
Abstract
In mammals, the suprachiasmatic nucleus (SCN) is the central pacemaker organizing circadian rhythms of behavior and physiology. At the cellular level, the mammalian clock consists of autoregulatory feedback loops involving a set of “clock genes,” including the Cryptochrome (Cry) genes, Cry1 and Cry2. Experimental evidence suggests that Cry1 and Cry2 play distinct roles in circadian clock function. In mice, Cry1 is required for sustained circadian rhythms in dissociated SCN neurons or fibroblasts but not in organotypic SCN slices or at the behavioral level, whereas Cry2 is not required at any of these levels. It has been argued that coupling among SCN cellular oscillators compensates for clock gene defects to preserve oscillatory function. Here we test this hypothesis in Cry1−/− mice by first disrupting intercellular coupling in vivo using constant light (resulting in behavioral arrhythmicity) and then examining circadian clock gene expression in SCN slices at the single cell level. In this manner, we were able to test the role of intercellular coupling without drugs and while preserving tissue organization, avoiding the confounding influences of more invasive manipulations. Cry1−/− mice (as well as control Cry2−/− mice) bearing the PER2::LUC knock-in reporter were transferred from a standard light:dark cycle to constant bright light (~650 lux) to induce arrhythmic locomotor patterns. In SCN slices from these animals, we used bioluminescence imaging to monitor PER2::LUC expression in single cells. We show that SCN slices from rhythmic Cry1−/− and Cry2−/− mice had similarly high percentages of functional single-cell oscillators. In contrast, SCN slices from arrhythmic Cry1−/− mice had significantly fewer rhythmic cells than SCN slices from arrhythmic Cry2−/− mice. Thus, constant light in vivo disrupted intercellular SCN coupling to reveal a cell-autonomous circadian defect in Cry1−/− cells that is normally compensated by intercellular coupling in vivo.
The mammalian circadian system is a collection of cellular clocks located throughout the body that are synchronized by a primary pacemaker within the suprachiasmatic nucleus (SCN) (Dibner et al., 2010; Mohawk et al., 2012; Welsh et al., 2010). Non-SCN cells are autonomous oscillators but, in the absence of the SCN, may desynchronize from one another (Nagoshi et al., 2004; Welsh et al., 2004), as they appear to lack local coupling within tissues (Stratmann and Schibler 2006). In contrast, the neuronal network within the SCN couples its component single-cell oscillators to produce a coherent circadian oscillation at the tissue level (Aton and Herzog 2005; Welsh et al., 2010). Unlike the independent oscillations of dissociated SCN neurons, which exhibit various periods and phases, SCN neurons within a slice adopt identical circadian periods and relatively stable phase relationships (Herzog et al., 2000; Welsh et al., 1995). Such coordinated rhythms can be measured longitudinally in SCN slices in vitro by single-cell bioluminescence imaging of clock gene expression using Per1-luc or PER2::LUC reporters (Evans et al., 2011; Foley et al., 2011; Yamaguchi et al., 2003; Yan et al., 2007). The precise relative phasing of individual cells is preserved from cycle to cycle, in a complex pattern of distinct phases and amplitudes, suggesting that SCN coupling is mediated by specific neural circuits, rather than by homogeneous neuronal interactions. The mechanism of this coupling involves neuronal firing and chemical synapses, with vasoactive intestinal polypeptide (VIP) playing a particularly important role (Aton and Herzog 2005; Welsh et al., 2010).
Within individual cells, circadian oscillations are driven by a delayed negative feedback loop in which CLOCK/BMAL1 heterodimers activate transcription of Period (Per) and Cryptochrome (Cry) genes. After transcriptional and posttranscriptional delays, the PERs and CRYs form complexes that inhibit CLOCK/BMAL1 activity, thus inhibiting their own transcription (Mackey 2007; Takahashi et al., 2008). In time, proteasomal degradation of PERs and CRYs relieves the inhibition and allows the cycle to begin anew. The roles of putative molecular clock components have traditionally been evaluated by testing behavioral rhythms of knockout animals. Both Cry1−/− and Cry2−/− mice have intact rhythms of locomotor activity in constant darkness, with shorter period in Cry1−/− mice and longer period in Cry2−/− mice relative to wild-type (van der Horst et al., 1999; Vitaterna et al., 1999). At the level of single cells and non-SCN tissues, however, Cry1−/− produces a more profound defect (Liu et al., 2007). Whereas non-SCN tissues, fibroblasts, and dissociated SCN neurons cultured from Cry2−/− mice are self-sufficient oscillators, only rare, transient rhythms are observed in single cells and non-SCN tissues from Cry1−/− mice. In contrast, SCN slices from both Cry1−/− and Cry2−/− mice are strongly rhythmic with short and long period, respectively, in parallel with behavior. Thus, some property of the SCN in vivo, which is preserved in slices but not in dispersed cell culture, compensates for the Cry1−/− genetic defect to produce strong rhythms. Since an important distinction between an SCN slice and dispersed SCN neurons and non-SCN tissues is the presence of coupling in the former, this suggests that Cry1−/− cells may require intercellular communication across the network to generate sustained rhythms and that SCN coupling rescues oscillatory function in the face of this genetic defect.
Here we test the hypothesis that coupling among SCN single-cell oscillators, rather than some other aspect of tissue organization, is responsible for the compensatory effect observed in Cry1−/− SCN slices. We reasoned that if SCN coupling is required to compensate for a Cry1−/− defect, then disrupting coupling in Cry1−/− SCN slices should impair rhythmicity. Although SCN coupling can be disrupted by genetic or pharmacological manipulations targeting cellular coupling mechanisms (e.g., VIP gene knockout, or toxins to block synaptic transmission), such manipulations can also adversely affect oscillatory function of individual SCN neurons (Aton et al., 2005; Deery et al., 2009). We wished to avoid such confounding effects on cellular circadian function by disrupting coupling in a less invasive manner.
Specifically, SCN coupling can be altered in vivo by manipulation of environmental lighting conditions, such as exposure to constant bright light (LL). In nocturnal rodents, LL lengthens the free-running period and decreases the duration and level of locomotor activity (Aschoff 1960). Further, some rodents held under LL develop arrhythmic activity patterns that reflect desynchrony among SCN neurons (Ohta et al., 2005). Importantly, LL disrupts coupling without otherwise altering the characteristics of single-cell rhythms (Ohta et al., 2005). Therefore, to test whether SCN coupling is required to compensate for Cry1 deficiency, we disrupted SCN coupling in Cry1−/− or Cry2−/− mice through in vivo exposure to LL and then tested in vitro for defects in PER2::LUC rhythms displayed by SCN neurons within slices collected from these mice. We predicted that if Cry1−/− rhythmicity depends on SCN coupling, then disrupting this coupling should impair single-cell rhythmicity in Cry1−/− slices, but not in Cry2−/− slices.
Materials and Methods
Animals
Homozygous Cry1−/− and Cry2−/− mice (van der Horst et al., 1999) were crossed with homozygous PER2::LUC knock-in mice (Yoo et al., 2004) to generate homozygous reporter knockout mice as in Liu et al. (2007). Prior to use in experiments, mice were group-housed without running wheels and entrained to a 14-h light/10-h dark cycle (LD 14:10, lights on: 0600 PST). Ambient temperature was maintained at 22 ± 2 °C, with food (Purina Rodent Chow #5001, St. Louis, MO) and water available ad libitum. At 7 to 9 weeks of age, male Cry1−/− and Cry2−/− mice were transferred to individual wheel-running cages located within light-tight chambers and exposed to constant bright light (LL, 650 lux at cage level). All animal studies were conducted in accordance with the regulations of the Committee on Animal Care and Use at the University of California, San Diego.
Behavioral analyses
Wheel-running activity rhythms were analyzed visually to identify animals that had become arrhythmic and remained arrhythmic for at least 2 weeks prior to dissection. Locomotor activity patterns were submitted to χ2 periodogram analyses (ClockLab; Actimetrics, Wilmette, IL), and animals were verified to be arrhythmic when the χ2 periodogram failed to detect a significant period in the circadian range (18-30 h, p < 0.05, 1-tailed). Animals that maintained coherent circadian rhythms under LL were selected as rhythmic controls for each genotype. For animals with coherent locomotor rhythms, circadian time (CT) of dissection was calculated by use of a linear regression fit to activity onsets (defined as CT12) displayed over the preceding 6 to 7 days.
Explant culture
Animals were humanely killed by decapitation. For all mice, SCN dissection was performed at a standard clock time (1000 PST). SCN slices (200 µm) were cut in the coronal plane with a motorized vibratome. Each slice was trimmed near the edges of the SCN and cultured on a membrane (Millicell-CM, PICMORG030-50; Millipore, Billerica, MA) in a sealed dish containing 1.2 mL HEPES-buffered serum-free explant medium (Dulbecco’s modified Eagle’s medium [DMEM]; GIBCO 12100-046; GIBCO, Carlsbad, CA) supplemented with 1.2 g/L NaHCO3, 10 mM HEPES, 4 mM glutamine, 25 U/mL penicillin, 25 µg/mL streptomycin, 2% B-27 (GIBCO 17504-044), and 1 mM luciferin (L-8220; BioSynth, Staad, Switzerland).
Bioluminescence imaging
To study rhythmic circadian clock gene expression in SCN slices at single-cell resolution, PER2::LUC bioluminescence imaging was performed for 3 to 5 days, with 30 min temporal resolution (Welsh et al., 2005). Immediately after dissection, SCN slices were transferred to the stage of an inverted microscope (Olympus IX71; Olympus, Center Valley, PA) housed within a dark room and maintained at 36 °C within a heated lucite chamber (Solent Scientific, Segensworth, UK). Images were collected with an Olympus 10× UPlanSApo objective and transmitted to a CCD camera (Spectral Instruments SI800, Tucson, AZ) cooled to −90 °C. To assess long-term changes in bioluminescence rhythmicity, a subset of SCN slices from arrhythmic Cry1−/− mice were transferred from the camera to a LumiCycle luminometer (Actimetrics) kept inside a standard tissue culture incubator at 36 °C. Bioluminescence from each dish was continuously recorded with a photomultiplier tube (PMT) for 70 sec every 10 min. Slices were retained within the luminometer for at least 4 weeks, and the medium was changed once a week.
Data analysis
Bioluminescence images were analyzed with MetaMorph software (Molecular Devices, Sunnyvale, CA). Spurious events were removed by using the minimum value in pixel-wise comparisons of 2 consecutive images. Images were corrected for bias and dark current by background subtraction. For single-cell analyses, selected cells were those that were easily discriminated from adjacent cells and continued to exhibit biolumines-cence over the entire 3- to 4-day imaging experiment. Luminescence intensity for each cell was measured within a manually defined region of interest that was adjusted to accommodate cell movement. Data were processed further using Excel (Microsoft, Redmond, WA), where luminescence values were converted to photons/min based on rated quantum efficiency and gain of the camera. Luminescence time series were then imported into LumiCycle Analysis software (Actimetrics). The first 12 h of each time series was excluded due to high transient luminescence upon dissection. A linear baseline was subtracted from the raw data (polynomial order = 1), and the baseline-subtracted data were fit by a sine wave from which period and phase were determined. Single cells were defined as rhythmic using a best-fit sin wave with criteria of a goodness-of-fit (percent of total variance accounted for by the fitted curve) ≥25%, a circadian period (18-36 h), and amplitude ≥15 (Liu et al., 2007). Phases were plotted and analyzed with Oriana software (Kovach Computing Services, Anglesey, UK). All other statistical tests were conducted with JMP software (SAS Institute, Cary, NC). Data in figures are means ± SEM.
Results
Cry1−/− and Cry2−/− mice were both highly susceptible to constant bright light, such that a great majority of animals of each genotype (87% of Cry1−/−, 70% of Cry2−/−) developed arrhythmic locomotor patterns within 7 weeks after transfer to LL (Figure 1). Cry1−/− and Cry2−/− mice did not differ significantly in the proportion that developed arrhythmic behavioral activity patterns during the first 7 weeks in LL (Figure 1; Fisher’s exact test χ2(1) = 1.04, p = 0.36). Of animals that became arrhythmic, genotype did not significantly influence the rate at which arrhythmia developed (Figure 1; t(23) = −0.39, p = 0.69).
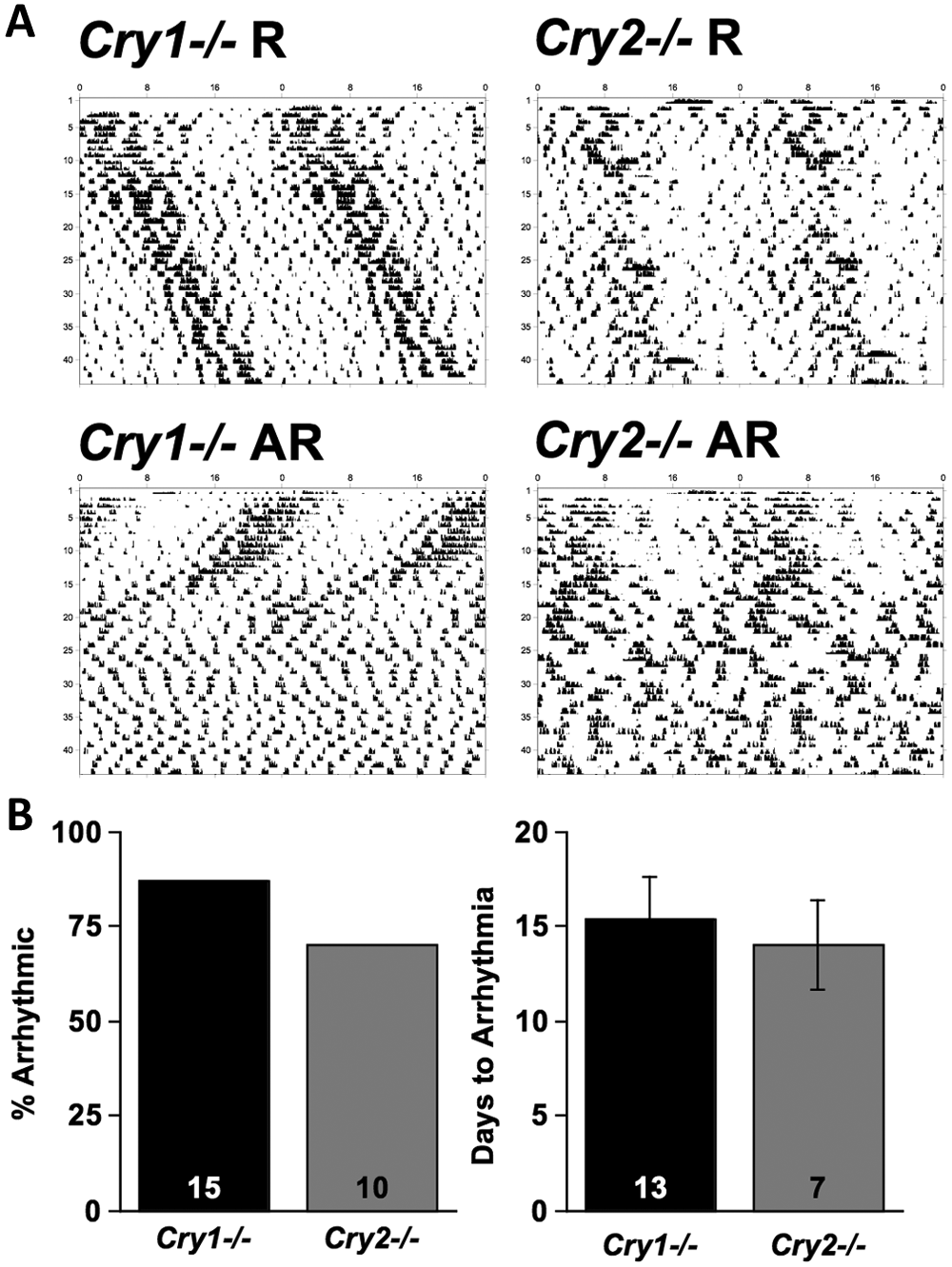
Effects of LL on locomotor behavior of Cry-deficient mice. (A) Representative double-plotted wheel-running actograms of Cry1−/− and Cry2−/− mice that were transferred to LL on the first day of the record. The majority of Cry-deficient mice quickly became arrhythmic after transfer to LL, but a minority of animals retained circadian rhythms for the first few weeks under LL. (B) Proportion of Cry1−/− and Cry2−/− animals that developed arrhythmic activity patterns within 7 weeks under LL (left) and latency to develop arrhythmia (right). Numbers within columns represent total sample sizes for animals held under LL for at least 7 weeks and used for behavioral analyses. R = rhythmic mice; AR = arrhythmic mice.
Regardless of behavioral phenotype, all SCN slices displayed PER2::LUC circadian rhythms in vitro (Figure 2). For rhythmic mice of each genotype, PER2::LUC rhythm phases were clustered according to in vivo circadian phase (Figure 2A; Rayleigh test, p < 0.01) rather than by dissection time (Figure 2B; Rayleigh test, p > 0.05). SCN slices from rhythmic Cry1−/− and Cry2−/− mice did not differ in peak time (t(6) = 1.8, p = 0.12) or rhythm amplitude (t(5) = −2.1, p = 0.09). In contrast, for arrhythmic mice of each genotype, PER2::LUC rhythm phases were clustered according to dissection time (Figure 2C; Rayleigh test, p < 0.01). Relative to SCN slices from arrhythmic Cry2−/− mice, SCN slices from arrhythmic Cry1−/− mice peaked 9 h earlier (t(9) = 20.4, p < 0.0001) and displayed lower amplitude rhythms (t(9) = −2.4, p = 0.04).
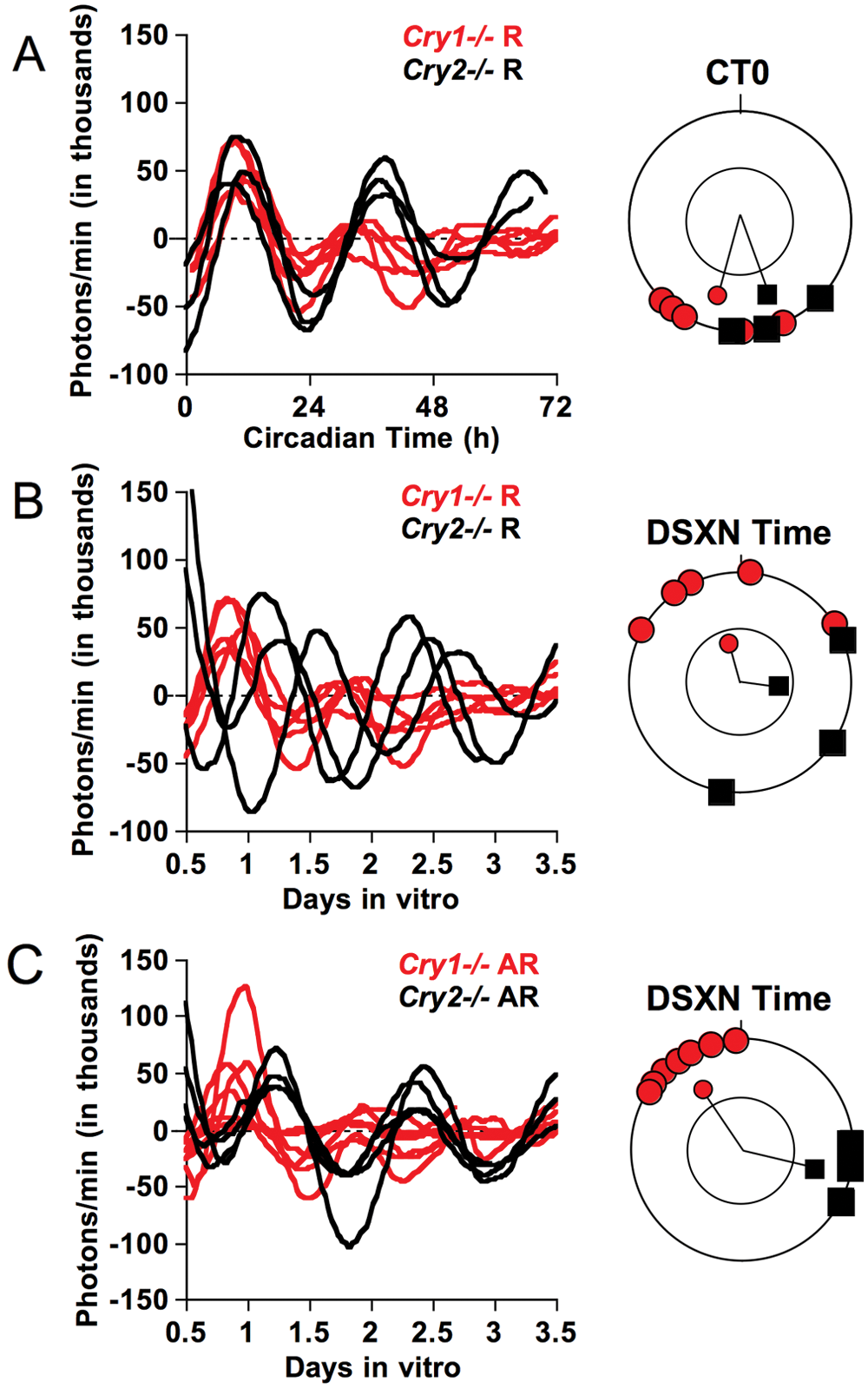
Effects of genotype and LL-induced behavioral arrhythmicity on PER2::LUC rhythms of SCN slices in vitro illustrated by baseline-subtracted time series. (A, B) PER2::LUC rhythms of SCN slices from rhythmic Cry1−/− (n = 5) and Cry2−/− (n = 3) mice were significantly clustered according to circadian time (A) but not dissection time (B). Rayleigh plots depict the phase distribution of peak PER2::LUC on the first cycle in vitro, plotted in angular degrees. Lines represent mean angular vectors, and those extending outside the inner circle indicate significant clustering for that group as determined by the Rayleigh test. (C) PER2::LUC rhythms of SCN slices from arrhythmic Cry1−/− (n = 7) and Cry2−/− (n = 5) mice were clustered according to dissection time and genotype. R = rhythmic mice; AR = arrhythmic mice; DSXN = dissection.
As a test of the correspondence between in vivo and in vitro rhythms, we next analyzed the period of PER2::LUC rhythms displayed by SCN slices and neurons. Circadian period displayed by SCN slices and single cells reflected the previously described effects of Cry genotype, even in slices from mice rendered behaviorally arrhythmic (Table 1). Overall, Cry1−/− SCN slices had significantly shorter periods than Cry2−/− SCN slices (Genotype: F(1, 14) = 114.4, p < 0.0001), and this difference was accentuated in SCN slices from arrhythmic mice (Genotype × Phenotype: F(1, 14) = 8.9, p = 0.0098). Of single cells displaying a period in the circadian range (18-36 h), Cry1−/− cells had a significantly shorter period than Cry2−/− cells (Genotype: F(1,14) = 53.9, p < 0.0001), regardless of behavioral phenotype (Genotype × Phenotype: F(1, 14) = 2.3, p = 0.15).
Period of PER2::LUC rhythms reflects
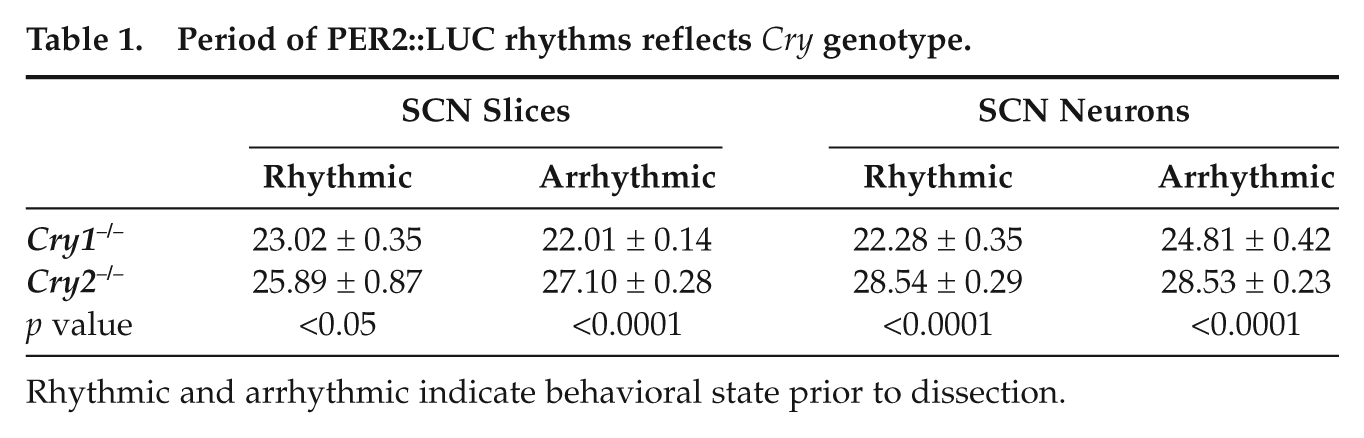
Rhythmic and arrhythmic indicate behavioral state prior to dissection.
To assess synchrony among cells, we analyzed PER2::LUC rhythms of single neurons in SCN slices from rhythmic and arrhythmic Cry2−/− mice (Figure 3A). Relative to slices from rhythmic mice, slices from arrhythmic mice tended to have more variance in phase among cells, less concentration of phase, and a shorter vector radius for the cells (χ2(1) = 3.33, p < 0.07 for each test). Although not statistically significant at the p = 0.05 criterion level for individual tests, the consistency of the trend across all 3 measures indicates that coupling was compromised by LL, although not completely abolished.
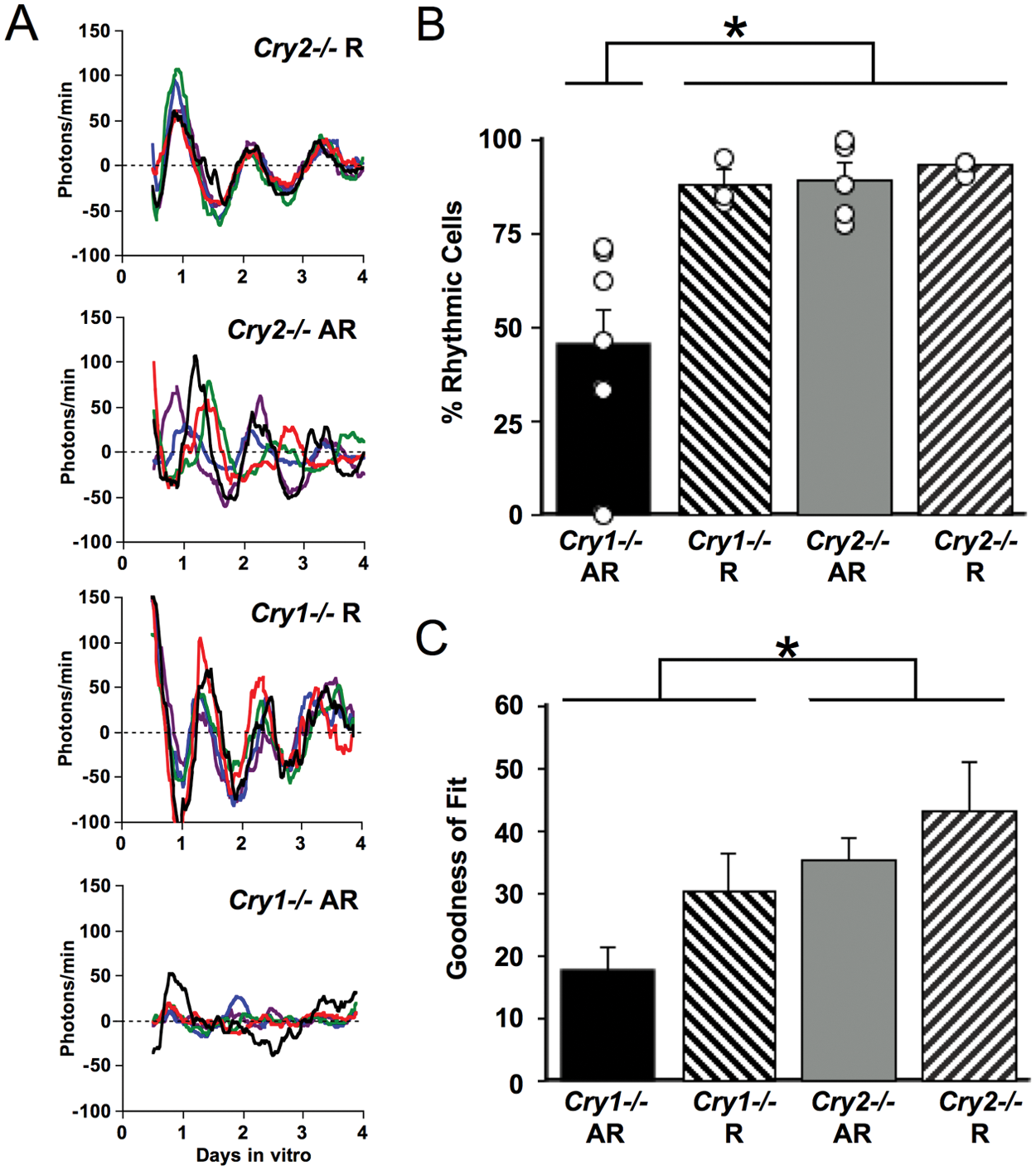
Reduced PER2::LUC rhythmicity of single cells in SCN slices from behaviorally arrhythmic Cry1−/− mice. (A) Representative background-subtracted time series of PER2::LUC rhythms displayed by SCN neurons from rhythmic and arrhythmic Cry1−/− and Cry2 −/− mice (5 cells per panel, each cell in a different color). More examples are shown in Supplementary Figures S1 to S4. (B) SCN slices from arrhythmic Cry1−/− mice had fewer rhythmic neurons relative to slices from rhythmic Cry1−/− mice, arrhythmic Cry2−/− mice, or rhythmic Cry2−/− mice. (C) The goodness of fit (percentage of total variance in the PER2::LUC trace accounted for by a best-fit sine wave) was reduced in SCN cells from Cry1−/− mice and was lowest in Cry1−/− cells from arrhythmic mice. Average number of cells extracted per slice: 40 cells. R = rhythmic mice; AR = arrhythmic mice. *Tukey’s HSD (honestly significant difference) test, p < 0.01.
Cry genotype and LL-induced behavioral phenotype of an animal interacted to influence the prevalence and robustness of rhythms displayed by neurons within an SCN slice (Figure 3A,B and Suppl. Figures S1-S4; F(1, 14) = 4.6, p < 0.05). SCN slices from rhythmic Cry1−/− and Cry2−/− mice displayed a high percentage of rhythmic neurons (88% and 93%, respectively). SCN slices from arrhythmic Cry2−/− mice had a similarly high percentage of rhythmic neurons (89%). In contrast, SCN slices from arrhythmic Cry1−/− mice had a much lower percentage of rhythmic neurons (45%; Tukey’s honestly significant difference [HSD], p < 0.01). For rhythmic cells, Cry genotype and behavioral phenotype also affected the robustness of rhythms as measured by “goodness of fit,” or percentage of variance accounted for by a fitted sine wave (Figure 3C). Cry1−/− cells displayed a lower goodness of fit than Cry2−/− cells (F(1, 14) = 8.88, p < 0.01), and goodness of fit tended to be lower in cells from arrhythmic mice than in those from rhythmic mice (F(1, 14) = 3.94, p = 0.067). SCN cells from arrhythmic Cry1−/− mice had the least robust rhythms compared with SCN cells from each of the 3 other groups (Figure 3C; Tukey’s HSD, p < 0.01).
SCN slices from arrhythmic Cry1−/− animals were viable, even though they had fewer rhythmic cells. Average brightness of PER2::LUC bioluminescence requires ATP and is one indicator of tissue viability. As indicated by PER2::LUC expression averaged over the first 2 days in vitro, SCN slices from Cry1−/− mice were at least as bright as those from Cry2−/− mice (Figure 4A) and much brighter in the case of rhythmic Cry1−/− mice (Genotype: F(1, 14) = 7.5, p = 0.016; Phenotype: F(1, 14) = 6.1, p = 0.027; Genotype × Phenotype: F(1, 14) = 6.94, p = 0.0196). Also, SCN slices from arrhythmic Cry1−/− animals eventually recovered robust PER2::LUC rhythms after a few medium changes (Figure 4B).
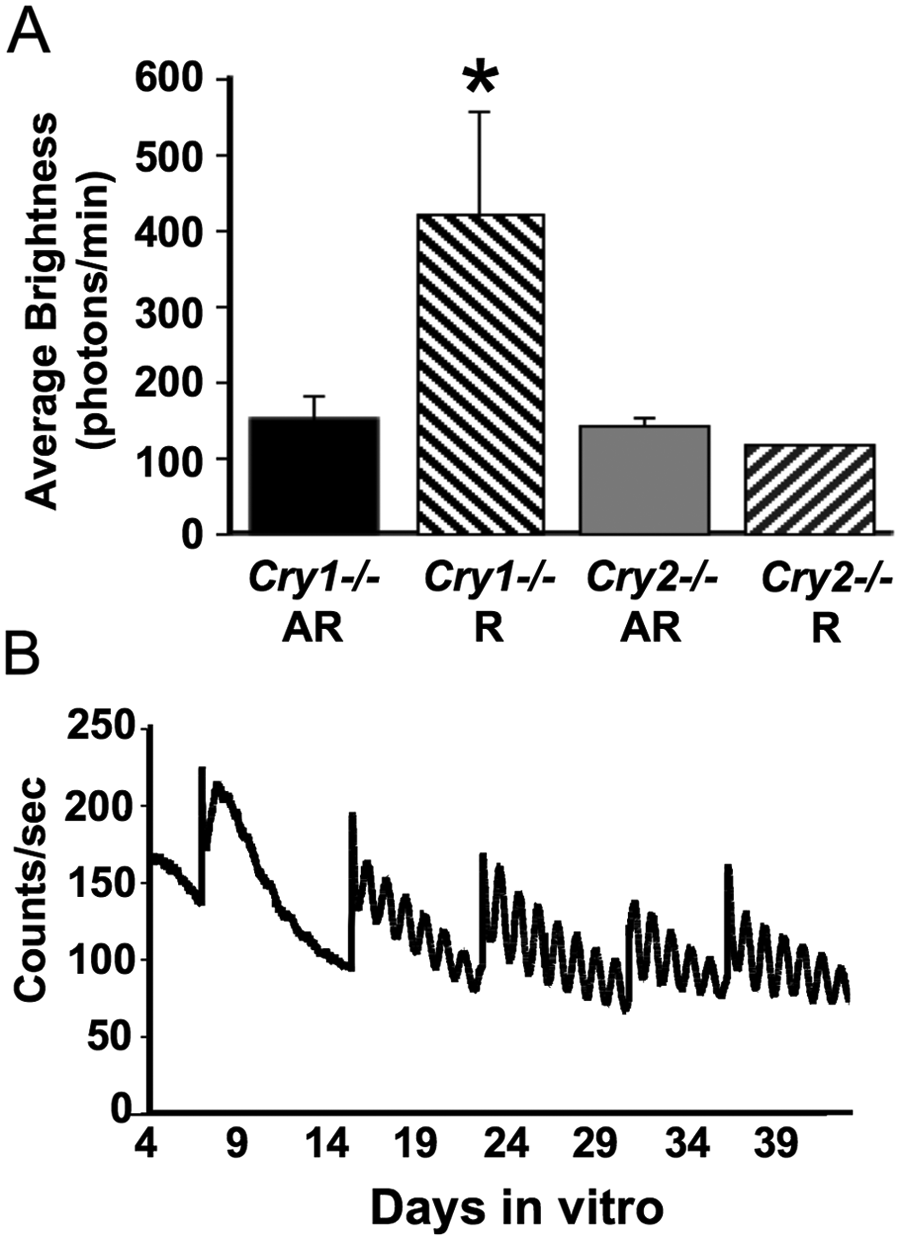
Viability of Cry1−/− SCN slices from arrhythmic mice. (A) SCN slices from Cry1−/− animals were at least as bright as Cry2−/− slices. *Different from all other groups, Tukey’s HSD (honestly significant difference) test, p < 0.01. (B) Example of an SCN slice from an arrhythmic Cry1−/− mouse that initially displayed very low amplitude rhythms but then high amplitude and sustained rhythms after 2 medium changes. R = rhythmic mice; AR = arrhythmic mice.
Discussion
SCN neurons deficient in Cry1 exhibit prominent and persistent circadian rhythms of PER2 expression in SCN slice preparations but not in dispersed culture (Liu et al., 2007). We hypothesized that this is because sustained circadian rhythmicity in Cry1−/− cells requires intercellular coupling, rather than some other property of the intact SCN slice that is missing in dispersed cultures. In the present study, we test this hypothesis by disrupting SCN coupling in vivo through exposure to LL and then assessing oscillatory function of individual cells in SCN slices by PER2::LUC imaging. LL can suppress locomotor activity rhythms in Cry1−/− and Cry2−/− mice, as in wild-type mice (Spoelstra and Daan 2008). In wild-type mice, LL-induced behavioral arrhythmia is due to desynchronization among SCN neurons (Ohta et al., 2005), and we hypothesized that the same would be true for Cry knockout mice. If so, then we reasoned that Cry2−/− neurons, which (like wild-type neurons) are competent single-cell oscillators in dispersed culture, should exhibit strong but desynchronized rhythms in SCN slices taken from arrhythmic animals. We further reasoned that disrupting SCN intercellular coupling through in vivo exposure to LL should cripple the oscillatory function of Cry1−/− but not Cry2−/− neurons, mimicking the results of cell dispersal in culture. We therefore predicted that LL would (1) produce behavioral arrhythmicity of Cry knockout mice, (2) preserve rhythmicity but reduce synchrony of SCN neurons in slices from arrhythmic Cry2−/− mice, and (3) reduce rhythmicity of SCN neurons in slices from arrhythmic Cry1−/− mice. The present results are consistent with all 3 predictions and support the hypothesis that Cry1−/− cells are not cell-autonomous oscillators but require SCN coupling to maintain rhythmic expression of PER2.
We considered the possibility that Cry1−/− and Cry2−/− mice might respond differently to LL than wild-type mice, because previous studies found altered responses to light in both genotypes: decreased induction of Per1 in SCN (Thresher et al., 1998; Vitaterna et al., 1999) but also larger phase delays of behavioral rhythms (Spoelstra and Daan 2008; Thresher et al., 1998). In the only previous study examining effects of LL in Cry knockout mice (Spoelstra and Daan 2008), locomotor activity rhythms were suppressed similarly in Cry1−/−, Cry2−/−, and wild-type mice, but incidence of complete arrhythmia was not reported, and LL intensity and duration were limited. In the present study, we found that bright LL suppressed locomotor activity rhythms rapidly in both Cry1−/− and Cry2−/− mice: 87% of Cry1−/− mice and 70% of Cry2−/− mice became arrhythmic within 7 weeks in LL. In previous work, wild-type mice in LL have generally developed arrhythmia more slowly (Ohta et al., 2005; Pickard 1994; Sollars et al., 2002; Spoelstra and Daan 2008; Steinlechner et al., 2002; Vitaterna et al., 1993), but methodological differences (e.g., mouse strain, LL intensity and duration) limit direct comparisons with the present results. While it is uncertain whether LL is more disruptive for the behavior of Cry-deficient mice than that of wild-type mice, it is clear that Cry1−/− and Cry2−/− mice rapidly become arrhythmic in bright LL.
On the basis of previous work using the Per1-GFP mouse (Ohta et al., 2005), we predicted that LL-induced behavioral arrhythmicity would reflect desynchrony among SCN neurons. At the single cell level, PER2::LUC rhythm phases of single cells were indeed more broadly distributed in Cry2−/− SCN slices from arrhythmic mice than in Cry2−/− slices from rhythmic mice. However, even in SCN slices from behaviorally arrhythmic mice, cell phases were not randomly distributed. This suggests that our LL manipulation was sufficient to render mice behaviorally arrhythmic but did not completely abolish synchrony within SCN slices. Consistent with this, all SCN slices, even those from arrhythmic mice, had clearly discernible circadian PER2::LUC rhythms at the level of the whole slice. Since other studies have observed arrhythmicity in SCN slices from arrhythmic rodents exposed to long-term LL (Granados-Fuentes et al., 2004; Ohta et al., 2005), it may be that more time under LL is required to completely desynchronize SCN neurons. Another possibility is that desynchronized SCN rhythms in behaviorally arrhythmic mice are more susceptible to the resetting effects of dissection, which could partially restore synchronization in vitro. Consistent with this possibility, and as also found in a previous study of Per1-luc rats in LL (Yoshikawa et al., 2005), SCN slice phases of arrhythmic animals were determined by time of dissection, unlike the SCN slices of rhythmic animals, whose phases were clustered according to in vivo circadian time. We also found a nearly antiphasic relationship between slices from arrhythmic Cry1−/− and Cry2−/− mice, suggesting genotypic differences in phase-shifting effects of dissection, possibly related to the altered photic resetting responses previously reported for Cry1−/− and Cry2−/− mice (Spoelstra and Daan 2008; Thresher et al., 1998; Vitaterna et al., 1999). It is also of interest that differences in circadian period of SCN slices and cells from Cry1−/− and Cry2−/− mice were consistent with the period phenotypes described previously (Liu et al., 2007; van der Horst et al., 1999; Vitaterna et al., 1999). Finally, SCN slices from arrhythmic Cry1−/− mice showed reduced amplitude relative to SCN slices from arrhythmic Cry2−/− mice, which reveals a persistent influence of LL that is consistent with a reduced amplitude of individual cells and/or greater desynchrony among cells within Cry1−/− slices. In summary, both single-cell and whole SCN slice rhythm phenotypes in vitro are consistent with partial rather than complete desynchrony of cells by LL, with some genotype-specific effects on phase and period.
Despite the lack of complete desynchrony, LL-induced arrhythmia was sufficient to produce a genotypically specific reduction in the rhythmicity of Cry1−/− neurons in SCN slices. Wild-type mice were not included in this study, but Cry2−/− mice provided an excellent control group since Cry1−/− and Cry2−/− mice both lack a core clock gene and display rapid arrhythmia under LL but nevertheless differ fundamentally in the effects of LL on SCN neuron rhythmicity. The proportion of individual cells showing detectable PER2::LUC rhythms was reduced in SCN slices from behaviorally arrhythmic Cry1−/− mice, relative to arrhythmic Cry2−/− mice, rhythmic Cry2−/− mice, and rhythmic Cry1−/− mice. We also examined another measure of rhythmicity: the goodness of fit to a best-fit sine wave in the circadian period range. This goodness of fit was lower in Cry1−/− than in Cry2−/− cells, tended to be lower in cells from arrhythmic mice than in cells from rhythmic mice, and was lowest of all in Cry1−/− cells from arrhythmic mice. Thus, even though the LL manipulation used here did not appear to completely desynchronize SCN cells, partial disruption of SCN intercellular coupling was sufficient to reduce rhythmicity of Cry1−/− cells in SCN slices.
This reduced rhythmicity observed in cells of SCN slices from arrhythmic Cry1−/− mice does not appear to be related to reduced tissue viability. SCN slices from arrhythmic Cry1−/− mice did have lower amplitude PER2::LUC rhythms. However, if Cry1 deletion compromised cell viability, we would expect to see a similarly reduced amplitude in Cry1−/− slices from rhythmic mice, as well as a reduction in the average brightness of PER2::LUC bioluminescence in Cry1−/− slices. On the contrary, rhythms of Cry1−/− and Cry2−/− slices from rhythmic mice were of comparable amplitude, and Cry1−/− slices were either just as bright as Cry2−/− slices (for arrhythmic mice) or substantially brighter than Cry2−/− slices (for rhythmic mice). The greater average PER2::LUC brightness in Cry1−/− SCN slices from rhythmic mice was also observed previously for individual dispersed Cry1−/− cells (Liu et al., 2007), likely reflecting the strong repressor function of CRY1 protein at the Per2 gene promoter. The viability of SCN slices from arrhythmic Cry1−/− mice was also demonstrated by the recovery of robust rhythmicity observed after several medium changes.
Completely abolishing SCN intercellular coupling by mechanical dispersal of cells results in markedly reduced rhythmicity of Cry1−/− cells (Liu et al., 2007). In the present study, treating mice with LL was designed to disrupt SCN intercellular coupling without the concomitant destruction of other tissue properties of the SCN slice, thereby testing whether reduced rhythmicity is specifically due to disruption of circadian coupling. With the relatively gentle manipulation of LL exposure, coupling was only partially disrupted, so it is not surprising that the proportion of rhythmic Cry1−/− cells was not as low as in dispersed cells, where coupling is abolished altogether. Nevertheless, reduced rhythmicity of Cry1−/− SCN neurons by LL-induced disruption of intercellular coupling supports the hypothesis that Cry1−/− cells do indeed depend on such coupling to maintain robust circadian function.
It remains unknown what underlying mechanisms explain different CRY1 and CRY2 contributions to single-cell oscillatory function, that is, why uncoupled Cry1−/− cells are nearly arrhythmic, whereas Cry2−/− cells are strongly rhythmic with long period. Based on the behavior of Cry mutant mice in constant darkness, it was originally proposed that Cry1 and Cry2 serve redundant roles in maintaining the molecular oscillator but antagonistic roles in determining its pace (van der Horst et al., 1999; Vitaterna et al., 1999). While effects on period length are highly reproducible, other data suggest that Cry1 and Cry2 deletions do not have identical effects on rhythm strength and precision even at the behavioral level (van der Horst et al., 1999; Oster et al., 2002; Spoelstra and Daan 2008; Thresher et al., 1998). At the molecular level, both CRY1 and CRY2 are potent repressors of CLOCK:BMAL1 transcription that can induce nuclear translocation of PER (Griffin et al., 1999; Kume et al., 1999; Lee et al., 2001). CRY1 and CRY2 display an overall 70% to 80% similarity in amino acid sequence (Hirayama et al., 2003; Ozturk et al., 2007; Sancar 2004; Tamanini et al., 2007), but the C-terminal tail domain is generally less conserved (Chaves et al., 2006; Hirayama et al., 2003). CRY1 and CRY2 may differ in transcriptional and/or posttranslational modifications that produce different affinities for PER or other clock components. CRY1 and CRY2 may also have different levels of protein expression and/or amplitude of rhythms (Okamura et al., 1999; Vitaterna et al., 1999; Yagita et al., 2002). Finally, a recent study demonstrated that the precise phase of rhythmic Cry1 expression is essential to its function, whereas Cry2 expression need not be rhythmic for normal circadian function (Ukai-Tadenuma et al., 2011). Further studies are needed to delineate the specific mechanisms underlying the distinct roles of CRY1 and CRY2 in the intracellular oscillator.
Addendum
After the manuscript was accepted for publication, studies were published which indicate that CRY1 is a stronger repressor than CRY2, and that this can serve as a biochemical basis for differences in Cry knockout phenotypes (Khan et al., 2012; Hirota et al., 2012).
Footnotes
Acknowledgements
Supported by NIH grant R01 MH082945 (D.K.W.), a VA Career Development Award to D.K.W., and NIH grant R01 MH051573 (Dr. Steve Kay).
Conflict of Interest Statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.