
Abstract
Bipolar disorder (BD) and major depressive disorder (MDD) are heritable neuropsychiatric disorders associated with disrupted circadian rhythms. The hypothesis that circadian clock dysfunction plays a causal role in these disorders has endured for decades but has been difficult to test and remains controversial. In the meantime, the discovery of clock genes and cellular clocks has revolutionized our understanding of circadian timing. Cellular circadian clocks are located in the suprachiasmatic nucleus (SCN), the brain’s primary circadian pacemaker, but also throughout the brain and peripheral tissues. In BD and MDD patients, defects have been found in SCN-dependent rhythms of body temperature and melatonin release. However, these are imperfect and indirect indicators of SCN function. Moreover, the SCN may not be particularly relevant to mood regulation, whereas the lateral habenula, ventral tegmentum, and hippocampus, which also contain cellular clocks, have established roles in this regard. Dysfunction in these non-SCN clocks could contribute directly to the pathophysiology of BD/MDD. We hypothesize that circadian clock dysfunction in non-SCN clocks is a trait marker of mood disorders, encoded by pathological genetic variants. Because network features of the SCN render it uniquely resistant to perturbation, previous studies of SCN outputs in mood disorders patients may have failed to detect genetic defects affecting non-SCN clocks, which include not only mood-regulating neurons in the brain but also peripheral cells accessible in human subjects. Therefore, reporters of rhythmic clock gene expression in cells from patients or mouse models could provide a direct assay of the molecular gears of the clock, in cellular clocks that are likely to be more representative than the SCN of mood-regulating neurons in patients. This approach, informed by the new insights and tools of modern chronobiology, will allow a more definitive test of the role of cellular circadian clocks in mood disorders.
Major depressive disorder (MDD) and bipolar disorder (BD) are neuropsychiatric disorders of brain mechanisms governing mood, goal-directed activity, sleep, cognition, and appetite. MDD is characterized by episodes of depressed mood, whereas BD includes both depressive episodes and periods of abnormally elevated or irritable mood known as mania. Genetic factors account for much of the etiological variance in these disorders (heritability 37% for MDD, 85% for BD) (Sullivan et al., 2000; McGuffin et al., 2003). Moreover, MDD and BD likely share common genetic mechanisms: among relatives of BD probands, risk of BD is elevated 7-fold, but risk of MDD is also elevated 2-fold (Kelsoe, 2003). The pathophysiology of mood disorders is unknown, but disruptions in daily rhythms associated with BD and MDD have led to consideration of the circadian clock as a possible causal factor. For decades, the search for clock abnormalities in mood disorders has focused on disruptions of sleep/wake and endocrine cycles that pointed to defects of the master clock in the suprachiasmatic nucleus (SCN). However, recent work has revealed an abundance of circadian clocks in brain regions outside the SCN (Abe et al., 2002; Guilding and Piggins, 2007) as well as network features of the SCN clock that render it uniquely resistant to perturbation (Welsh et al., 2010). In considering this new evidence, we conclude that if clock gene defects cause mood disorders, the SCN may not be the affected brain region, and focusing on this region may obscure genetic defects that are manifest only outside the SCN.
Clock Organization
The SCN is the dominant circadian pacemaker in mammals (Welsh et al., 2010), synchronized to the environment by light input from melanopsin-containing, intrinsically photosensitive retinal ganglion cells (ipRGCs; Ecker et al., 2010), and in turn synchronizing subsidiary oscillators in other tissues. Circadian rhythmicity is cell-autonomous, in both SCN neurons (Welsh et al.,1995) and non-SCN cells (Welsh et al., 2004). At the cellular level, the clock is a network of “clock genes,” transcriptional regulators that maintain rhythmic expression of their target genes over ~24-h cycles (Takahashi et al., 2008). The core of this clock is a delayed negative feedback loop in which PER and CRY proteins (PER1/2/3, CRY1/2) inhibit their own expression by interfering with the positive transcription factors CLOCK and BMAL1 at E-box regulatory elements. Additional positive and negative regulators (RORA/B/C and Rev-Erbα/β, respectively) act at RRE sites to regulate rhythmic expression of BMAL1 and other clock components. Also crucial for period determination are posttranslational modifications of clock components by signaling molecules like casein kinases δ/ε and glycogen synthase kinase 3β (GSK3β) (Reischl and Kramer, 2011). While this core clock mechanism is ubiquitous, details may differ among tissues. Most notably, whereas certain genetic perturbations (e.g., Cry1 knockout) render most tissues arrhythmic, cellular coupling uniquely present in SCN mitigates these defects, allowing for maintenance of rhythms in SCN tissue and behavior (Liu et al., 2007). SCN network properties also confer resistance of the clock to temperature perturbations (Buhr et al., 2010). Thus, non-SCN cellular clocks are more vulnerable than the SCN network clock to genetic or environmental insults, and this may lead to impairments in processes normally regulated by the clock in non-SCN cells.
Clinical Features of Mood Disorders Suggest Clock Abnormalities
BD and MDD involve deficits in reward processing and motivation. In depression, perception of reward is blunted, with corresponding reductions in motivation to pursue hedonic goals. Conversely, in mania, motivation to pursue rewarding stimuli is pathologically increased. In healthy humans, mood and reward are modulated by circadian phase (Boivin et al., 1997; Birchler-Pedross et al., 2009) and hence may be sensitive to alterations of the circadian clock. It has been recognized for decades that BD and MDD patients commonly exhibit disturbed sleep, diurnal mood variation (Hall et al., 1964), and cyclicity of mood and sleep disturbances (Riemann et al., 2002), suggesting the possibility of clock dysfunction. The existence of seasonal affective disorder (SAD), in which mood episodes recur seasonally, also suggests a connection between light-sensitive pathological mood states and light-sensitive circadian clocks (Rosenthal et al., 1984). Recognition of these phenomena led to development of such therapeutic interventions as bright light (Lewy et al., 1998; Eastman et al., 1998; Terman et al., 1998), sleep deprivation (Boivin, 2000), and shifts of sleep timing (Wehr et al., 1979; Sack et al., 1985) as antidepressants. Similarly, in BD, sleep loss or bright light can trigger mania, whereas extended darkness can reduce mania (Wehr et al., 1998).
Clock Dysfunction in Mood Disorders
Sleep abnormalities such as short latency to REM sleep and early morning awakening (Benca et al., 1992) led to the “phase advance hypothesis” of depression, suggesting that the SCN is phase advanced relative to sleep time (Wehr et al., 1979). However, these sleep changes could also be explained by reduced sleep drive, independent of circadian phase (Borbely, 1987). Although some studies have found abnormally early rhythm phases in depression (Wehr et al., 1979), these studies examined clock function indirectly by measuring processes regulated by the clock (e.g., body temperature, melatonin), outputs that are influenced by noncircadian (“masking”) factors not well-controlled in these studies. Most problematic of the masking effects are the sleep/wake changes in depression that were the original basis for the phase advance hypothesis. Not only can these sleep changes be explained as effects of noncircadian factors (Borbely, 1987), but early morning awakening can confound phase measurements by altering patterns of activity and light exposure, thereby not only affecting clock outputs but also shifting the clock itself, that is, causing both illusory and real (but secondary) phase advances. For these reasons, many of the conclusions drawn from early studies in MDD and BD patients are suspect.
Masking can be controlled by using constant routine protocols (Duffy and Dijk, 2002), forced desynchrony protocols (Dijk et al., 1999), and/or dim light melatonin onset as a marker of circadian phase (Benloucif et al., 2008). From studies employing such precautions, a complex picture of depression emerges. In SAD, a majority of patients (~70%) with phase delays can be treated by advancing phase with morning light or evening melatonin (Avery et al., 1997; Lewy et al., 2006). But others have abnormal phase advances, and deviations from normal phase alignment in either direction are correlated with severity of depression (Lewy et al., 2006). In MDD, the clock has been reported as phase delayed, with the extent of delay correlating with the severity of depression (Emens et al., 2009). This predominance of phase delays in MDD is consistent with the tendency for depressed patients to prefer later schedules (evening chronotype) (Drennan et al., 1991; Chelminski et al., 1999). It is also consistent with elevated rates of depression in subjects with extreme evening chronotypes (Shirayama et al., 2003; Kitamura et al., 2010; Abe et al., 2011) and the therapeutic effects of morning light, which produces phase advances (Sack et al., 1985). Some studies have also found decreased rhythm amplitudes in depressed patients (Souetre et al., 1989), including one forced desynchrony study in SAD (Koorengevel et al., 2002). In BD, no constant routine or forced desynchrony studies have been reported, but both phase and amplitude disruptions have been found. An actigraphic study of 19 euthymic BD patients revealed instability in daily rhythms and lower rhythm amplitudes but no consistent differences in phase (Jones et al., 2005). One study of DSPS subjects showed that mania is more common in phase delayed subjects compared to controls (Lee et al., 2011), a finding consistent with reports that patients with BD have a greater evening preference relative to controls (Wood et al. 2009). In summary, phase misalignment is frequently present in mood disorders, with delays more common than advances. Rhythm amplitude may also be reduced. While effects are modest, this might be expected from a defect that is more effectively buffered in the SCN than in brain structures involved in mood regulation.
Moody Clocks: Trait or State?
While it is common to consider BD and MDD as episodic illnesses, the clinical reality is unclear. In many cases, symptoms rarely remit. Longitudinal studies of BD reveal that patients are symptomatic ~50% of the time (Judd et al., 2002, 2003). Even during intervals of recovery, or in unaffected first degree relatives, cognitive (Glahn et al., 2010; Burdick et al., 2011) and affective (Linke et al., 2012) abnormalities are present. While recoveries are more common in MDD, recovered patients have decreased levels of brain derived neurotrophic factor (Molendijk et al., 2010), cognitive deficits (Paelecke-Habermann et al., 2005), and altered reward processing (Dichter et al., 2012), relative to controls who have never been depressed, suggesting underlying trait vulnerabilities in BD and MDD.
We hypothesize that circadian clock dysfunction is another enduring trait marker of MDD and BD, encoded by genetic variants in the clock. While the influence of environmental stressors on mood could also be mediated by acute dysfunction of the clock (a clock state), we posit that genetic vulnerability in the clocks of mood disorders patients increases the likelihood and intensity of this occurrence. In other words, genetic variants may increase sensitivity to environmental insults, increasing the probability that stressors will induce pathological fluctuations in mood, or that mood fluctuations occur autonomously in the absence of stressors (Post, 1992). This implies that cells obtained from mood disorder patients, which harbor all of the genetic determinants of clock function, should reveal clock traits predisposing to mood disorders, regardless of whether the cells were collected during a mood episode.
While attractive as the basis for a model, the dissociation of state and trait may prove to be an oversimplification. Epigenetic factors affect transcription by way of gene silencing or activation, typically in response to features of an organism’s unique environmental and developmental history (Tsankova et al., 2007). As epigenetic states are induced and long lasting, they are neither fully traits nor states. Histone modifications are involved in clock gene expression rhythms (Etchegaray et al., 2003), and NPAS2 promoter methylation leads to enduring changes in expression (Suter et al., 2011), suggesting that clock function could be altered by epigenetic processes. A recent demonstration of imprinting on the clock is a study showing that photoperiod manipulations during development affect subsequent gene expression and behavior rhythms in inbred adult mice (Ciarleglio et al. 2011). While more evidence is needed, we anticipate that clock epigenetics could inform developmental hypotheses of mood disorder onset as well as the incomplete penetrance of the illnesses, issues that sit uneasily between state and trait.
Human Genetic Studies
Numerous studies have found clock gene polymorphisms associated with mood disorders (Table 1), but most have small sample sizes, and replication has been difficult due to inconsistent inclusion criteria and phenotypic measures. In genome-wide association studies (GWAS), sample sizes are larger, but the number of variants examined is also much larger. Conventional unfocused analysis of GWAS using high statistical stringency has not strongly implicated clock genes in BD or MDD, except that RORA is associated with depressive personality traits (Terracciano, 2010) and antidepressant treatment response (Garriock et al., 2010). But reexamination of 14 GWAS studies using a more focused approach revealed that less stringently defined genetic associations with BD, MDD, and 2 related disorders are enriched among clock genes and pervasively rhythmic clock-controlled genes (McCarthy et al., 2012). Similarly, combining GWAS findings with evidence from genetic linkage and gene expression studies also implicates several clock genes in BD, including ARNTL, RORB, and GSK3β (Patel et al., 2010).
Genetic associations between the circadian clock and mood disorders.
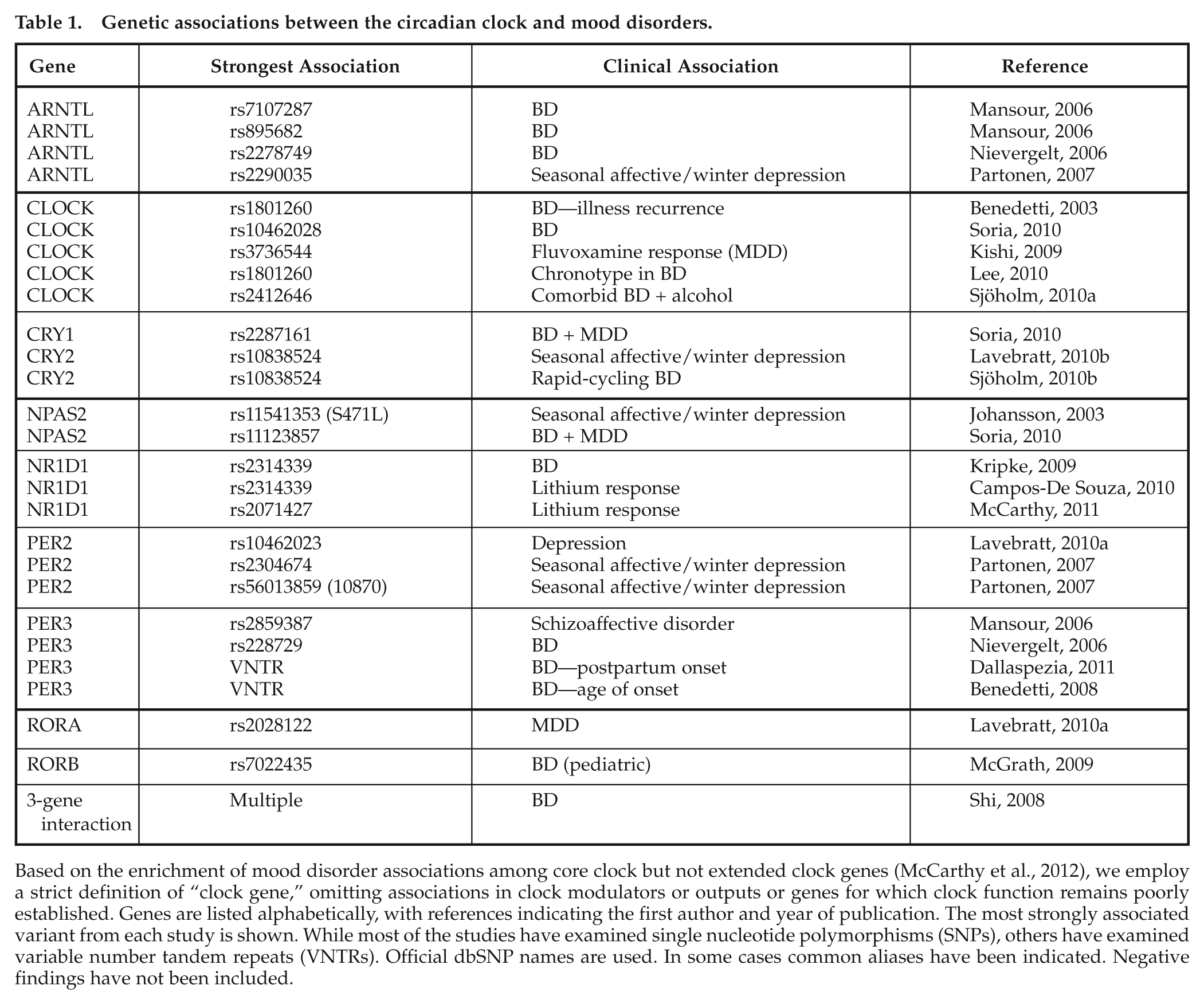
Based on the enrichment of mood disorder associations among core clock but not extended clock genes (McCarthy et al., 2012), we employ a strict definition of “clock gene,” omitting associations in clock modulators or outputs or genes for which clock function remains poorly established. Genes are listed alphabetically, with references indicating the first author and year of publication. The most strongly associated variant from each study is shown. While most of the studies have examined single nucleotide polymorphisms (SNPs), others have examined variable number tandem repeats (VNTRs). Official dbSNP names are used. In some cases common aliases have been indicated. Negative findings have not been included.
Clocks in Animal Models of Mood Disorders
While human studies remain a priority, animal studies are critical for determining how and where clock dysfunction affects mood in the brain. No animal model captures the full spectrum of mood phenotypes, particularly transitions among mania, depression, and normal mood (euthymia) seen in human patients, but diverse animal models have implicated clock genes in mood-related pathways. In the learned helplessness model of depression, rats exhibit long free-running circadian periods (Stewart et al., 1990). Per2-deficient mice show reduced monoamine oxidase A and increased dopamine (DA) in ventral striatum reward circuits and reduced depression- and anxiety-like behaviors (Hampp et al., 2008). Mice deficient in Rorb also show reduced depression and anxiety-like behaviors (Masana et al., 2007).
Clock-d19 mice, with a dominant-negative mutation of the gene Clock, have been proposed as a model of mania (Roybal et al., 2007). Compared to controls, Clock-d19 mice sleep less (Naylor et al., 2000), explore more, are motivated more by rewarding stimuli, and show decreased depression-like behavior in the forced swim test and learned helplessness models (Easton et al., 2003; Roybal et al., 2007). Some of these effects are reversed by lithium (Roybal et al., 2007) or viral transfer of wild-type Clock into the ventral tegmental area (VTA) (Roybal et al., 2007). At the cellular level, the Clock-d19 mutation increases excitability of VTA neurons, at least partly due to increased expression of tyrosine hydroxylase (McClung et al., 2005). RNAi knockdown of Clock in the VTA of wild-type mice enhances DA cell firing and produces mania-like hyperactivity in novel environments but also increased depression-like behavior in the forced swim and learned helplessness paradigms, a profile reminiscent of bipolar “mixed” states (Mukherjee et al., 2010).
In seasonally breeding animals, manipulations of day length affect reproduction, a photoperiodic response that depends on the SCN clock (Goldman, 2001) and is reminiscent of seasonal depression (Kripke, 1984). In diurnal rodents, shorter days induce depression-like behavior (Prendergast and Nelson, 2005), effects reversed by morning light or antidepressants (Ashkenazy et al., 2009; Krivisky et al., 2011). Such effects are less consistently found in nocturnal rodents (Prendergast and Kay, 2008).
Where Do Circadian Clocks Affect Mood?
The precise location of the brain dysfunction in MDD or BD is unknown (Krishnan and Nestler, 2010). The serotonergic neurons of the dorsal raphé and DA neurons of the VTA contribute to mood regulation and are targets for many antidepressant drugs. However, most antidepressants require weeks to work, and it is thought that their therapeutic effects may involve reorganization elsewhere in the brain, possibly in the hippocampus, anterior cingulate, or prefrontal cortex.
The SCN is the principal circadian pacemaker, but only limited evidence implicates the SCN in mood regulation. In postmortem brains of MDD patients, one study found lower levels of arginine vasopressin mRNA in the SCN relative to controls (Zhou et al., 2001). Also, rodents with SCN lesions have reduced depression-like behavior in the forced swim test (Tataroglu et al., 2004).
Effects of light on mood may be mediated through the SCN, but the SCN is not the only light-sensitive structure in the brain (Fig. 1). In mice, inappropriately timed light increases depression-like behavior, an effect that depends on input from ipRGCs (LeGates et al., 2010). While ipRGCs provide photic input to the SCN, anatomical studies have revealed that they also project to the lateral habenula (LHb), medial amygdala, and periaqueductal gray, areas important in mood regulation (Hattar et al., 2006; Ecker et al., 2010).
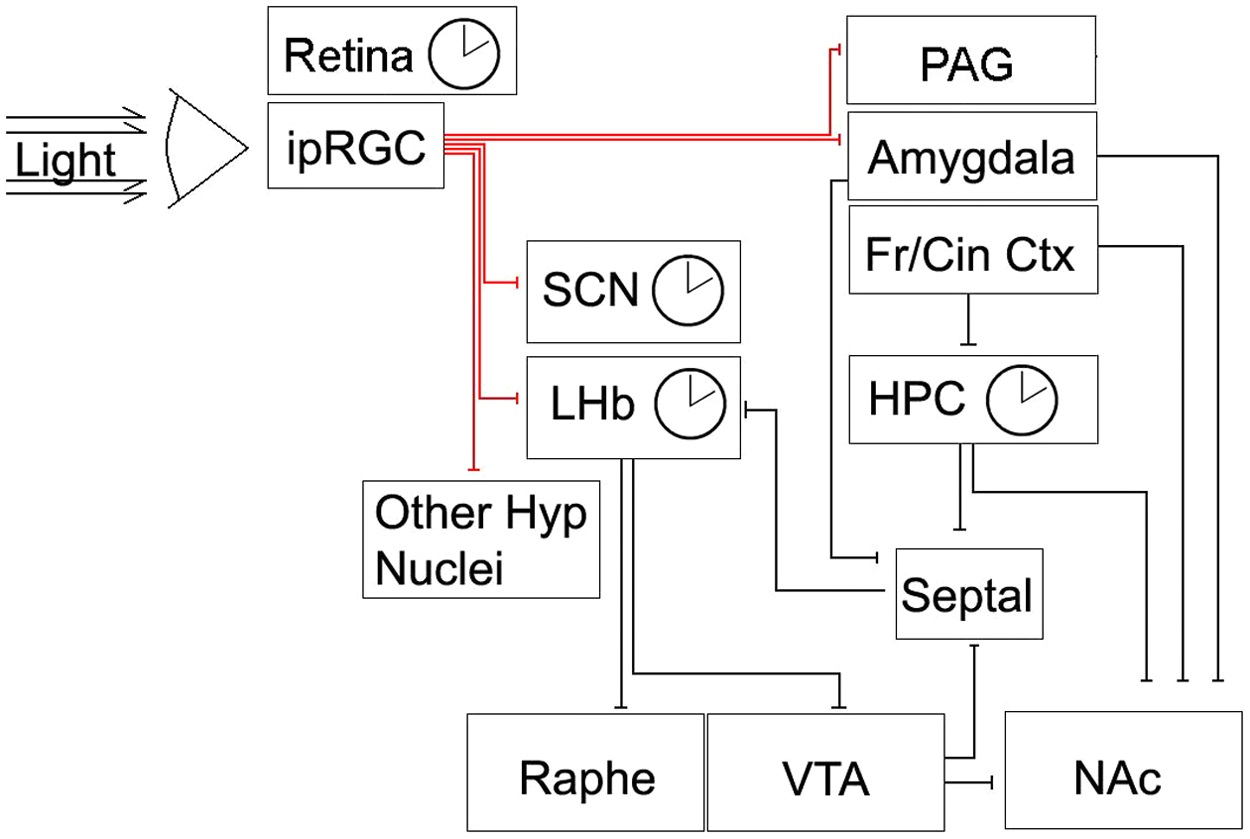
Light input to brain circuits implicated in mood regulation. A simplified diagram of the neural connections governing mood is shown. A number of structures in this circuit harbor circadian oscillators (indicated by clock symbol). For clarity, widespread serotonin and DA projections of the dorsal raphé and VTA, respectively, are not shown. Fr/Cin Ctx = frontal/anterior cingulate cortex; HPC = hippocampus; Hyp = hypothalamus; ipRGC = intrinsically photosensitive retinal ganglion cells; LHb =lateral habenula; NAc = nucleus accumbens; PAG = periaqueductal gray; Septal = septal nuclei; Raphe = dorsal raphé nuclei; SCN = suprachiasmatic nuclei; VTA = ventral tegmental area.
The midbrain DA system has been strongly implicated as a site where circadian clocks may alter mood. In this circuit, VTA “reward neurons” project to the nucleus accumbens (Nac) and prefrontal cortex and respond to rewarding stimuli including psychostimulant drugs (Nestler and Carlezon, 2006). The firing rates of some VTA neurons vary with time of day (Luo et al., 2008), and DA content in the NAc is increased in Per2 mutant mice (Hampp et al., 2008). As discussed, Clock-d19 mice show increased VTA DA neuron excitability (Roybal et al., 2007).
The LHb is another site where circadian clocks could alter mood, as it contains a light-sensitive circadian clock (Tavakoli-Nezhad and Schwartz, 2006) and plays a role in reward processing (Hikosaka et al., 2008). In the medial LHb, spontaneous and evoked FOS expression varies with time of day (Chastrette et al., 1991; Tavakoli-Nezhad and Schwartz, 2006), and LHb neurons show circadian firing (Zhao and Rusak, 2005) and PER2 rhythms (Guilding et al., 2010), even when disconnected from the SCN. Interestingly, many LHb neurons are light responsive, with preferential responses at night, similar to the circadian gating seen in the SCN (Zhao and Rusak, 2005). Omission of an expected reward results in increased firing of LHb “disappointment neurons” (Christoph et al., 1986; Matsumoto and Hikosaka, 2007), which then inhibit VTA DA neurons (Schultz et al., 1997). The LHb shows increased metabolic activity (Caldecott-Hazard et al., 1988) and neuronal firing during depression-like behavior in rats, which is reduced by LHb ablation (Yang et al., 2008) or inactivation (Li et al., 2011). Reduced LHb volume has been found in MDD and BD patients by MRI (Savitz et al., 2011) and in MDD patients postmortem (Ranft et al., 2010).
The hippocampus also has a circadian clock that could affect mood. Hippocampal size is reduced in MDD and BD and normalized by antidepressants or lithium (Hallahan, 2011; MacQueen and Frodl, 2011). Hippocampal clock gene expression is rhythmic in vitro (Wang et al., 2009) and may affect neurogenesis, which varies with time of day (Tamai et al., 2008; Gilhooley et al., 2011) and is increased in Per2Brdm1 mutants (Borgs et al., 2009). Neurogenesis is induced by antidepressants and suppressed by stress (Samuels and Hen, 2011), so hippocampal circadian clocks could affect mood by gating neurogenesis.
Autonomous or semi-autonomous circadian clocks also exist in the retina (Yoo et al., 2004,), olfactory bulb (Granados-Fuentes et al., 2006), and other regions (Abe et al., 2002; Guilding and Piggins, 2007) where clock defects could affect mood. Brain reward circuitry may also contain additional, unconventional circadian clocks. Rewarding stimuli such as feeding or methamphetamine induce circadian activity rhythms in rodents, even in the absence of the SCN and even in animals rendered otherwise arrhythmic by deletion of clock genes (Stephan et al., 1979; Storch and Weitz, 2009; Mohawk et al., 2009a). These food-entrainable or methamphetamine-associated oscillators could contribute to mood disorders. Interestingly, lithium and its molecular target GSK3β modulate methamphetamine’s effects on behavioral rhythms (Mohawk et al., 2009b).
Clock Effects of Antidepressants and Mood Stabilizers
Many medications used to treat mood disorders also affect the circadian clock. In rodents, the selective serotonin reuptake inhibitor (SSRI) antidepressant fluoxetine advances SCN phase (Sprouse et al., 2006) and modulates serotonin- or light-induced phase shifts (Prosser et al., 2006; Cuesta et al., 2008), whereas other SSRIs shorten circadian period in SCN and fibroblasts (Nomura et al., 2008). The mood stabilizer valproic acid shifts phase and increases amplitude of clock gene expression rhythms in SCN and fibroblasts (Johansson et al., 2011) but has less consistent effects on circadian period (Chansard et al., 2007; Klemfuss and Kripke, 1995).
Of particular interest is lithium, which delays phase or lengthens the period of free-running rhythms in rodents (Kripke and Wyborney, 1980), monkeys (Welsh and Moore-Ede, 1990), and humans (Johnsson et al., 1983). Lithium also increases PER2 rhythm amplitude (Johansson et al., 2011; Li et al., 2012). It has been suggested that lithium’s effects on the clock may contribute to its therapeutic properties (Johnsson et al., 1983). One molecular target of lithium that might mediate its circadian effects is GSK3β. Lithium inhibits GSK3β (Klein and Melton, 1996), which phosphorylates the clock components BMAL1 (Sahar et al., 2010), CRY2 (Harada et al., 2005), PER2 (Iitaka et al., 2005), and REV-ERBα (Yin, 2006). Specific GSK3β inhibition shortens (rather than lengthens) period in mammalian cells (Hirota et al., 2008; Vougogiannopoulou et al., 2008), so inhibition of GSK3β cannot account for lithium’s period-lengthening effects. But like lithium and valproic acid, GSK3β inhibition increases rhythm amplitude, an effect that is nonadditive with lithium (Li et al., 2012). Hence, the amplitude-enhancing effect of lithium might be mediated through GSK3β.
Advantages of a Cell-Based Approach
For decades, evidence has accumulated suggesting an association between mood disorders and circadian clock dysfunction. However, clinical studies of circadian function in psychiatric patients are difficult and expensive and have been limited to very few subjects. Moreover, measures of SCN function are indirect in clinical studies, and masking effects are problematic. Most importantly, it has become clear from animal studies that circadian clocks outside the SCN may be important for mood regulation and that these clocks may be more vulnerable to perturbation. That is, non-SCN circadian phenotypes are typically more extreme than SCN phenotypes. Thus, even the best measures of SCN rhythms may be misleading if they are insensitive to defects affecting other circadian clocks more relevant to mood.
Fortunately, recent developments allow a more direct approach to studying non-SCN circadian clocks. It is now known that the core molecular mechanism of the circadian clock is intracellular, that it is similar in brain and peripheral cells (Yagita et al., 2001), and that bioluminescent reporters can be used for longitudinal measurements of clock gene expression (Welsh et al., 2005). Skin fibroblast cells are fully competent autonomous circadian oscillators (Welsh et al., 2004; Leise et al., 2012). Circadian phenotypes of mouse clock gene mutants measured in fibroblasts are similar to those in SCN or behavior but often more extreme (Brown et al., 2005; Liu et al., 2007). Remarkably, the circadian phenotype of familial advanced sleep phase syndrome (fASPS) can be reproduced by introducing a mutated PER2 gene and measuring gene expression rhythms in fibroblasts entrained to a temperature cycle (Vanselow et al., 2006). Thus, in principle, peripheral cells could be used to assess clock phenotypes in human patients and might be more sensitive than clinical methods for assessing defects in non-SCN cellular circadian clocks (Fig. 2).
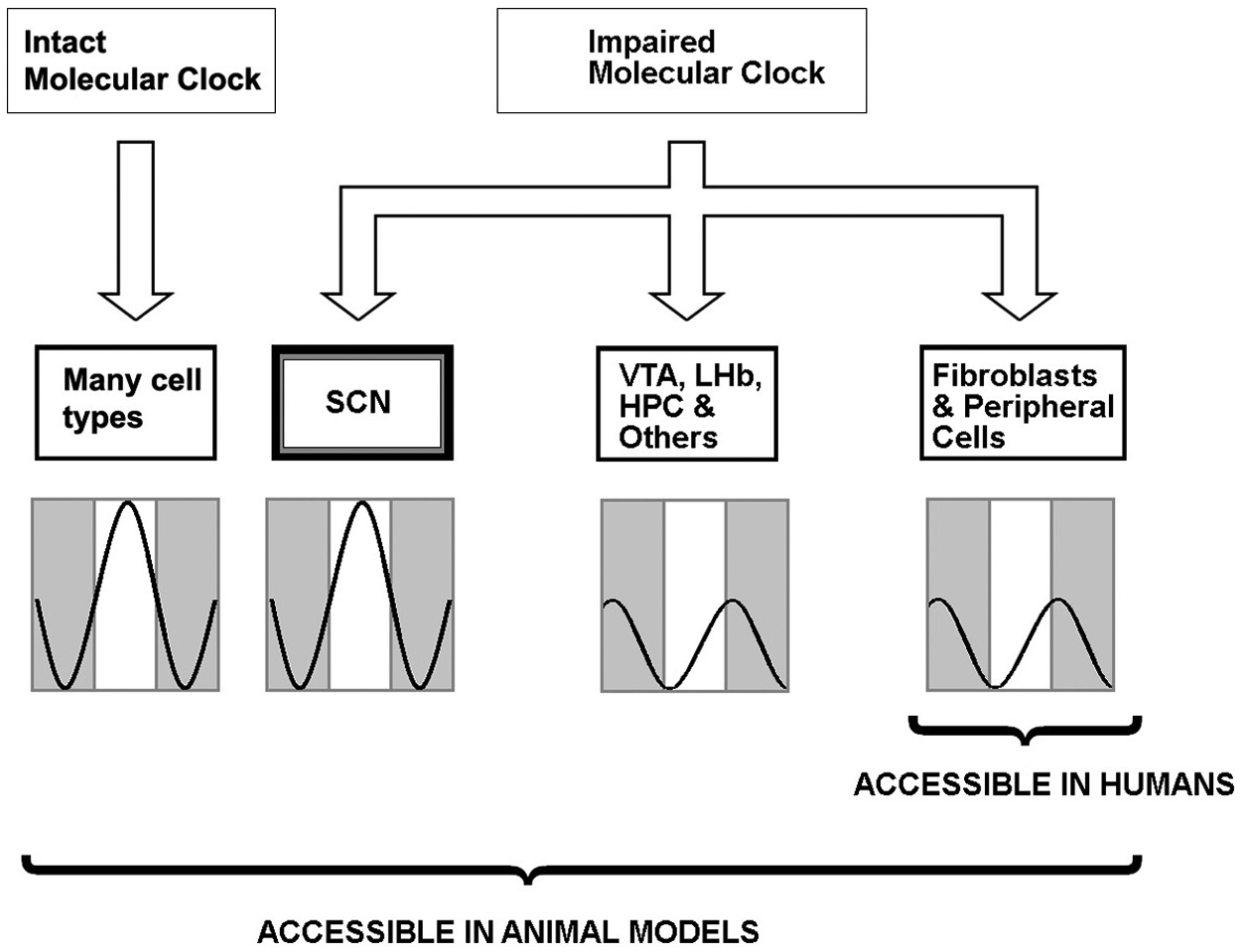
Conceptual basis for using peripheral cells in circadian clock phenotyping. In the healthy state, peripheral and SCN phenotypes are similar. When the clock has been altered (e.g., genetically), SCN rhythms may show subtle or no differences due to unique compensatory factors. In contrast, non-SCN cells are profoundly affected. Of the affected cell types, peripheral cells such as skin fibroblasts are readily accessible from living humans, whereas studies of neuronal cell types can be conducted in animals. In psychiatric illnesses, where the SCN is not likely to be the site of primary pathology, peripheral cells may be a better model of clock function than indirect measures of SCN rhythms (e.g., temperature or melatonin rhythms).
Initial studies exploring this idea used leukocytes obtained by serial blood sampling and measured gene expression by quantitative polymerase chain reaction (qPCR). Among 24 subjects sampled over 40 h, Archer et al. (2008) found that PER3 phase in leukocytes correlated with phase of sleep/wake, melatonin, and cortisol rhythms. Further progress has been made using fibroblasts. Using a lentiviral Bmal1-luc reporter, Brown et al. (2005) measured rhythms in fibroblasts cultured from 19 subjects and found that circadian period varied across subjects but was reproducible across replicate biopsies from the same individual. In a study of 28 subjects with extreme chronotypes, the same group found that morning preference correlated with shorter circadian periods (Brown et al., 2008). Among a subset of subjects with similar periods, cells from morning types (vs. evening types) had lower amplitude Rev-erbα rhythms and larger phase shifting responses to forskolin. Two recent studies compared circadian period estimated by Bmal1-luc in fibroblasts with period of melatonin rhythms in the same subjects. The measures were found to be correlated in one study (Pagani et al, 2010) but uncorrelated in the other, despite reproducibility across samples from a given subject (Hasan et al., 2012). It is not yet clear whether this discrepancy reflects differences between in vivo versus in vitro assays or differences between SCN versus non-SCN oscillators. Finally, in the only study to date of circadian rhythms in cells from mood disorders patients, Yang et al. (2009) obtained fibroblasts from 12 BD patients and 12 controls and used qPCR to measure rhythms over 60 h. The investigators found no evidence of period differences but did report lower amplitudes of BMAL1, REV-ERBα, and DBP rhythms in cells from BD donors.
Conclusion
MDD and BD are disorders defined in large part by uniquely human features. Therefore, delineation of the mechanisms underlying these illnesses has been challenging. Animal models do not adequately capture all important features. Living human brains are accessible only through imaging approaches. Postmortem human brains vary in age, agonal state, postmortem interval, and exposure to psychoactive substances and cannot be used for physiological studies. For these reasons, cell-based approaches like those used successfully to study the molecular basis of cancer have failed to translate to BD and MDD. However, unlike other genes implicated in mood disorders, clock genes operate as a well-characterized circuit that is functional and accessible in peripheral cells from patients or neurons from rodent models and can be studied quantitatively in a single cell. This vastly simplifies the task of identifying clock gene defects which, once characterized, can be studied in the context of brain circuits in animals. We believe that this cell-based approach will allow a definitive test of the hypothesis that circadian clocks contribute to the pathophysiology of mood disorders, potentially breaking an impasse in the study of these illnesses.
Footnotes
Acknowledgements
M.J.M. is supported by a VA Career Development Award (1IK2BX001275). D.K.W. receives support from a Veterans Affairs Merit Award (1I01BX001146), NIH (R01 MH082945), and a NARSAD Young Investigator Award. The funders had no role in the analysis, decision to publish, or preparation of the manuscript. Thanks to Drs. Daniel Kripke and Alfred Lewy for their comments on early drafts of the manuscript.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.