
Abstract
The suprachiasmatic nucleus (SCN) drives and synchronizes daily rhythms at the cellular level via transcriptional-translational feedback loops comprising clock genes such as Bmal1 and Period (Per). Glycogen synthase kinase 3 (GSK3), a serine/threonine kinase, phosphorylates at least 5 core clock proteins and shows diurnal variation in phosphorylation state (inactivation) of the GSK3β isoform. Whether phosphorylation of the other primary isoform (GSK3α) varies across the subjective day-night cycle is unknown. The purpose of this study was to determine if the endogenous rhythm of GSK3 (α and β) phosphorylation is critical for rhythmic BMAL1 expression and normal amplitude and periodicity of the molecular clock in the SCN. Significant circadian rhythmicity of phosphorylated GSK3 (α and β) was observed in the SCN from wild-type mice housed in constant darkness for 2 weeks. Importantly, chronic activation of both GSK3 isoforms impaired rhythmicity of the GSK3 target BMAL1. Furthermore, chronic pharmacological inhibition of GSK3 with 20 µM CHIR-99021 enhanced the amplitude and shortened the period of PER2::luciferase rhythms in organotypic SCN slice cultures. These results support the model that GSK3 activity status is regulated by the circadian clock and that GSK3 feeds back to regulate the molecular clock amplitude in the SCN.
At the cellular level, 24-h timing is maintained by transcriptional-translational feedback loops resulting in rhythmic expression of core “clock genes,” in which the activators (CLOCK and BMAL1) are expressed in antiphase to the suppressors (PERs and CRYs; Partch et al., 2014, for review). A secondary feedback loop consists of CLOCK-BMAL1 activation of a nuclear orphan receptor REV-ERBα, which feeds back to transcriptionally repress Bmal1. The length of the molecular clock cycle can be regulated by posttranslational events, including phosphorylation by kinases such as casein kinase I enzymes (Partch et al., 2014). We know much less about how other kinases, such as glycogen synthase kinase 3 (GSK3), regulate the period and/or amplitude of the molecular clock, especially in neurons of the central circadian pacemaker, the suprachiasmatic nucleus (SCN) of the hypothalamus. Given that dysregulated GSK3 signaling has been implicated in psychiatric and neurological disorders (Hooper et al., 2008; Li and Jope, 2010; Duncan and Zee, 2013), and many therapeutic drugs target GSK3 (e.g., lithium, valproate, risperidone, etc.; Li and Jope, 2010), important questions to address are how GSK3 activity varies over the course of the day and how the molecular clock is affected by these rhythms.
A key feature of GSK3 is that it is active in its default state and that its 2 isoforms are inactivated by phosphorylation (at Ser-21 for GSK3α [S21-GSK3α] and Ser-9 for GSK3β [S9-GSK3β]). This critically important phosphorylation state balance of S9-GSK3β changes across the light-dark cycle in the SCN (Iwahana et al., 2004; Iitaka et al., 2005). In some brain regions, the 2 distinct GSK3 isoforms can have functional differences, such as differences in substrate recognition, axonal growth, regulation of synaptic plasticity, and formation of senile plaques in an Alzheimer mouse model (Castaño et al., 2010; Soutar et al., 2010; Hurtado et al., 2012; Shahab et al., 2014). Therefore, it is important to determine whether phosphorylation of S21-GSKα also varies across the subjective day-night cycle in the SCN. Because rhythmic phosphorylation of S9-GSK3β is dependent on the molecular clock in peripheral tissues such as the heart (Durgan and Young, 2010), we hypothesized that rhythmic phosphorylation of GSK3α and GSK3β persists in constant darkness in the SCN.
Using Western blot analysis (as in Paul et al., 2012), we investigated p-GSK3α and p-GSK3β levels in mice housed in constant darkness (DD) for 14 to 20 days. Both p-GSK3α and p-GSK3β (as a percentage of total GSK3α or GSK3β, respectively) displayed a statistically significant rhythm (n = 27/time course; p < 0.05; Figure 1A,C,E) during DD conditions. Interestingly, p-GSK3α peaked during the late subjective night (circadian time or CT phase, 20.94 ± 0.47, where CT 12 refers to activity onset; mesor, 1.02 ± 0.51; amplitude, 0.41 ± 0.08). In contrast, p-GSK3β peaked during the early subjective day (circadian time or CT phase, 2.93 ± 1.45; mesor, 1.51 ± 0.16; amplitude, 0.61 ± 0.22). There were no significant differences in total GSK3α or GSK3β expression with respect to time (cosinor nonlinear regression, p > 0.05; Figure 1A,D,F), consistent with previous reports in the SCN and liver from mice housed in LD (Iitaka et al., 2005). The persistence of rhythmic GSK3α/β inactivation in the absence of light cues provides evidence for intrinsic clock regulation of GSK3 inactivation state.
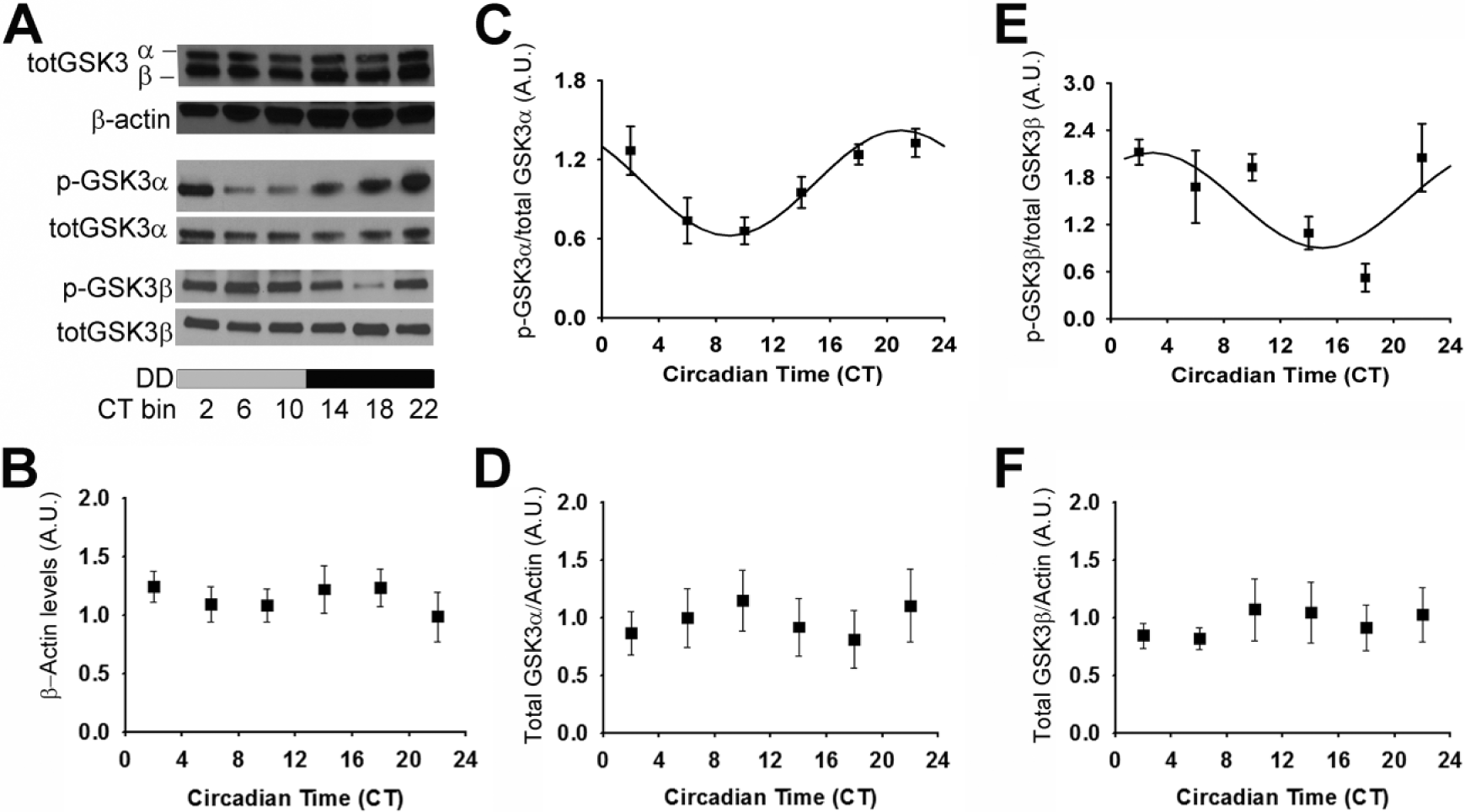
Rhythmic phosphorylation of GSK3 in the SCN persists in constant darkness. (A) Representative Western blots of total GSK3α/β reblotted for β-actin (top), p-GSK3α reblotted for GSK3α (middle), and p-GSK3β reblotted for GSK3β (bottom). Quantification (mean ± SEM per CT bin) of (B) β-actin levels, (C) p-GSK3α to total GSK3α ratio, (D) total GSK3α (normalized to β-actin), (E) p-GSK3β to total GSK3β ratio, and (F) total GSK3β (normalized to β-actin) when sampled at various times across DD. Cosinor nonlinear regression: (C) R 2 = 0.53, F(2, 24) = 13.73, p < 0.05, n = 27/time course; (D) R 2 = 0.25, F(2, 24) = 3.96, p < 0.05; n = 27/time course; (E) R 2 = 0.21, F(2, 22) = 2.91, p > 0.05; n = 26/time course.
Previous evidence suggests that GSK3 directly phosphorylates at least 5 core clock proteins: PER2, CRY2, CLOCK, BMAL1, and REVERBα (Kaladchibachi et al., 2007; Spengler et al., 2009; Kurabayashi et al., 2010; Sahar et al., 2010). Given that phosphorylation of BMAL1 by GSK3β affects Bmal1 translation and protein stability in vitro (Yin et al., 2006; Sahar et al., 2010; Valnegri et al., 2011), we tested the initial hypothesis that the time-dependent balance of phosphorylated to de-phosphorylated GSK3 is critical for BMAL1 expression rhythms in the SCN. Specifically, p-GSK3α and p-GSK3β rhythms were eliminated using a double transgenic mouse model of chronic GSK3 activity (GSK3-KI mice) in which 2 serine-alanine mutations (GSK3αS21A/S21A and GSK3βS9A/S9A) render both isoforms of GSK3 constitutively active (but at endogenous levels; McManus et al., 2005; Paul et al., 2012). Wheel-running rhythms of these mice have decreased rhythmic amplitude, lengthened α (active period), increased activity bouts per day, and increased SCN excitability at night compared with wild-type (WT) controls (Paul et al., 2012). This circadian phenotype was not observed in mice bearing single KI mutations (GSK3αS21A/S21A or GSK3βS9A/S9A), likely due to functional redundancy between the GSK3 isoforms, and therefore, only mice with both α and β isoform mutations were investigated in the current studies. Using Western blot analysis of isolated SCN from individual animals, we quantified BMAL1 expression (as a percentage of α-Tubulin expression) over a 24-h period in isolated SCN after GSK3-KI or WT control mice were housed in DD for at least 2 weeks.
Cosinor nonlinear regression revealed a significant BMAL1 expression rhythm in WT mice (n = 27/time course; cosinor nonlinear regression analysis; p < 0.05 for SCN) with peak BMAL1 expression in the subjective night (mesor, 0.82 ± 0.08; amplitude, –0.30 ± 0.11; phase, 14.85 ± 10.61; Figure 2). In GSK3-KI mice, however, BMAL1 expression did not exhibit a significant 24-h rhythm (n = 25/time course; as determined by cosinor nonlinear regression analysis, p = 0.91; Figure 2). There were no significant differences in α-tubulin expression levels with respect to time or genotype (p > 0.05). These results indicate that constitutive GSK3 activation disrupts circadian expression of BMAL1 in the central pacemaker. Based on phosphorylation status, we predict that GSK3β activity is highest in WT SCN neurons at approximately CT16, when BMAL1 protein levels peak. GSK3α activity likely peaks around CT9, a time at which BMAL1 protein levels are increasing. These observations are consistent with the conceptual model that GSK3-mediated phosphorylation is an early event, which primes BMAL1 for subsequent degradation via ubiquitin/proteosomal degradation (Figure 2; Sahar et al., 2010). The possibility also remains that chronic GSK3 activation influences BMAL1 protein levels indirectly, through phosphorylation of other clock components (e.g., REV-ERBα, which in turn represses Bmal1 transcription).
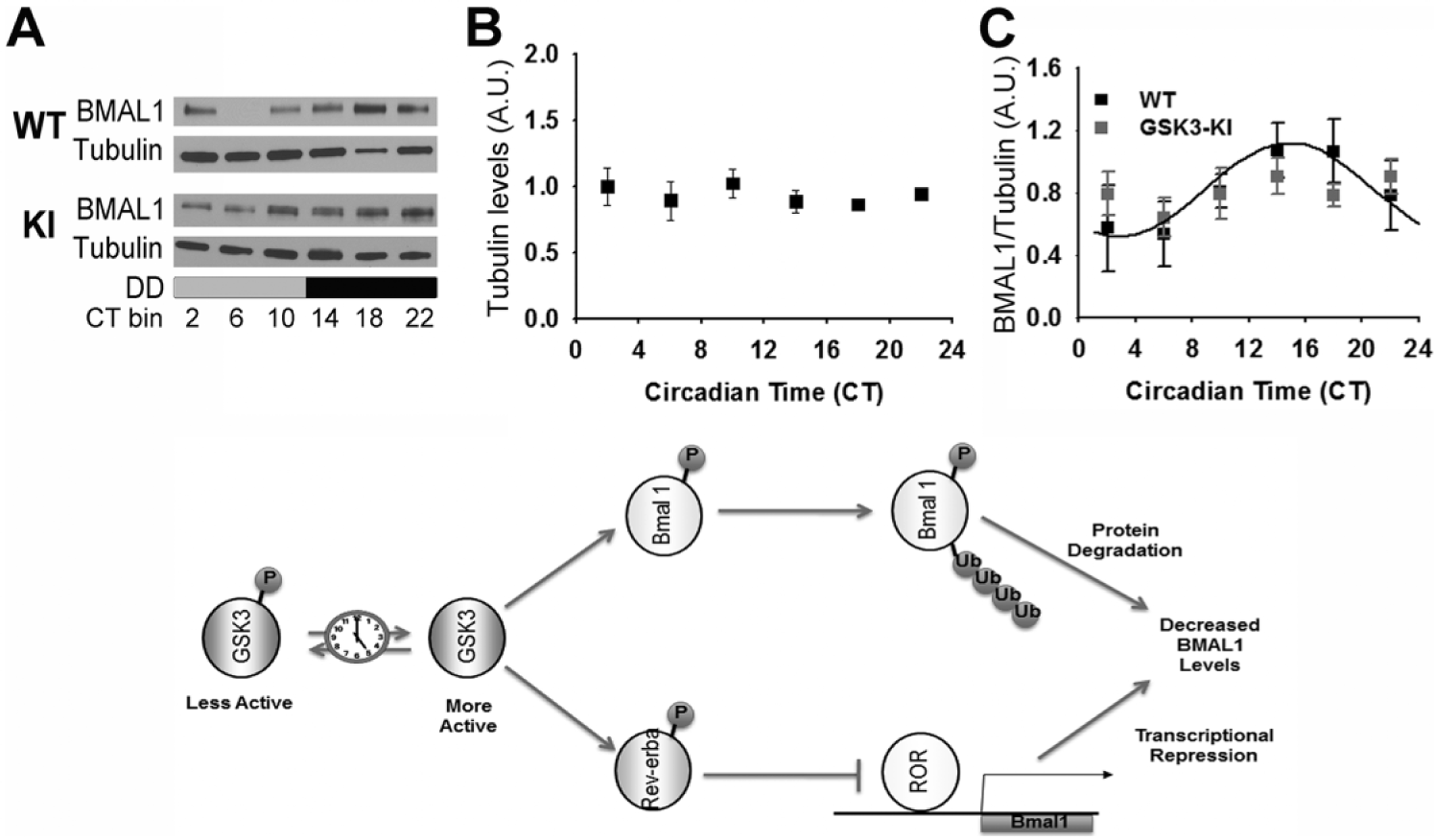
Rhythmic BMAL1 expression in mice with chronic GSK3 activation. (A) Representative Western blots of BMAL1 reblotted for α-tubulin. Quantification (mean ± SEM per CT bin) of (B) α-tubulin levels and (C) BMAL1 levels (normalized to α-tubulin) when sampled across DD for wild-type (WT; black; n = 27/time course) and GSK3-KI (gray; n = 25/time course) mice. Cosinor nonlinear regression: WT, R 2 = 0.23, F(2, 24) = 3.50, p < 0.05; GSK3-KI, R 2 = 0.01, F(2, 22) = 0.57, p = 0.91. (Bottom) Schematic representing hypothetical model for the interrelationship between GSK3 and the circadian clock. Circadian clock–dependent regulation of GSK3 results in time-of-day–dependent oscillations in target proteins, including BMAL1 and REV-ERBα. Phosphorylation of BMAL1 primes this clock component for ubiquitination and subsequent degradation, while phosphorylation of REV-ERBα promotes nuclear translocation and inhibition of ROR-mediated Bmal1 transcription. Increased BMAL1 protein degradation coupled with diminished Bmal1 transcription will result in diminished BMAL1 protein levels and therefore attenuated circadian clock output.
While it is appreciated that Bmal1 is a necessary molecular clock component for behavioral rhythmicity (Bunger et al., 2000), whether or not BMAL1 protein level rhythmicity is necessary for a functional pacemaker is unclear (Shi et al., 2010). To test the idea that GSK3 activation rhythms regulate molecular clock amplitude and periodicity, we used the Period2Luciferase (Per2Luc) mouse model as a readout of real-time molecular clock function (Yoo et al., 2004). Specifically, a selective GSK3 inhibitor (20 µM CHIR-99021) was chronically applied to the media of organotypic tissue cultures of SCN from mPer2Luc+/– reporter mice (Yoo et al., 2004; Besing et al., 2012). This compound inhibits both GSK3 isoforms by competing with ATP for the ATP-binding site of the kinase (Cline et al., 2002; Ring et al., 2003). Application of this small-molecule GSK3 inhibitor nearly doubled the amplitude of the chi-squared periodogram analysis and significantly shortened the period length of SCN explant PER2::LUC rhythms (n = 9/group; t tests, p < 0.05; Figure 3A,B), suggesting that chronic GSK3 inhibition enhances the amplitude and speeds up the periodicity of the molecular clock. These results are consistent with previous in vitro studies that show that molecular clock amplitude is enhanced and period is shortened by pharmacological GSK3 inhibition in fibroblasts and mammalian cell lines (Hirota et al., 2008; Vougogiannopoulou et al., 2008; Li et al., 2012). On the other hand, molecular clock amplitude is reduced by GSK3 overexpression (Sahar et al., 2010; Li et al., 2012). Although it is well documented that the GSK3 inhibitor lithium lengthens the period of circadian rhythms (Mason and Biello, 1992; Abe et al., 2000; Hirota et al., 2008; Mohawk et al., 2009; Osland et al., 2011), our results with a more selective GSK3 inhibitor raise the possibility that lithium-induced effects may be due to nonspecific targets, perhaps via inositol phosphatase inhibition (Quiroz et al., 2004).
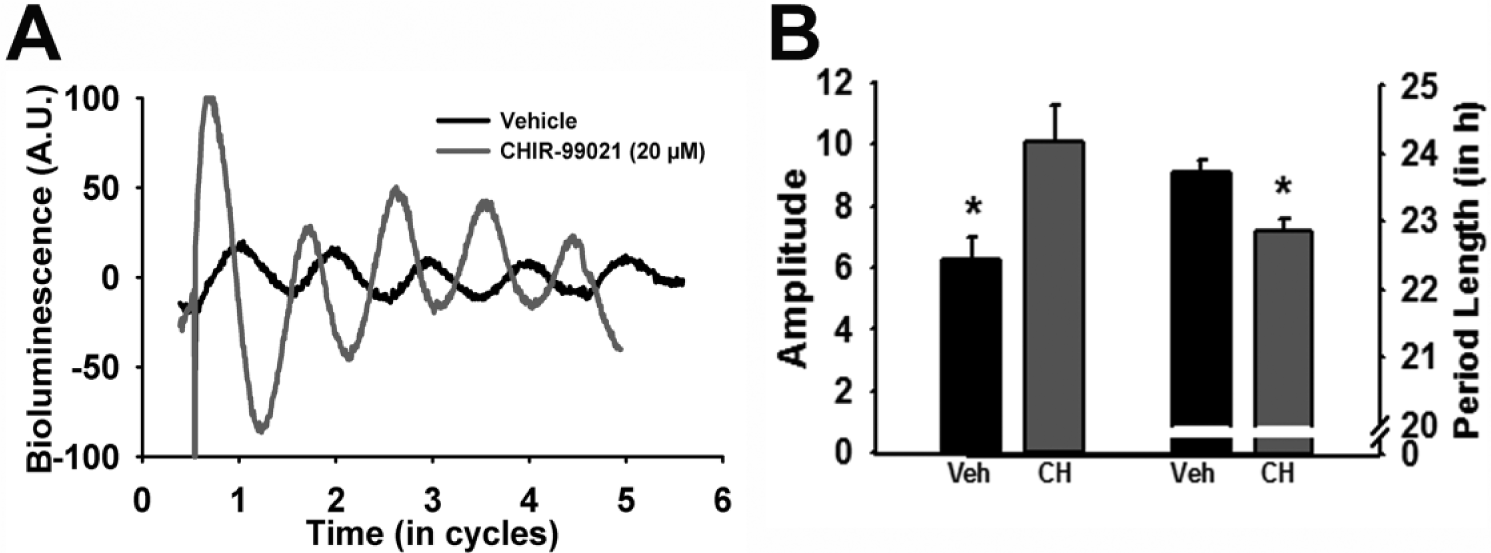
GSK3 inhibition modulates the molecular clock. (A) Representative bioluminescent traces from PER2::LUC organotypic SCN cultures treated with vehicle (black) or CHIR-99021 (20 µM; gray). (B) Bar graph (mean ± SEM) of amplitude (left; t(16) = 2.68; *p < 0.05) and period length (right; t(16) = −3.83, *p < 0.05).
The present study shows that both GSK3 isoforms are rhythmically inactivated in the SCN in constant conditions, consistent with previous evidence in LD (Iwahana et al., 2004; Iitaka et al., 2005). These results support the model that GSK3 activity is clock controlled (Durgan and Young, 2010), potentially regulated by rhythmic phosphorylation of AKT (Panda et al., 2002; Durgan and Young, 2010), a primary upstream kinase of GSK3 (Hur and Zhou, 2010). In addition, our data suggest that GSK3 feeds back to affect molecular clock function in SCN neurons. Specifically, when both GSK3 isoforms were constitutively activated, BMAL1 rhythms within the SCN became arrhythmic. It is interesting that BMAL1 levels do not fall below baseline levels in mice with chronic GSK3 activity. Future studies should investigate whether there is a pool of BMAL1 that is resistant to degradation via the GSK3-E3-ubiquitin pathway and whether chronic GSK3 activation affects transcriptional clock gene rhythms and/or cellular compartmentalization of clock proteins. Our study also found that disruption of GSK3 activation rhythms with chronic pharmacological inhibition nearly doubled PER2::LUC rhythms in SCN organotypic cultures. Investigation of the effects of phase-specific inhibition of GSK3 on the molecular clock will be important to address in the future. Taken together, our results support the conceptual model that disruption of GSK3 activity rhythms affects the molecular clock such that chronic activation impairs rhythmicity of some molecular clock components (such as BMAL1; Figure 2), while chronic inhibition enhances rhythmicity of the molecular clock (as indicated by real-time PER2::LUC rhythms).
Footnotes
Acknowledgements
This work was supported by National Institutes of Health Grants R00GM086683 and R01NS082413 to K.L.G., F31NS086282 to J.R.P., F31NS084683 to L.M.H., R01HL123574 to M.E.Y., and T32NS061788—Training Program in the Neurobiology of Cognition and Cognitive Disorders. We thank Rita M. Cowell for technical assistance.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.