, 2 mM EDTA, 0.1% sodium dodecyl sulfate [SDS], 1% Nonidet P-40, and 0.5% sodium deoxycholate, pH 7.4) containing protease inhibitors. Protein concentration was determined using a bicinchoninic acid assay (BCA) and prepared in Laemmli sample buffer (BioRad). Proteins were resolved on a gradient SDS gel and transferred to nitrocellulose paper. Following blocking in 5% nonfat dry milk or 5% bovine serum albumin for 1 h at room temperature, membranes were incubated with primary antibody overnight at 4 °C. The following primary antibodies were used: BMAL1 (Abcam 231793), TnI (Phosphosolutions 2010-TNI), phospho-mTOR (Ser2448) (Cell Signaling 5536), total mTOR (Cell Signaling 2972), phospho-AKT (Ser473) (Cell Signaling 9271), total AKT (Cell Signaling 9272), phospho-S6RP (Ser235/236) (Cell Signaling 2211), total S6RP (Cell Signaling 2217), phospho-4E-BP1 (Cell Signaling 9459), and total 4E-BP1 (Cell Signaling 9452). Membranes were washed and incubated with secondary antibody for 1 h at room temperature. Protein bands were visualized using chemiluminescent substrate and autoradiography. Even loading of proteins was verified by β-actin and Ponceau S staining. For quantification of phospho/total protein ratios, we first quantified phospho-specific protein expression. Membranes were stripped, blocked, and re-probed for total protein.
Real-Time RT-PCR
RNA was extracted from the LV using standard Trizol protocols and reverse-transcribed (iScript cDNA; BioRad). Real-time reverse transcription polymerase chain reaction (RT-PCR) was performed using SYBR Green Supermix (BioRad). Data were normalized to the housekeeping gene β-actin. Delta-delta Ct was calculated within sex and genotype, and data were expressed as fold change. Oligonucleotide sequences were as follows: Bmal1 (forward: 5′-ATCAGCGACTTCATGTCTCC-3′; reverse: 5′-CTCCCTTGCATTCTTGATCC-3′), Per2 (forward: 5′-GCCAAGTTTGTGGAGTTCCTG-3′; reverse: 5′-CTTGCACCTTGACCAGGTAGG-3′), Clock (forward: 5′-TTGCTCCACGGGAATCCTT-3′; reverse: 5′-GGAGGGAAAGTGCTCTGTTGTAG-3′), Ppar-α (forward: 5′-GACAGTGACAGACAACGGCA-3′; reverse: 5′-GTGGCAGGAAGGGAACAGAC-3′), Mstn (forward: 5′-TCACGCTACCACGGAAACAA-3′; reverse: 5′-AGGAGTCTTGACGGGTCTGA-3′), Pcg1-α (forward: 5′-AGCCTCTTTGCCCAGATCTT-3′; reverse: 5′-GGCAATCCGTCTTCATCCAC-3′), Glut4 (forward: 5′-GCCCCATTCCCTGGTTCATT-3′; reverse: 5′-GACCCATAGCATCCGCAACA-3′), Cox4 (forward: 5′-TCCCCACTTACGCTGATCG-3′; reverse: 5′-GATGCGGTACAACTGAACTTTCT-3′), Pdk4 (forward: 5′-TGAACACTCCTTCGGTGCAG-3′; reverse: 5′-TGTCTACAAACTCTGACAGGGC-3′), Cpt1 (forward: 5′-ATCATGTATCGCCGCAAACT-3′; reverse: 5′-GGGATGCGTGTAGTGTTGAAC-3′).
Histology
Whole hearts were removed, washed, and embedded in optimum cutting temperature (OCT) and frozen at −80 °C. Hearts were transversely cut (6 μm). H&E staining was used to visualize the heart. Briefly, slides were placed into Bouin’s solution (picric acid) for perfusion fixation. Slides were dehydrated using alcohol and vitrified in dimethylbenzene. Sections were stained with hematoxylin, differentiated with 0.3% acid alcohol, and stained with eosin. Cleared and mounted slides were imaged at 4× magnification.
Lectin staining was used for quantification of myocyte cell size. LVs were frozen in OCT and cut on a pre-cooled cryostat (Leica Biosystems) at 4-5 μm thickness. Sections were fixed with 3% paraformaldehyde for 10 min and permeabilized with 1% Triton X-100 for 10 min. Sections were stained with wheat germ agglutinin-fluorescein isothiocyanate (Sigma-Aldrich) for 25 min in the dark and mounted for imaging. Slides were imaged by fluorescent Olympus IX71 inverted microscope (Olympus) and CoolSnap HQ2 CCD camera (Roper Scientific). The image-plane pixel dimension was 1.3 μm for 40× magnification. ImageJ (version 1.53a, National Institutes of Health) was used to analyze the images and calculate cardiomyocyte-projected area in a blinded manner. Each group had at least 3 biological replicates and 6 views for analysis.
For assessment of fibrosis, picrosirius red staining was used. OCT frozen LV samples were cut at 6-7 μm. Slides were rehydrated in distilled water, followed by picrosirius red solution (Abcam). After incubation, slides were washed with 1% acetic acid solution and dipped in absolute alcohol. Slides were dehydrated and cleared for imaging. Slides were imaged at 6× magnification (Olympus). Red fibrotic area was quantified in a blinded manner using ImageJ. Each group had at least 3 biological replicates and 6 views for analysis.
Statistical Analyses
Data were analyzed by 3-way analysis of variance (ANOVA) (genotype × sex × exercise) with post hoc Student’s t test to compare within-genotype and sex differences. Significance was set a priori at α < 0.05. Statistical tests were performed using SPSS Statistics Version 23 (IBM Corporation). Data are expressed as mean ± SEM.
Results
Cardiac-specific deletion of Bmal1 was confirmed by reverse transcription quantitative polymerase chain reaction (RT-qPCR) and Western blotting. The expression of Bmal1 mRNA and protein was ~50% lower in Bmal1 cKO hearts compared with WT controls (p < 0.05) (Figure 2a-2c), consistent with previous reports of inducible Bmal1 deletion (Liang et al., 2022). We then assessed the expression of circadian genes at ZT0 and ZT10, when Bmal1 expression is highest and lowest, respectively (Dierickx et al., 2022). As expected, the expression of Per2, Rev-erbα, and Clock varied by time of day in WT mice. However, time-of-day variation was blunted or even opposite with Bmal1 deletion (Figure 2d-2f). To confirm the tissue specificity of deletion of Bmal1, we compared Bmal1 mRNA and protein expression in hind limb muscle (soleus, gastrocnemius, and plantaris) and found that the expression did not differ between WT and Bmal1 cKO mice (Figure 2g).
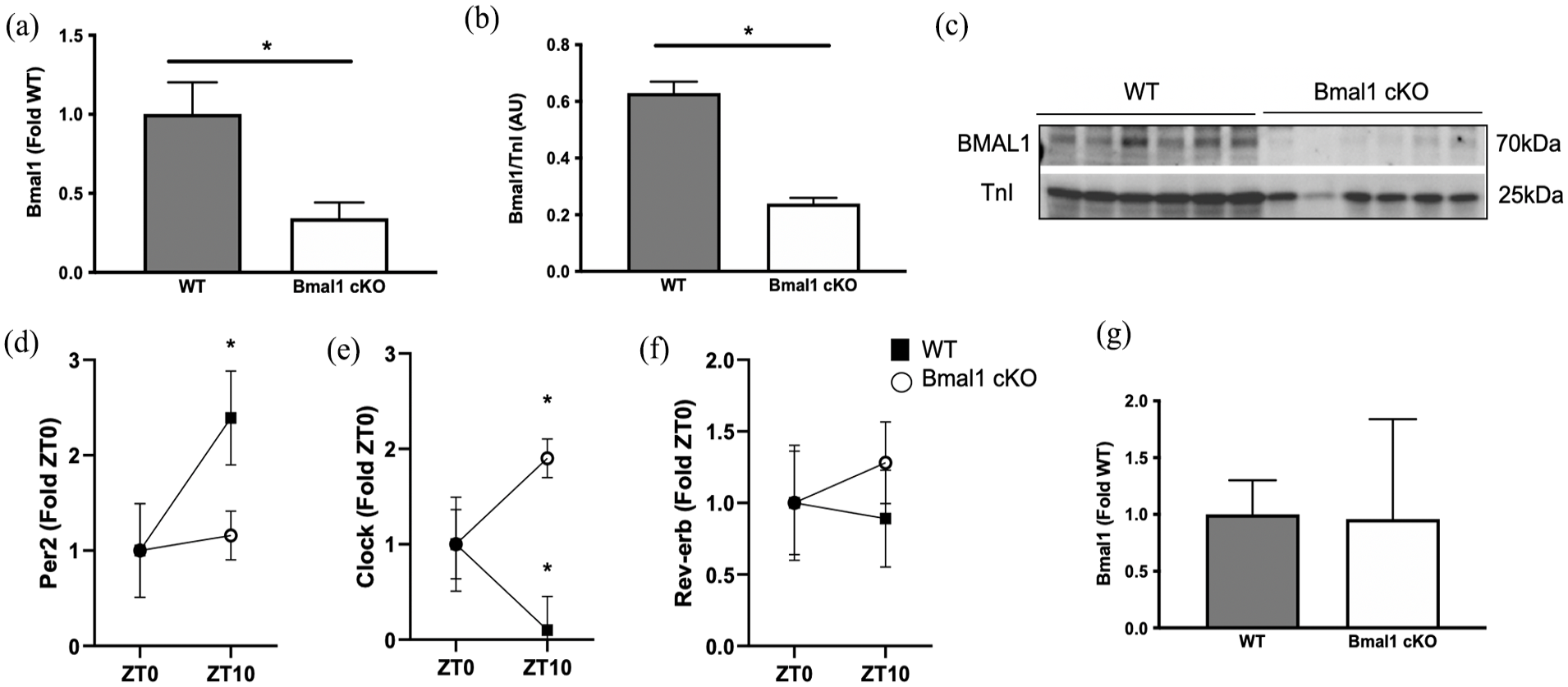
Cardiomyocyte-specific deletion of Bmal1. The expression of (a) mRNA and (b) protein was lower in the LV from Bmal1 cardiac knockout (Bmal1 cKO) mice compared with WT mice. (c) Representative immunoblot. Gene expression was assessed by qRT-PCR and expressed as fold relative to WT using ΔΔCt calculations. *p < 0.05 as assessed by Student’s t test to compare Bmal1 cKO and WT. (d) The expression of Per2 and (e) Clock in the LV was differentially expressed by day-night in WT animals but was not by deletion of Bmal1. (f) Rev-erb was not differentially expressed in WT or Bmal1 cKO by ZT. (g) The expression of Bmal1 in hind limb skeletal muscle was unchanged with cardiac deletion of Bmal1. Gene expression was assessed by qRT-PCR and expressed as fold relative to ZT0 using ΔΔCt calculations. Data are presented as mean ± SEM. *p < 0.05 as assessed by Student’s t test to compare ZT0 and ZT10 within genotype. n = 6-8 per group. Abbreviations: LV = left ventricle; WT = wild-type; qRT-PCR = quantitative reverse transcription polymerase chain reaction.
Total engagement in voluntary wheel running did not differ between WT and Bmal1 cKO mice (Figure 3a), nor did maximal exercise time to exhaustion (Figure 3b), suggesting total aerobic capacity was similar between genotypes. However, when analyzed on an hourly basis, both male and female Bmal1 cKO mice displayed shifted running patterns compared with WT mice (Figure 3c), with a higher percentage of running activity performed during the light period (Figure 3d). Since running patterns also serve as a measure of circadian rhythm (Aguilar-Arnal et al., 2013), we validated the disrupted diurnal running pattern with assessment of body temperature. BMAL1 cKO mice exhibited disruption of entrained circadian rhythms of body temperature compared with WT mice (Figure 4a). For the hour-by-hour analysis of body temperature, a 2-way repeated-measures ANOVA revealed significant genotype × time interaction effect, F23,92 = 2.529, p = 0.0009. Sidak post hoc tests revealed a significantly lower body temperature (p = 0.0321) in Bmal1 cKO mice compared with WT mice at ZT23 (ZT0 = lights-on) after adjusting for multiple comparisons. Furthermore, unpaired t tests (1-tailed) revealed that the bathyphase (daily trough) of body temperature was significantly earlier in BMAL1 cKO mice compared with WT mice (t = 2.142, df = 4, p = 0.0495) (Figure 4a′).
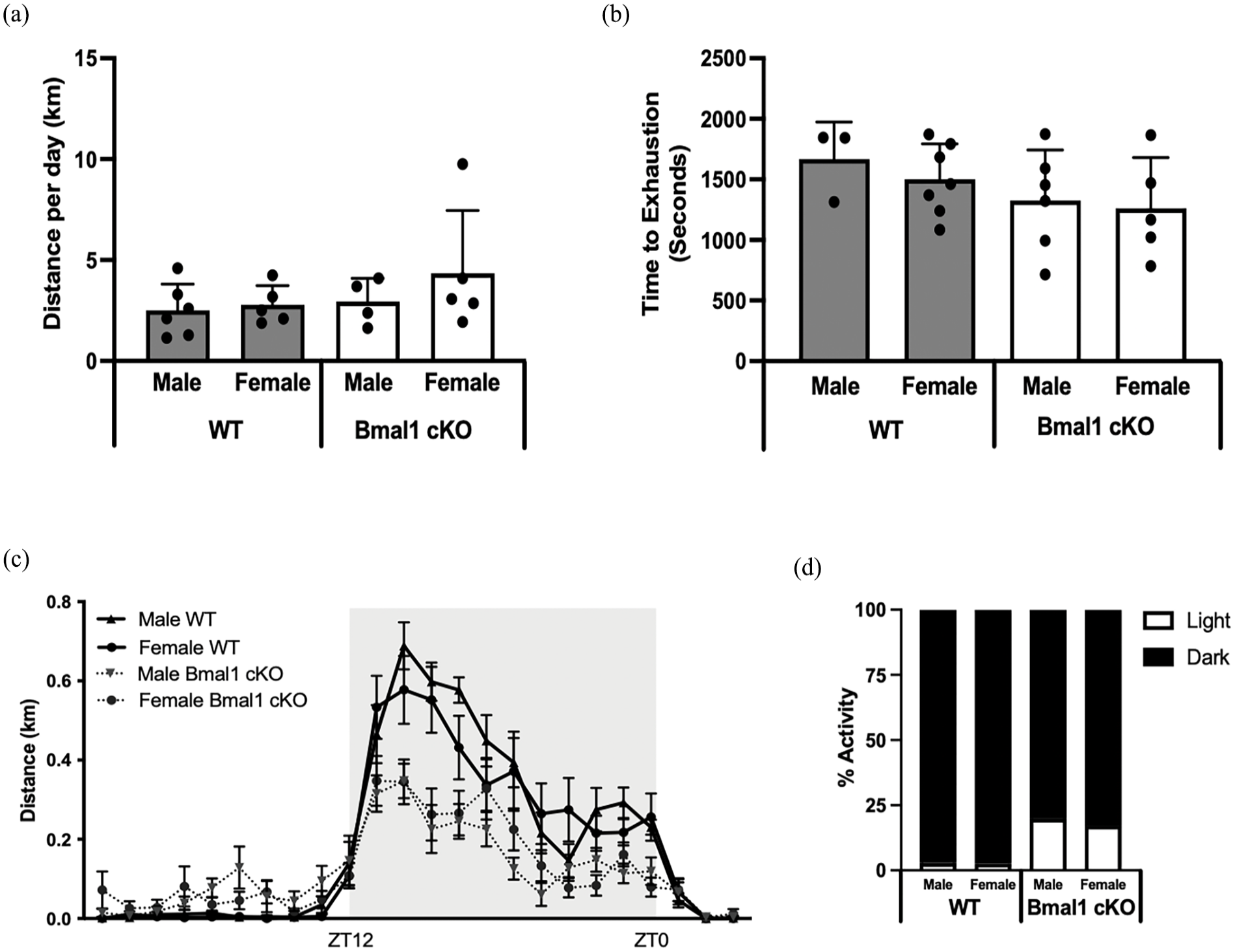
Engagement in voluntary wheel running in Bmal1 cKO and wild-type mice. (a) Daily voluntary running distance and (b) treadmill time to exhaustion did not differ between Bmal1 cKO and wild-type mice. (c) Hourly running distance with ZT12 indicating lights-off and ZT0 indicating lights-on. (d) Percent activity during light and dark periods demonstrating that Bmal1 cKO mice are more active in the light period compared with WT. n = 4-6 per group. Abbreviations: WT = wild-type; cKO = cardiac knockout.
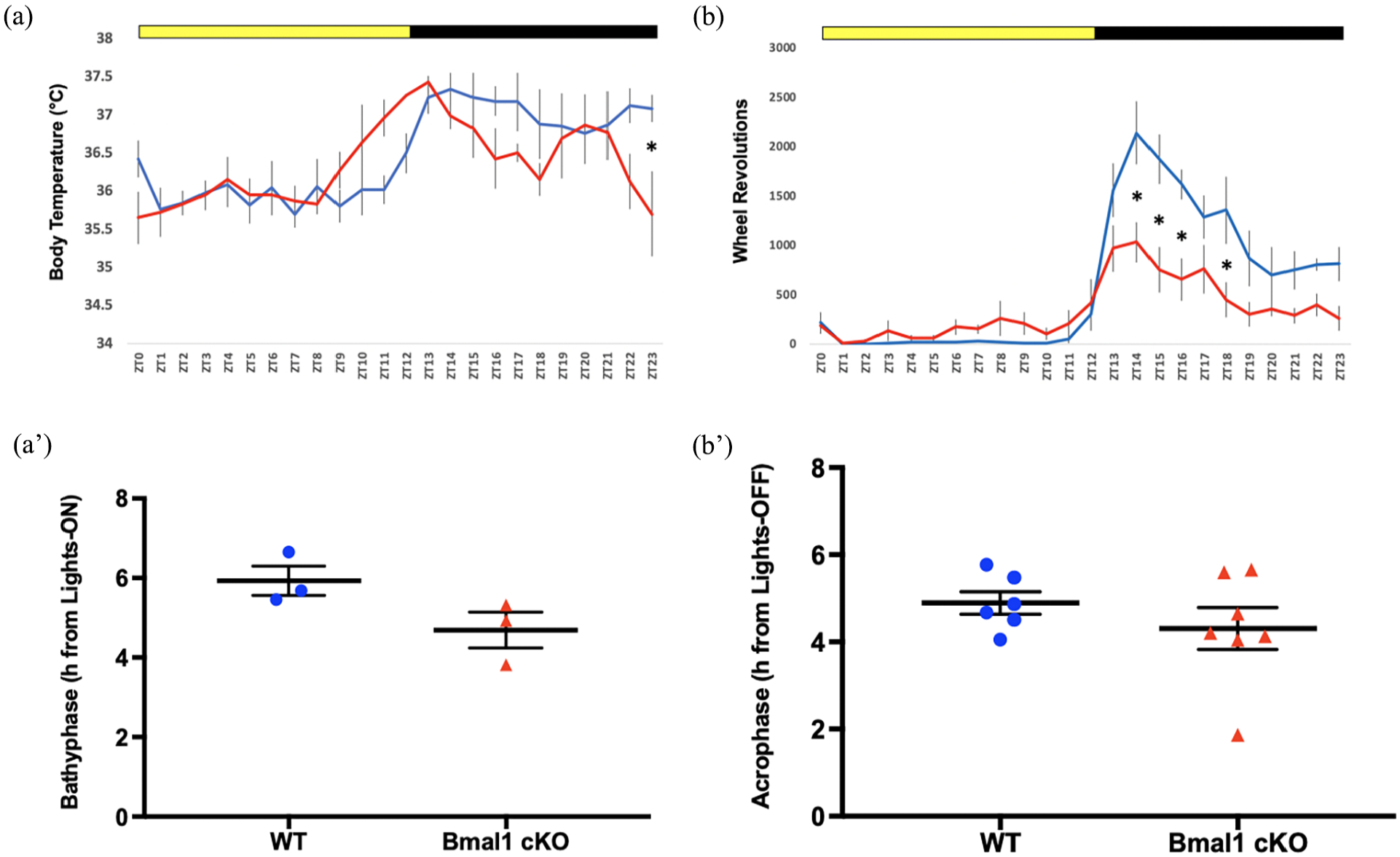
Circadian profiles in Bmal1 cKO and wild-type mice. (a) BMAL1 cKO mice exhibited disruption of entrained circadian rhythms of body temperature compared with WT mice and (a′) bathyphase (daily trough) of body temperature was significantly earlier in BMAL1 cKO mice compared with WT mice. (b) BMAL1 cKO mice also exhibited disruption of entrained circadian rhythms of wheel running, with hour-by-hour analysis of wheel running revealing a significant genotype × time interaction effect; however, (b′) the acrophase (daily peak) of wheel running was not significantly different between genotypes. n = 3-6 mice per group. Abbreviations: WT = wild-type; cKO = cardiac knockout.
BMAL1 cKO mice also exhibited disruption of entrained circadian rhythms of wheel running. For the hour-by-hour analysis of wheel running, a 2-way repeated-measures ANOVA revealed a significant genotype × time interaction effect, F23,253 = 5.521, p < 0.0001 (Figure 4b). Sidak post hoc tests revealed significantly lower wheel running in Bmal1 cKO mice compared with WT mice at ZT14 (p < 0.0001), ZT15 (p < 0.0001), ZT16 (p = 0.0005), and ZT18 (p = 0.0014) after adjusting for multiple comparisons. However, the acrophase (daily peak) of wheel running was not significantly different between genotypes (t = 1.019, df = 11, unpaired, 1-tailed, p = 0.165) (Figure 4b′). Actograms comparing differences in body temperature (Figure 5a) and wheel running (Figure 5b) also show the blunted wheel running rhythms and body temperature discrepancies.
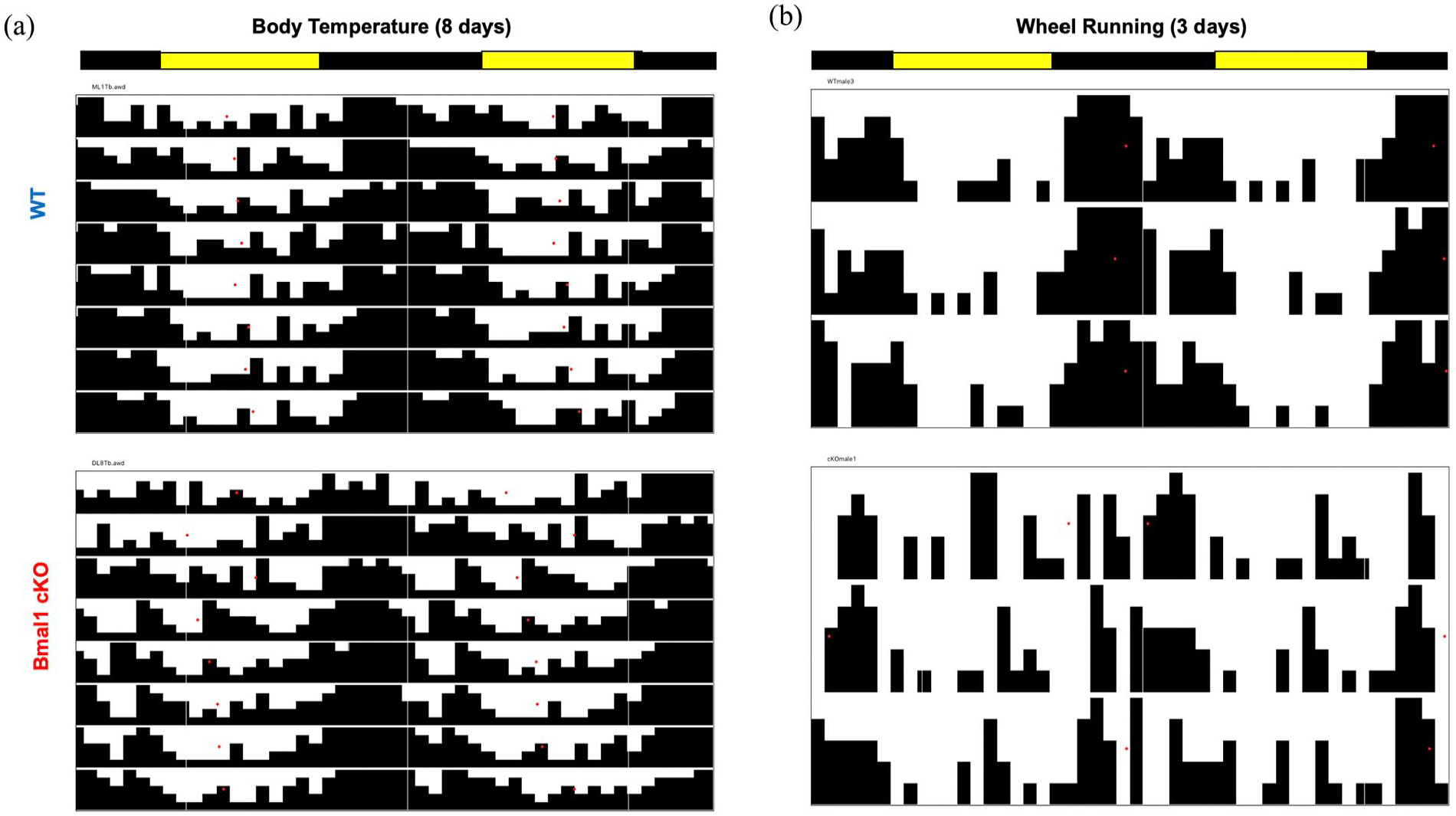
(a) Actograms comparing differences in body temperature between WT and Bmal1 cKO mice and (b) wheel running also showing the blunted wheel running rhythms and body temperature fluctuations. Abbreviations: WT = wild-type; cKO = cardiac knockout.
We then analyzed cardiac remodeling to determine the impact of Bmal1 deletion on cardiac structure and function. Cardiac-specific deletion of Bmal1 resulted in higher cardiac and LV mass as normalized to tibia length (Figure 6a and 6b). As expected, female WT mice underwent elevations in cardiac and LV mass with voluntary wheel running. Running wheel engagement also resulted in elevated cardiac and LV mass in Bmal1 cKO mice and this was exacerbated compared with WT animals as evidenced by the significant interaction between genotype and running. Cardiac myocytes were larger in Bmal1 cKO and this was exacerbated by wheel running, confirming hypertrophic remodeling (Figure 6c). Interestingly, cardiac fibrosis was also higher in hearts from Bmal1 cKO compared with WT mice (Figure 6d) and this was exacerbated in Bmal1 cKO mice. Bmal1 cKO mice developed impaired systolic function as evidenced by lower EF, FS, and fractional area change (FAC) compared with WT (Figure 7a-7d). While running modestly improved systolic function in WT animals, it did not impact systolic function in the setting of Bmal1 cardiac deletion. Together, these data suggest that exercise-induced cardiac remodeling is impaired in the setting of Bmal1 deletion and that running is likely not cardioprotective in this model. All echocardiographic analyses are presented in Table 1.
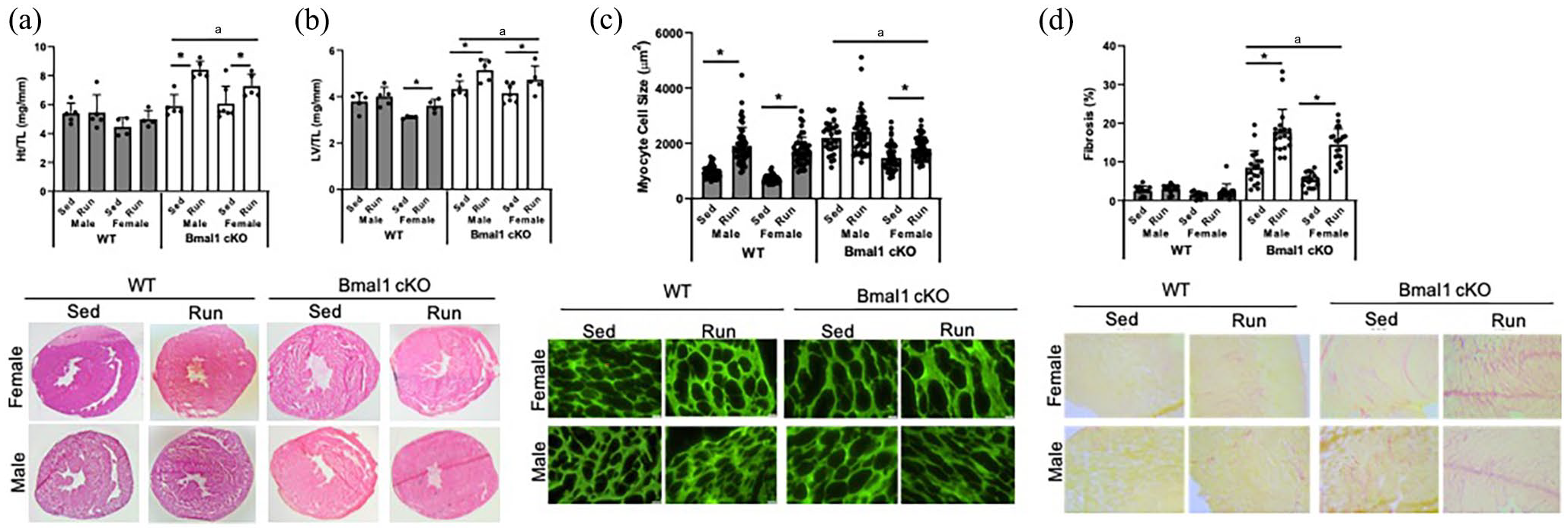
Cardiac remodeling in Bmal1 cKO mice and the impact of exercise on cardiac remodeling. (a) Heart weight normalized to tibia length (HW/TL) differed by genotype with higher cardiac mass in Bmal1 cKO mice. (b) LV weight normalized to TL (LV/TL) differed by genotype and sex. H&E-stained images of whole hearts from male and female WT and Bmal1 cKO mice. (c) Cardiac myocyte cell size differed by genotype, sex, and genotype × sex interaction. Representative images (40× magnification) from lectin staining of LV sections. (d) Fibrosis differed by genotype and sex. Representative images (6× magnification) from Picrosirius red staining. Data are presented as means ± SEM. Effect of genotype, sex, and running was assessed by 3-way ANOVA with post hoc Student’s t test.Abbreviations: WT = wild-type; TL = tibia length; LV = left ventricle; ANOVA = analysis of variance; cKO = cardiac knockout; Sed = sedentary; Run = wheel running.
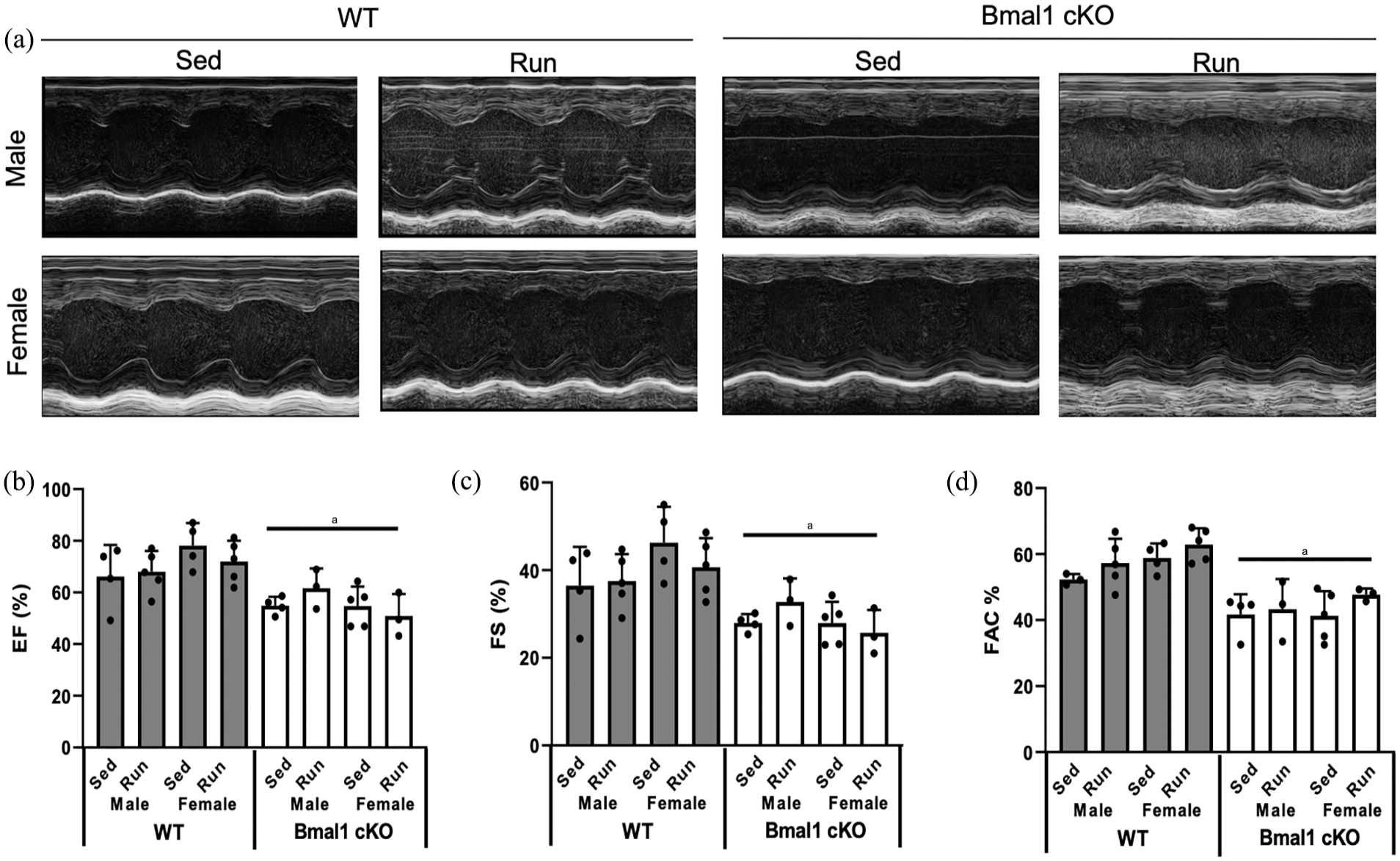
Cardiac function in WT and Bmal1 cKO mice. (a) Representative images from M-mode echocardiography in sedentary and wheel running WT and Bmal1 cKO mice; (b) EF: ejection fraction; (c) FS: fractional shortening; (d) FAC: fractional area change. Effect of genotype, sex, and running was assessed by 3-way ANOVA with post hoc Student’s t test. n Abbreviations: WT = wild-type; ANOVA = analysis of variance; cKO = cardiac knockout.
Echocardiography in sedentary (Sed) and wheel running (Run) Bmal1 cardiac knockout (Bmal 1cKO) and wild-type (WT) mice.
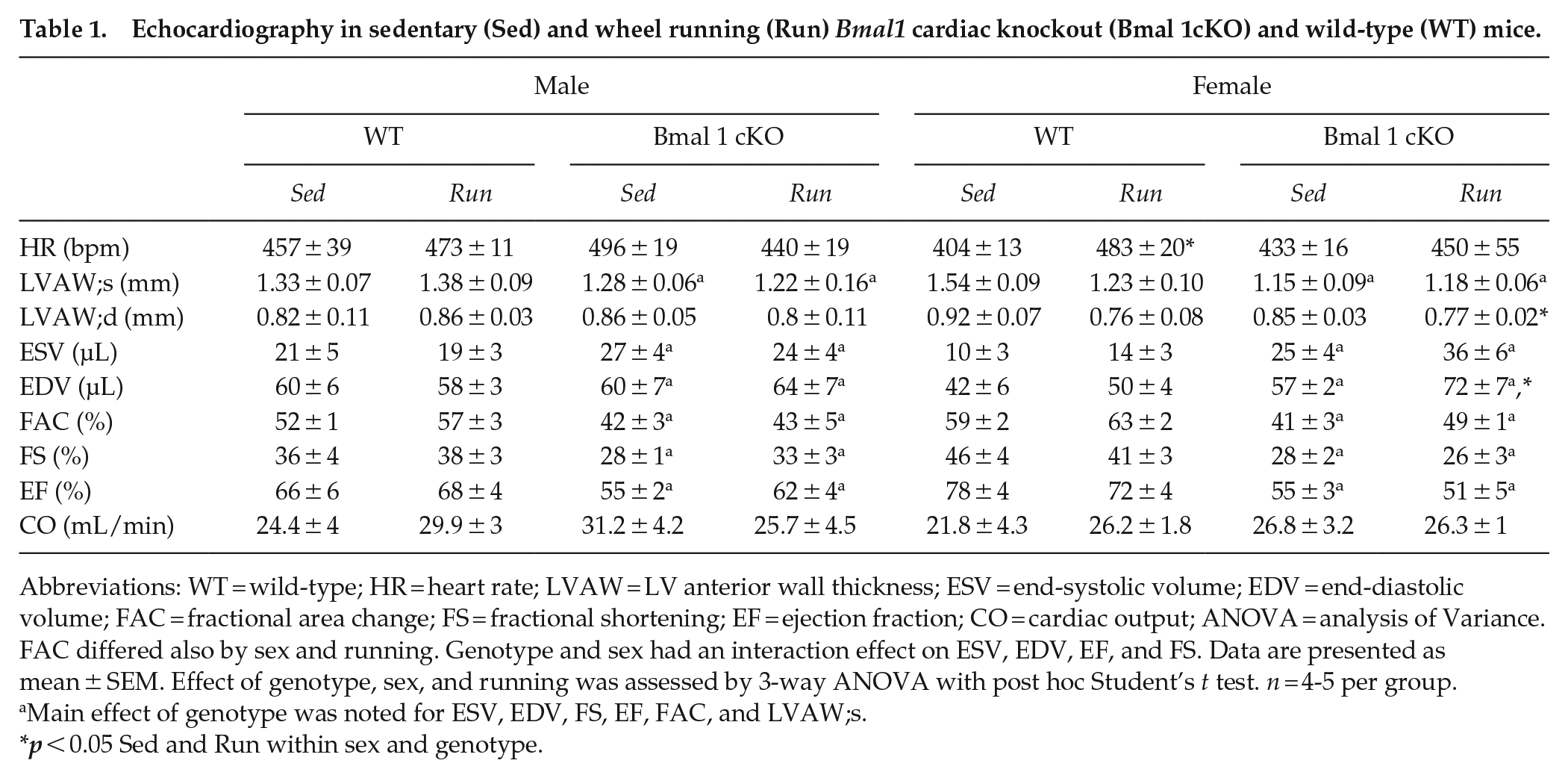
Abbreviations: WT = wild-type; HR = heart rate; LVAW = LV anterior wall thickness; ESV = end-systolic volume; EDV = end-diastolic volume; FAC = fractional area change; FS = fractional shortening; EF = ejection fraction; CO = cardiac output; ANOVA = analysis of Variance. FAC differed also by sex and running. Genotype and sex had an interaction effect on ESV, EDV, EF, and FS. Data are presented as mean ± SEM. Effect of genotype, sex, and running was assessed by 3-way ANOVA with post hoc Student’s t test. n = 4-5 per group.
Main effect of genotype was noted for ESV, EDV, FS, EF, FAC, and LVAW;s.
p < 0.05 Sed and Run within sex and genotype.
To begin to determine the mechanisms by which cardiac deletion of Bmal1 results in cardiac hypertrophy, we quantified several regulators of cardiac growth. Cardiac natriuretic peptides correlate with cardiac stretch in disease (Wang et al., 2004); thus, we analyzed the protein expression of atrial naturetic factor (ANP). Cardiac deletion of Bmal1 resulted in higher expression of ANP (Figure 8a and 8b). The expression of myostatin, a negative regulator of cardiac mass (Morissette et al., 2006), tended to be higher in Bmal1 cKO compared with WT mice (p = 0.09), but the expression was not impacted by running (Figure 8c). Mammalian target of rapamycin (mTOR) is a potent regulator of cardiac growth (Sciarretta et al., 2018); thus, we assessed its activation by phosphorylation and downstream targets S6 ribosomal protein (RPS6) and 4E-BP1. mTOR activation was generally unchanged by deletion of Bmal1 (Figure 8e) although running inhibited mTOR in WT mice, but activated mTOR in male Bmal1 cKO. mTOR target 4E-BP1 was inhibited in Bmal1 cKO mice (Figure 8f), while rps6 was not different between genotypes (Figure 8g). The protein kinase Akt alters mTOR activity to regulate cardiac hypertrophy (Shiojima et al., 2005); however, phosphorylation of Akt was largely unchanged by Bmal1 deletion or running (Figure 8h). Quantification of these regulators of the hypertrophic phenotype occurred at ZT0, the time at which mTOR signaling has been previously demonstrated to be most active (Khapre et al., 2014). However, the activity of mTOR and its targets at ZT12 were also not different between Bmal1 cKO and WT mice, suggesting that activation of this pathway is likely not contributory to the hypertrophic phenotype in our model (data not shown).
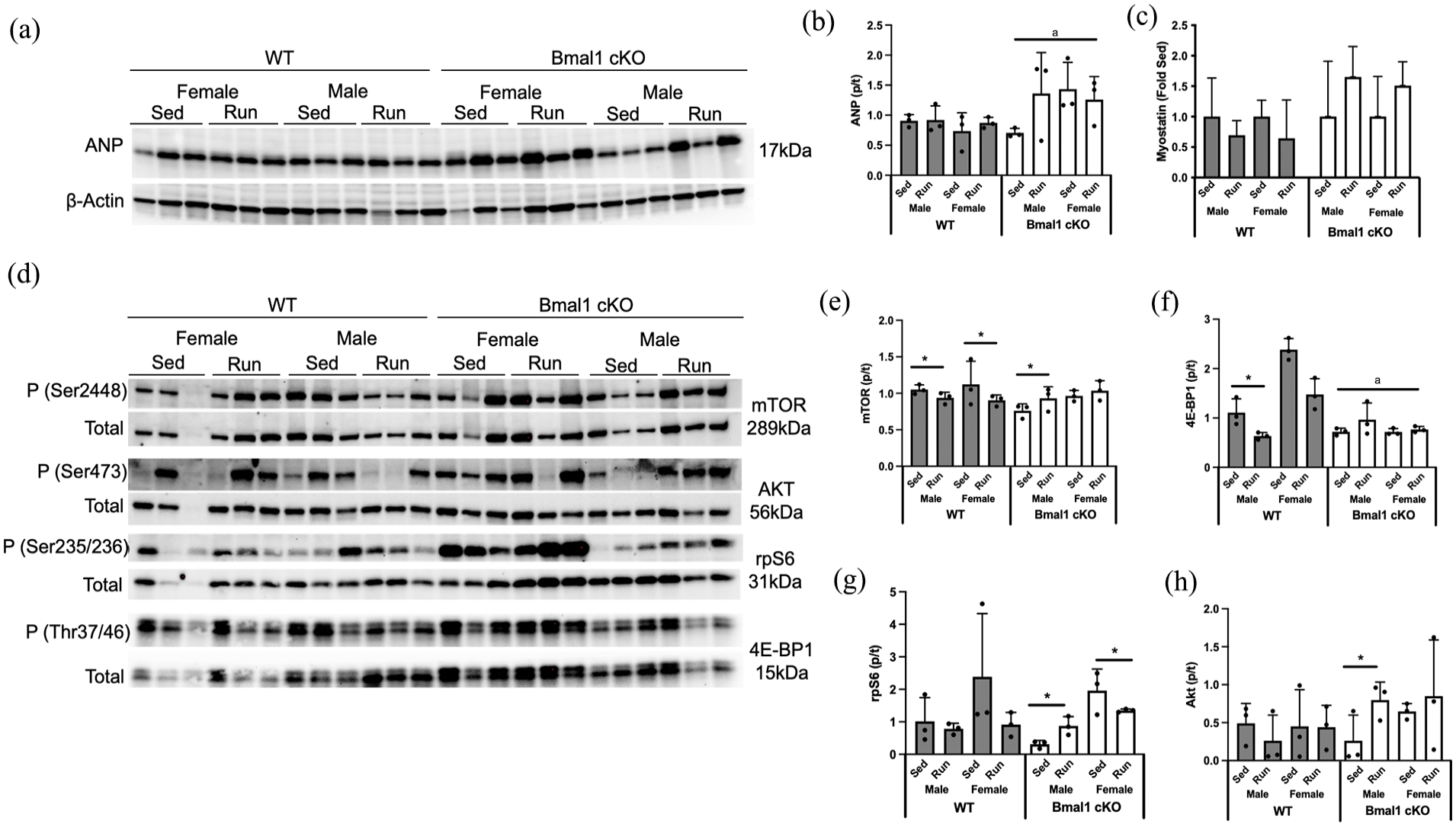
Mechanisms of cardiac hypertrophy in sedentary (Sed) and wheel running (Run) wild-type (WT) and Bmal1 cardiac knockout (Bmal1 cKO) mice. (a, b) ANP expression was higher with deletion of BMAL1. (c) Myostatin gene expression tended to be lower with Bmal1 cKO (p = 0.09) but was not impacted by running. (d) Regulators of cardiac growth also differed by genotype. (e) mTOR did not differ by genotype but was lower in WT mice in response to running and activated in male Bmal1 cKO mice. mTOR target (f) Rps6 was higher in female mice compared with male mice but did not differ by genotype or running, while (g) 4EBP1 was lower in Bmal1 cKO compared with WT mice, higher with running, and demonstrated a significant interaction between genotype × run. (h) Akt activation did not differ by genotype, sex, or running. Effect of genotype, sex and running was assessed by 3-way ANOVA with post hoc Student’s t test. Abbreviations: ANP = atrial naturetic factor; mTOR = mammalian target of rapamycin; ANOVA = analysis of variance.
Given the strong relationship between metabolic remodeling and cardiac hypertrophy (Gibb and Hill, 2018), we quantified the expression of key metabolic regulators. Fold difference within sex and genotype sedentary is presented to reflect differences in gene expression with running. However, we also performed 2-way ANOVA on ΔCt values to determine the impact of genotype on expression. Peroxisome proliferator-activated receptor-α (PPARα) expression was lower in Bmal1 cKO hearts compared with WT (Figure 8a). Peroxisome proliferator-activated receptor-gamma coativator-1α (PGC-1α), glucose transporter 4 (GLUT4), pyruvate dehydrogenase kinase (PDK4), cytochrome c oxidase 4 (COX4), and carnitine palmitoyltransferase 1 (CPT1) were all unchanged by Bmal1 deletion or by running (Figure 9b-9e), together suggesting modest changes in metabolic gene expression that likely do not significantly contribute to the phenotype observed in our model.
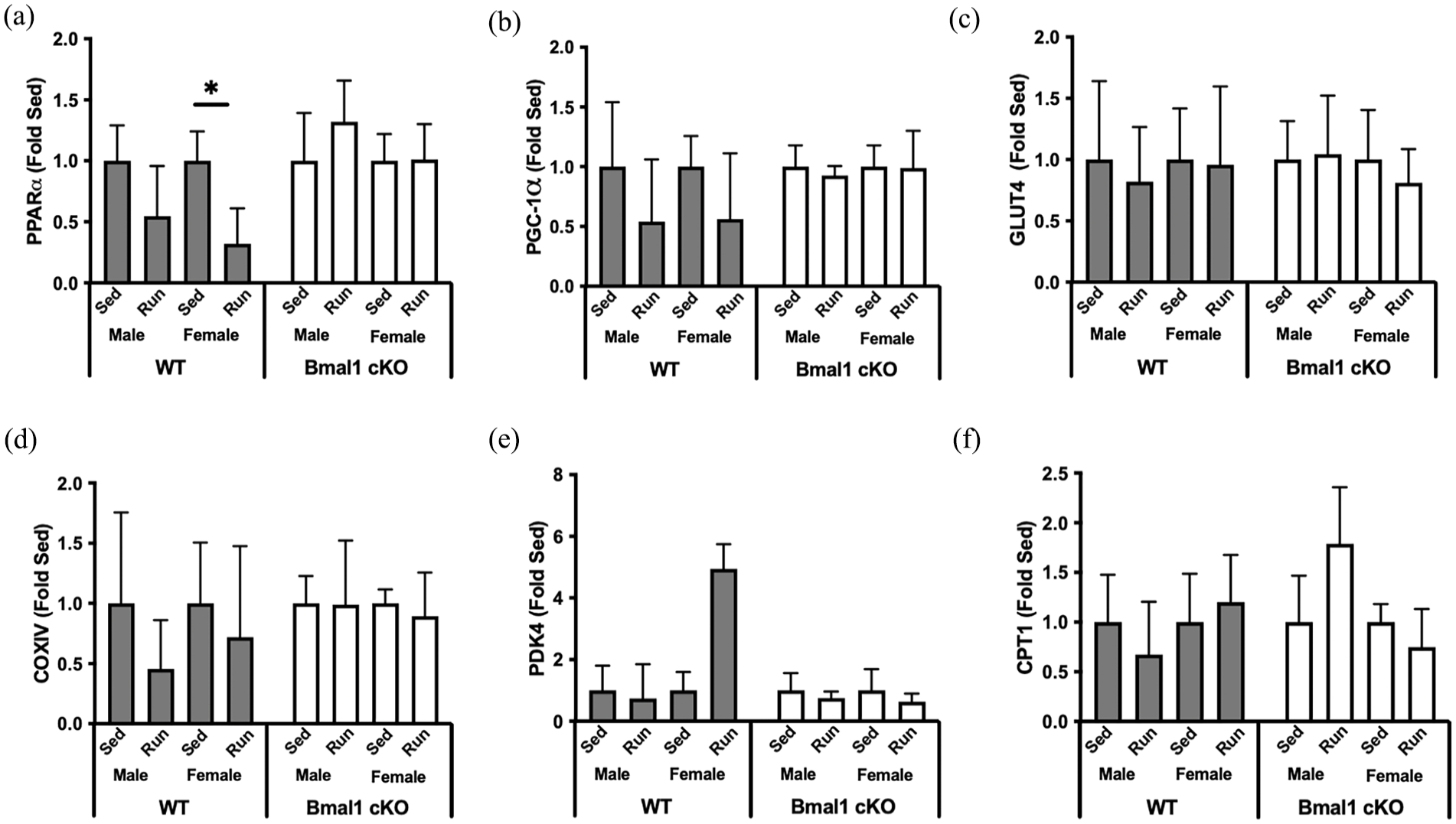
Metabolic regulators of cardiac remodeling in sedentary (Sed) and wheel running (Run) wild-type (WT) and Bmal1 cardiac knockout (Bmal1 cKO) mice. (a) The expression of PPARα was lower in Bmal1 cKO mice compared with WT mice and was lower in WT female runners. Main effect of genotype, run, and genotype × run. (b) PGC-1α, (c) GLUT4, (d) COXIV, (e) PDK4, and (f) CPT1 were unchanged by BMAL1 deletion or by running. Effect of genotype, sex, and running was assessed by 3-way ANOVA with post hoc Student’s t test. n = 4 per group. Abbreviations: PPARα = peroxisome proliferator-activated receptor-α; PGC-1α = peroxisome proliferator-activated receptor-gamma coativator-1α; GLUT4 = glucose transporter 4; COXIV = cytochrome c oxidase 4; PDK4 = pyruvate dehydrogenase kinase; CPT1 = carnitine palmitoyltransferase 1; ANOVA = analysis of variance.
Discussion
Critical biological processes in the heart are under circadian control. Disruption of circadian rhythm is strongly linked to cardiovascular diseases (Rudic and Fulton, 2009; Young and Bray, 2007). In the current work, we demonstrated that postnatal deletion of Bmal1 in cardiac myocytes results in a hypertrophic and fibrotic phenotype concomitant with impaired systolic function. Interestingly, cardiac deletion of Bmal1 also disrupted systemic circadian rhythm, suggesting for the first time that the heart may regulate circadian activity. Voluntary wheel running did not rescue cardiac structure or function and in some cases exacerbated cardiac remodeling, suggesting that in the setting of a heart with a broken clock, exercise may not be cardioprotective. Given the growing number of individuals experiencing chronic disruption of circadian rhythm, understanding the cardiac clock and identification of therapeutics to fix it is warranted.
Targeted loss-of-function studies have attempted to elucidate the role of the cardiac circadian clock in regulating cardiac function. The first of such models, the CCM mouse (Durgan et al., 2011), demonstrated higher cardiac mass alongside septal wall thickening and myocyte hypertrophy but with no differences in cardiac fibrosis compared with WT controls (Durgan et al., 2011). However, despite elevated cardiac mass, systolic function as measured by FS and EF was not impaired. CLOCK and BMAL1 co-transcriptionally regulate circadian gene expression; thus, subsequent studies aimed to identify whether cardiac deletion of Bmal1 resulted in similar phenotypes as Clock deletion. Myocyte cross-sectional area was unchanged in CBK mice at 12 and 36 weeks of age with elevated myocardial fibrosis only evident by 36 weeks (Young et al., 2014). CBK mice developed diastolic dysfunction, though interestingly these mice also showed impaired systolic function as evidenced by a reduced EF (Ingle et al., 2015). Myocyte-specific deletion of nuclear receptor and repressive clock component Rev-erb also resulted in mild hypertrophy, systolic impairment, and premature mortality (Dierickx et al., 2022). However, recent work with inducible Bmal1 deletion in the postnatal heart did not result in hypertrophy or fibrosis (Liang et al., 2022). Collectively, although these reports demonstrate that deletion of cardiac clock genes causes cardiac dysfunction, the cardiac phenotypes and ultimately mechanisms by which clock genes cause cardiac dysfunction clearly lack consensus. The present work adds to this collective knowledge by demonstrating that inducible deletion of cardiac Bmal1 in adulthood causes cardiac hypertrophy, fibrosis, and systolic dysfunction. The reasons for the differences in cardiac phenotype in these various models are currently not clear, but are likely due to differences in the timing of gene deletion (constitutive or inducible) as well as the temporal nature of the disease.
The mechanisms by which deletion of a core circadian gene causes cardiac remodeling are still without clarity. Early microarray experiments in the CBK heart identified 19 differentially expressed genes that contained putative BMAL1-binding sites. Of these, 16 genes were also enriched in the CCM heart. Gene ontology analysis suggested that these genes enriched 3 main categories: clock function, metabolism, and cell signaling, none of which have direct hypertrophic roles (Young et al., 2014). However, a few more recent lines of work have attempted to identify hypertrophic pathways, with efforts focused on the master regulator of cellular growth mTOR. BMAL1 is a negative regulator of mTOR, and mTOR peaks at ZT2 in the heart in a BMAL1-dependent fashion (Khapre et al., 2014). CBK mice have elevated mTOR activation compared with wild-type mice (McGinnis et al., 2017). Additional proof of concept for mTOR-mediated hypertrophy in the CBK mouse comes from the rescue of the hypertrophic phenotype by short-term inhibition of mTOR by rapamycin (McGinnis et al., 2017). However, in our hands, we did not demonstrate elevated mTOR activity, despite measuring activation at ZT0 when mTOR activity is high (McGinnis et al., 2017). Furthermore, elevated mTOR activity was not noted in the hearts from mice that engaged in wheel running, despite exacerbated hypertrophy in these hearts, suggesting that other mechanisms are likely involved. Therefore, we also measured metabolic signaling, given the robust causative or at least correlative link between remodeling and cardiac metabolism (Gibb and Hill, 2018). However, we did not note robust differences in metabolic gene expression between Bmal1 cKO mice and WT, nor did exercise impact expression. More recent work in the CBK mouse has demonstrated elevated growth hormone sensitivity in the heart (Sonkar et al., 2022), suggesting this may be a mechanism of hypertrophy. Further adding to the complexity of circadian-induced cardiac remodeling is the role of fibrosis. Previous work (Liang et al., 2022) and the data we present here demonstrate that cardiac-specific deletion of Bmal1 results in fibrotic remodeling. Silencing of Bmal1 in H9c2 cells exacerbates myofibroblast activation phenotype via proposed secretory mechanisms (Liang et al., 2022). Therefore, it is highly plausible that other cardiac cell types such as fibroblasts contribute to the cardiac remodeling phenotype noted with cardiac circadian disruption. Taken together, deletion of a clock gene in the heart induces distinct cardiac phenotypes, yet understanding how these phenotypes emerge is still in its infancy.
The cardiac circadian clock influences the responsiveness of the heart to external stimuli, though to date most efforts have focused on pathological stresses (Durgan et al., 2011; Durgan et al., 2010; Liang et al., 2022). Here, we aimed to understand how a physiological stress would induce cardiac remodeling and impact circadian rhythm. Regular endurance exercise is among the most cardioprotective interventions identified to date. Voluntary wheel running stimulates physiological cardiac remodeling, characterized by a ~10% increase in cardiac mass concomitant with improved cardiac function (Allen et al., 2001). In addition to stimulating beneficial cardiac remodeling, exercise is an emerging zeitgeber (Wolff and Esser, 2012), although this finding lacks consensus (Maier et al., 2022). Based on the cardioprotective properties of exercise and the suggestion that exercise can act as a zeitgeber, we expected that voluntary wheel running in Bmal1 cKO mice would be beneficial. However, we were surprised to find that exercise exacerbated maladaptive cardiac remodeling as evidenced by elevated myocyte size and cardiac fibrosis, and without improvements in cardiac function. These changes were also accompanied by elevated ANP expression, suggesting that the changes to the exercised heart were not likely adaptive (McMullen and Jennings, 2007). An important consideration for exercise-mediated health benefits is the volume of exercise activity. Previous reports have demonstrated that CCM mice have lower total wheel running activity and shorter bout length than WT mice. The authors proposed that this lower activity was due to cardiac-related decrements in exercise capacity (Ko et al., 2011), but capacity was not directly tested. Our data show that Bmal1 cKO mice have similar exercise capacity as evidenced by treadmill time to exhaustion, consistent with the observation that Bmal1 cKO mice ran similar distance as WT mice. We acknowledge that we did not measure run time or speed; thus, it is possible that Bmal1 deletion impacts exercise intensity. However, we did note a profound shifted hourly pattern of running wheel engagement, with Bmal1 cKO mice engaging in a high amount of activity during the light period. We hypothesize that the shift in activity from dark to light, despite equal engagement in activity, precludes exercise from being cardioprotective. That is, given that the heart undergoes repair during the subjective night when activity is normally lowest and physiological stress is at a minimum (Sole and Martino, 2009), the loss of this rest period with cardiac Bmal1 deletion interferes with the beneficial adaptation to exercise. Future work should aim to understand the health benefits of timing of exercise, particularly with respect to emerging understanding of circadian regulation of cardiac health.
In addition to impairing cardiac function, deletion of cardiac Bmal1 also resulted in aberrant circadian rhythm of running wheel engagement and core body temperature. These data suggest that the cardiac clock also regulates entrained circadian rhythm, likely independent of suprachiasmatic nucleus (SCN) control, because in our model, the SCN is presumably intact. Given that we did see a blunting of wheel running rhythms, as well as differences in body temperature, the lack of a significant difference in wheel running acrophase may reflect that sensory feedback from wheel running is masking underlying circadian dysfunction. Indeed, at least 2 previous studies have suggested that wheel running may not track as well with circadian phase compared with body temperature or general cage locomotor activity when mice have altered circadian phenotypes (Todd et al., 2020; Hannibal et al., 2011). Taken together, these data demonstrate that Bmal1 cKO mice have greater variability in circadian cycle core temperature contributing to a dampened amplitude in period power.
Although light is the primary regulator of circadian entrainment via the SCN, peripheral tissues such as the heart can be synchronized by food (Mistlberger, 1994) and temperature (Brown et al., 2002), among other zeitgebers (Durgan et al., 2005). Based on this finding, we propose that the heart can function as a driver of the molecular clock even though current paradigm suggests that the SCN acts as the master clock. Support for this hypothesis comes from non-cardiac models where the hepatocyte clock has been shown to control the expression of non-hepatocyte circadian genes, as evidenced by deletion of hepatocyte-specific Rev-erb-shifting rhythmic behavior of neighboring endothelial cells (Guan et al., 2020). Furthermore, liver-specific deletion of Bmal1 subtly increased the amplitude of clock gene expression in muscle (Lamia et al., 2008). Thus, while the field to date has not yet recognized the heart as a regulator of non-cardiac rhythm, we believe new details will emerge as more data are gathered about the molecular circadian crosstalk between tissues, independent of the SCN.
Limitations and Conclusions
Single deletion of most clock genes results in compensatory changes in others, thus limiting the impact of single-gene manipulation on circadian function. Bmal1 is the only single-gene mutation that fully eliminates circadian clock function (Bunger et al., 2000). In contrast to previous work that utilized constitutive deletion of Bmal1, the expression of Bmal1 in our mice was inducible. Therefore, Bmal1 expression was not manipulated until just before our experimental paradigms at 6-8 weeks of age. Given that early-in-life exposures are emerging as significant regulators of adult phenotypes including circadian disruption (Smarr et al., 2017) and that cardiac myocytes are non-proliferative during adult life (Soonpaa and Field, 1998), our temporal approach is a more robust translational model of adulthood circadian disruptions like shift work, jet lag, and aging. We also note that temporal differences may underlie the differences reported here in our data compared with the literature. That is, in the constitutive models, assessments were made from 8 to 36 weeks of age. Here, we quantified circadian and cardiac functions at ~12 weeks of age, but with recent Bmal1 deletion only 4 weeks prior. Therefore, it is feasible that were our mice to age, the phenotype may change, given that temporal outcomes of cardiac clock deletion are still unclear. Mice in this study were bred and raised in Laramie, Wyoming at 7200 feet. Altitude not only impacts running distances, but also contributes to differences in body weight, cardiac remodeling, and other physiological outcomes. Although the data are not yet clear, it also appears as though altitude may impact circadian rhythm (Mortola and Seifert, 2000). These biological outcomes of moderate altitude and hypoxia are currently under investigation in our lab, and thus we caution direct comparison of future studies to the data presented here without consideration of local oxygen tension.
The focus of the current work was on the cardiac clock and cardiac function. However, we acknowledge that non-cardiac systems are also critical with respect to both circadian rhythm and cardiac function. Perhaps the most obvious example is the brain which coordinately controls circadian rhythm input to peripheral tissues. We did not measure molecular signatures in the brain and it is possible that cardiac myocyte deletion of Bmal1 resulted in compensatory changes in clock function in the SCN or other areas responsible for regulating rhythm and cardiac function. Future experimental approaches are required to address these fundamental questions.
Footnotes
Acknowledgements
This publication was made possible by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grant P20GM121310-06. The authors thank Zackery Fullerton for technical assistance.
Author Contributions
DRB and EES designed the study; MY, AY, and SMP performed the research; MY, WDT, EES, and DRB wrote the manuscript. All authors analyzed the data, and revised and approved the manuscript.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.