
Abstract
Like neurons in the suprachiasmatic nucleus (SCN), the master circadian pacemaker in the brain, single fibroblasts can function as independent oscillators. In the SCN, synaptic and paracrine signaling among cells creates a robust, synchronized circadian oscillation, whereas there is no evidence for such integration in fibroblast cultures. However, interactions among single-cell fibroblast oscillators cannot be completely excluded, because fibroblasts were not isolated in previous work. In this study, we tested the autonomy of fibroblasts as single-cell circadian oscillators in high- and low-density culture, by single-cell imaging of cells from PER2::LUC circadian reporter mice. We found greatly reduced PER2::LUC rhythmicity in low-density cultures, which could result from lack of either constitutive or rhythmic paracrine signals from neighboring fibroblasts. To discriminate between these 2 possibilities, we mixed PER2::LUC wild-type (WT) cells with nonluminescent, nonrhythmic Bmal1–/– cells, so that density of rhythmic cells was low but overall cell density remained high. In this condition, WT cells showed clear rhythmicity similar to high-density cultures. We also mixed PER2::LUC WT cells with nonluminescent, long period Cry2–/– cells. In this condition, WT cells showed a period no different from cells cultured with rhythmic WT cells or nonrhythmic Bmal1–/– cells. In previous work, we found that low K+ suppresses fibroblast rhythmicity, and we and others have found that either low K+ or low Ca2+ suppresses SCN rhythmicity. Therefore, we attempted to rescue rhythmicity of low-density fibroblasts with high K+ (21 mM), high Ca2+ (3.6 mM), or conditioned medium. Conditioned medium from high-density fibroblast cultures rescued rhythmicity of low-density cultures, whereas high K+ or Ca2+ medium did not consistently rescue rhythmicity. These data suggest that fibroblasts require paracrine signals from adjacent cells for normal expression of rhythmicity, but that these signals do not have to be rhythmic, and that rhythmic signals from other cells do not affect the intrinsic periods of fibroblasts.
In mammals, circadian rhythms of physiology and behavior depend on a master biological clock located in the suprachiasmatic nucleus (SCN) of the hypothalamus. The SCN output mechanism that synchronizes circadian rhythms of the whole body includes both neuronal and humoral pathways (Hastings et al., 2007; Saper et al., 2005). Within SCN and other cells, a transcription-translation negative feedback loop underlies circadian oscillations. This core loop involves activation of Period (Per1, Per2, Per3) and Cryptochrome (Cry1 and Cry2) gene transcription by a BMAL1/CLOCK heterodimer and delayed inhibition of this process by complexes containing PER and CRY proteins (Mohawk and Takahashi, 2011).
Within the SCN, which consists of about 20,000 neurons, cells communicate with one another through diffusible messengers (Maywood et al., 2011; Silver et al., 1996), synaptic signaling (Aton et al., 2005; Yamaguchi et al., 2003), and electrical coupling via gap junctions (Colwell, 2000; Jiang et al., 1997) to create coherent rhythms that are more robust to genetic and environmental perturbations than those of single cells (Buhr et al., 2010; Liu et al., 2007). Although individual SCN neurons can function as independent oscillators (Webb et al., 2009; Welsh et al., 1995), the proportion of rhythmic cells is decreased when SCN cells are cultured at very low density. These results suggest the importance of cellular communication in generation of single-cell circadian rhythms (Webb et al., 2009).
Some tissues outside the SCN, such as the olfactory bulb (Abraham et al., 2005) and peripheral tissues such as liver (Yoo et al., 2004), have been shown to sustain rhythmicity without signals from the SCN, suggesting that local cell communication could contribute to generating coherent and robust rhythms even outside the SCN. However, the mechanisms by which this might occur are obscure.
Single fibroblasts, including some established cell lines, exhibit cell-autonomous circadian rhythms (Nagoshi et al., 2004; Welsh et al., 2004) just as SCN neurons do. A wide variety of signals, including serum shock (Balsalobre et al., 1998), glucocorticoids (Balsalobre et al., 2000; Izumo et al., 2003), and temperature changes (Buhr et al., 2010), can synchronize peripheral oscillators. However, in contrast to SCN neurons, cultures of fibroblasts damp rapidly when disconnected from the control exerted by the SCN (Hastings et al., 2007; Nagoshi et al., 2004). This damping is explained by loss of synchrony among cells with diverse periods (Nagoshi et al., 2004; Pulivarthy et al., 2007; Ukai et al., 2007; Welsh et al., 2004). Interestingly, more mature fibroblasts cultured at higher density tend to produce higher amplitude circadian oscillations (O’Neill and Hastings, 2008). However, when cell populations with different periods have been mixed (Nagoshi et al., 2004; Noguchi et al., 2008), or single-cell rhythms examined directly (Welsh et al., 2004), no evidence has been found for local circadian communication in fibroblast cultures.
Here, we examined the possibility of local circadian communication among non-SCN cells by culturing fibroblasts at low density, so that single fibroblasts are relatively isolated from other cells. We used primary fibroblasts from PER2::LUC knockin mice expressing a fusion of PER2 and firefly luciferase (Yoo et al., 2004) to monitor clock gene expression rhythms in single fibroblasts by single-cell bioluminescence imaging (Welsh et al., 2005). In low-density cultures, PER2::LUC rhythms of fibroblasts were largely abolished. Co-culturing fibroblasts with either rhythmic wild-type or nonrhythmic Bmal1–/– fibroblasts rescued rhythmicity. Co-culturing with longer period Cry2–/– fibroblasts also rescued rhythms but did not affect the period of rescued rhythms. Furthermore, conditioned medium from high-density fibroblast cultures rescued rhythmicity of fibroblasts in low-density culture. These results suggest that diffusible paracrine signals are necessary to generate robust circadian rhythms in single fibroblasts but that these signals do not have to be rhythmic and do not regulate the periods of fibroblasts.
Materials and Methods
Animals
Generation of mPer2Luciferase (PER2::LUC) knockin mice was described previously (Yoo et al., 2004). For this study, we used an alternative PER2::LUC mouse line incorporating an SV40 polyadenylation site to enhance expression levels (Welsh et al., 2004). The mice were developed at Northwestern University using the same method as was used in the original strain of knockin mice (Yoo et al., 2004). Nonluminescent wild-type (WT) mice (C57BL/6J) were purchased from Jackson Laboratory (Bar Harbor, ME). Bmal1–/– mice were generously donated by Dr. Pamela L. Mellon at the University of California, San Diego. This line was created by crossing Bmal1-floxed mutant mice (Jackson Laboratory, stock number 7668) (Storch et al., 2007) and zona pellucida 3 (Zp3)-cre mice (Lewandoski et al., 1997) to produce mice lacking Bmal1 in all cells. Cry2–/– mice were generously donated by Dr. Akira Yasui at Erasmus University in the Netherlands (van der Horst et al., 1999). Mice were bred as homozygotes and maintained in LD 12:12 light cycles (12 h light, 12 h dark) throughout gestation and from birth until used for experiments. Mouse studies were conducted in accordance with regulations of the Committee on Animal Care and Use at University of California, San Diego.
Cell Culture
Primary fibroblasts were prepared from neonatal (1-7 days old) PER2::LUC knockin mice. Tails from about 6 pups were dissected into small pieces by surgical knives in 1 mL of ice-cold 0.25% trypsin (Life Technologies 15090-046, Carlsbad, CA) in phosphate buffered saline (PBS). Dissected tails were then incubated in 5 mL of 0.25% trypsin at 37 °C for 30 min. After the addition of 5 mL of growth medium (DMEM [Life Technologies 11995-065] supplemented with 25 U/mL penicillin, 25 µg/mL streptomycin, and 10% fetal bovine serum [Life Technologies 26140-079]) at 20 °C, fibroblasts were triturated by mild pipetting. Remaining tissue fragments were removed by a cell strainer (70-µm nylon mesh, Fisher Scientific, Pittsburgh, PA). Strained fibroblasts were centrifuged for 5 min at 1000 rpm. Pelleted fibroblasts were resuspended in 10 mL of growth medium. Fibroblasts (2×105 cells/dish) were plated in 35-mm culture dishes with 2 mL of growth medium and maintained in a standard tissue culture incubator at 37 °C, equilibrated with 5% CO2. For mixing experiments, fibroblasts of different genotypes (PER2::LUC, WT, Bmal1–/–, Cry2–/–) were independently isolated in parallel. About 1×104 cells/dish of PER2:LUC fibroblasts were mixed at 20 °C with about 2×105 cells/dish of fibroblasts of another genotype, and the mixture was plated and cultured at 37 °C.
When fibroblasts became confluent, samples were cultured for 2 to 12 weeks in explant medium: DMEM (Life Technologies 12100046), serum-free, 1.2 g/L sodium bicarbonate, with phenol red, pH 7.4, supplemented with 10 mM HEPES, 25 U/mL penicillin, 25 µg/mL streptomycin, and 2% B-27 (Life Technologies 17504-044). For culture in serum-free, HEPES-buffered explant medium, 35-mm culture dishes with fibroblasts were covered by 40-mm circular coverslips (Thermo Scientific 40CIR1, Waltham, MA), sealed in place with vacuum grease to prevent evaporation, and maintained at 37 °C without CO2. Low-density culture was created by waiting for confluent fibroblasts to reduce cell density after long-term culture in serum-free explant medium or by removing loosely attached fibroblasts by pipetting. In high-density culture, 1% to 5% PER2::LUC fibroblasts were mixed with nonluminescent WT (C57/BL6J) fibroblasts to facilitate identification and tracking of single cells. Conditioned medium was explant medium used to culture confluent fibroblasts for 1 to 2 weeks.
Bioluminescence Imaging
For imaging, medium was replaced with fresh explant medium containing 1 mM luciferin potassium salt (BioSynth L-8220, Staad, Switzerland). The imaging was performed as previously described (Welsh et al., 2005; Welsh and Noguchi, 2012). Briefly, the culture was placed on the stage of an inverted microscope (Olympus IX71, Tokyo, Japan). A heated lucite chamber custom-engineered to fit around the microscope stage (Solent Scientific, Segensworth, UK) kept the cells at a constant 36 °C. The microscope rested on an antivibration table (TMC, Peabody, MA) in a dark, windowless room, isolated by black curtains. After the microscope was focused carefully with bright-field illumination, lights were turned off, and stray light was eliminated by covering the dish with a small black lucite box and draping the microscope with black plastic sheeting (Thorlabs BK5, Newton, NJ). Light from the sample was collected by an Olympus 4x XLFLUOR objective (NA 0.28) and transmitted directly to a cooled charge-coupled-device (CCD) camera mounted on the bottom port of the microscope. We used the Series 800 camera made by Spectral Instruments (Tucson, AZ), containing a back-thinned CCD thermoelectrically cooled to –90 °C with a rated quantum efficiency of 92% at 560 nm. We measured read noise of 2.5 electrons at 50 KHz readout and dark current of 0.0002 electrons/pixel/sec. The signal-to-noise ratio was increased by 8 × 8 binning of the 1056 × 1032 pixel array. Images were typically collected at intervals of 30 min, with 29.5-min exposure duration, for 5 to 7 days. Images were acquired to computer with SI Image SGL D software (Spectral Instruments), saved to hard disk, and analyzed with MetaMorph (Molecular Devices, Sunnyvale, CA).
Image Processing
In MetaMorph, cosmic ray artifacts were removed by pixel-wise comparison of pairs of consecutive images, that is, using the minimum value of each pixel to construct a new image from every pair of consecutive images. Thus, data were effectively smoothed by a running minimum algorithm, with a temporal window twice the duration of a single exposure (i.e., a smoothing window of 60 min for 30 min-exposures). In the resulting stack of images, luminescence intensity was measured within a region of interest defined manually for each cell. The position of the region was adjusted if necessary to accommodate movements of cells during the experiment, but its size was kept constant across the time series. Data were logged to Microsoft Excel (Redmond, WA) files for plotting and further analysis. Luminescence intensity values were corrected for bias and dark current by subtracting the minimum intensity of a background region devoid of cells and were then converted to photons per minute based on the quantum efficiency and gain of the camera.
Rhythm Data Analysis
Because of high initial transients of luminescence after medium change, the first 12 h of data were excluded from analysis. Overall brightness was calculated by averaging values from 12 h after the start of recording to the end of recording. A linear baseline was subtracted from raw data (polynomial order = 1). Single-cell circadian rhythmicity was assessed by spectral analysis using the FFT algorithm in LumiCycle Analysis (Actimetrics, Inc., Wilmette, IL). Cells were considered to show significant circadian rhythmicity when the spectral peak was in the circadian range (period 20-36 h), and at least 6% of total spectral power (in the period range 0-36 h) was contributed by the highest point in the circadian range, similar to methods described previously (Ko et al., 2010; Liu et al., 2007). For cells categorized as rhythmic, periods and amplitudes were computed from the best-fit sine wave (“Sin fit”) using LumiCycle Analysis. Cell density was estimated from a bright field image taken at the beginning of each experiment.
Results
Fibroblasts Lose PER2::LUC Rhythmicity in Low-Density Culture
To test whether cell density affects clock gene oscillations in fibroblasts as it does in SCN neurons (Webb et al., 2009), we cultured fibroblasts from PER2::LUC mice at various cell densities and imaged single-cell circadian rhythms. In the serum-free medium used during imaging, fibroblasts did not proliferate or migrate extensively. Over the course of 5- to 7-day imaging experiments, cell density decreased slightly in high-density cultures (average 18.1% decrease, n = 7 cultures) and did not change substantially in low-density cultures (average 8.9% increase, n = 3 cultures). In high-density culture, a majority of single fibroblasts showed clear rhythmicity (Fig. 1, B and C). However, in low-density culture, most cells did not show clear rhythmicity (Fig. 1A). The percentage of rhythmic cells in a culture increased dramatically as cell density increased from 27 cells/mm2 to 46 cells/mm2 (Fig. 2A). The average percentage of rhythmic cells was 18.4% in 4 low-density cultures (4-27 cells/mm2) and 75.4% in 8 high-density cultures (46-410 cells/mm2). These percentages were significantly different (Fig. 2B) (p < 0.01, Mann-Whitney U test). Although rhythmicity was lost in low-density culture, brightness of PER2:LUC expression was not significantly different from high-density culture (Fig. 2C) (n = 54 in 4 low-density cultures, n = 120 cells in 8 high-density cultures; p > 0.05, Mann-Whitney U test). For cells categorized as rhythmic, periods and amplitudes were not significantly different between low- and high-density cultures (Fig. 2, D and E) (n = 10 in 4 low-density cultures, n = 94 cells in 8 high-density cultures; p > 0.05, Mann-Whitney U test). Cells that remained rhythmic in low-density culture were not clustered with other rhythmic or nonrhythmic cells.
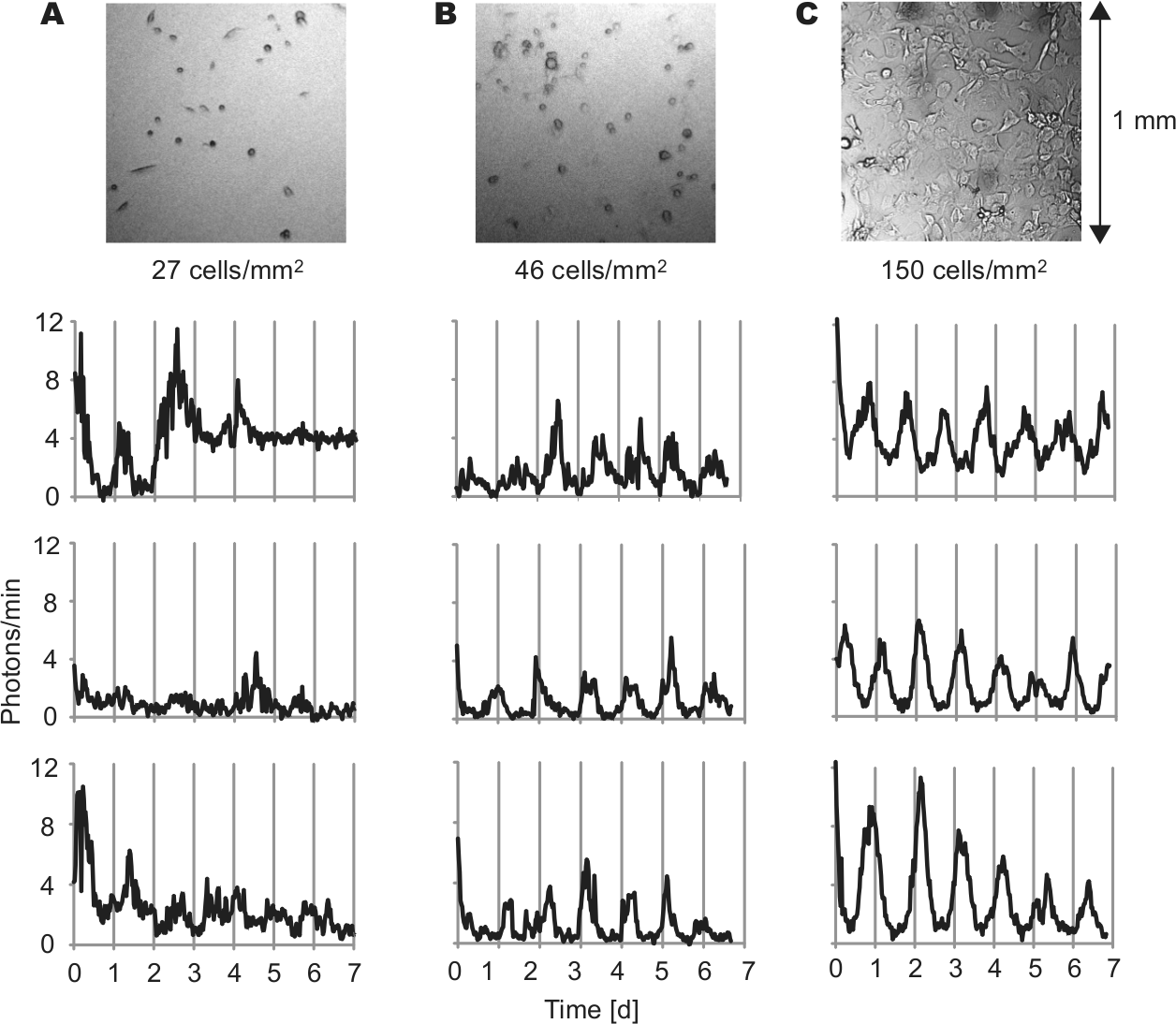
Fibroblasts lose PER2::LUC rhythmicity in low-density culture. Bright field images (above) and representative PER2::LUC bioluminescence intensity patterns (below) of fibroblasts in cultures with cell densities of 27 cells/mm2 (A), 46 cells/mm2 (B), and 150 cells/mm2 (C). Imaging began immediately following a change to fresh explant medium (day 0).
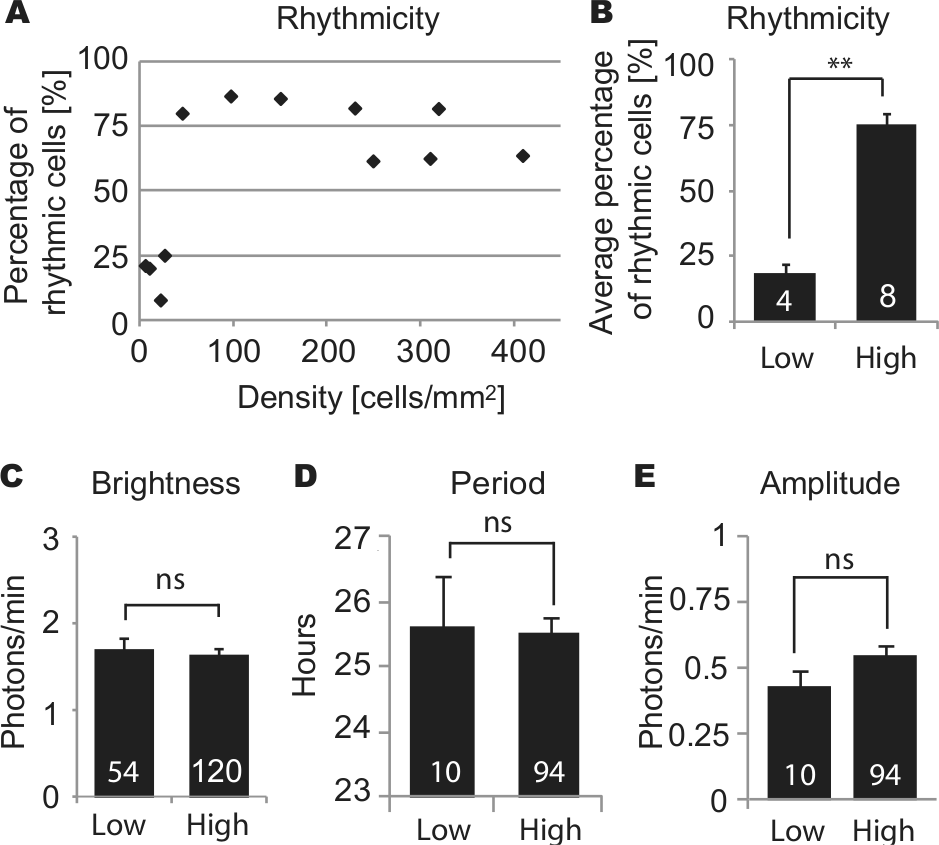
Circadian rhythm parameters in low- and high-density cultures: percentage of rhythmic cells for cultures of various cell densities (A), and percentage of rhythmic cells (B), brightness of cells (C), and (for rhythmic cells) period (D) and amplitude (E), in cultures of low density (<27 cells/mm2) and high density (>46 cells/mm2). Columns show average values ± SEM, and white numerals inside columns indicate numbers of cultures (B) or numbers of cells (C-E). ns = not significant; **p < 0.01 (Mann-Whitney U test).
Nonrhythmic Signals Rescue Rhythmicity of Fibroblasts in Low-Density Cultures
Reduced PER2::LUC rhythmicity in low-density cultures could result from lack of either constitutive or rhythmic paracrine signals from neighboring fibroblasts. To discriminate between these 2 possibilities, we mixed 1% to 5% luminescent WT cells with nonluminescent, nonrhythmic Bmal1–/– feeder cells (Liu et al., 2008), so that density of rhythmic cells was reduced to low levels (<20 cells/mm2) but overall cell density remained high. For comparison, we also used nonluminescent WT fibroblasts and longer period Cry2–/– fibroblasts (Liu et al., 2007) as feeder cells. With all 3 types of feeder cells, WT fibroblasts recovered rhythmicity (Fig. 3C). The average percentage of rhythmic WT cells with Bmal1–/– feeder cells (59.2%, n = 3 cultures) was slightly lower than the percentage with WT feeder cells (72.9%, n = 6 cultures) or Cry2–/– feeder cells (69.7%, n = 3 cultures). However, there was no statistically significant difference in percentage of rhythmic cells among the 3 types of cultures (n = 6, 3, 3 cultures with WT, Bmal1–/–, Cry2–/– feeder cells, respectively; p > 0.05, Kruskal-Wallis H test). Using different types of feeder cells also did not significantly alter PER2::LUC brightness (Fig. 3D) (n = 100, 42, 43 cells with WT, Bmal1–/–, Cry2–/– feeder cells, respectively; p > 0.05, ANOVA). For cells categorized as rhythmic, periods and amplitudes were analyzed. Although WT fibroblasts with Cry2–/– feeder cells showed ~1 h shorter mean period than cells with the other 2 feeder cell types, there was no statistically significant difference (n = 76, 27, 32 cells with WT, Bmal1–/–, Cry2–/– feeder cells, respectively; p > 0.05, ANOVA).
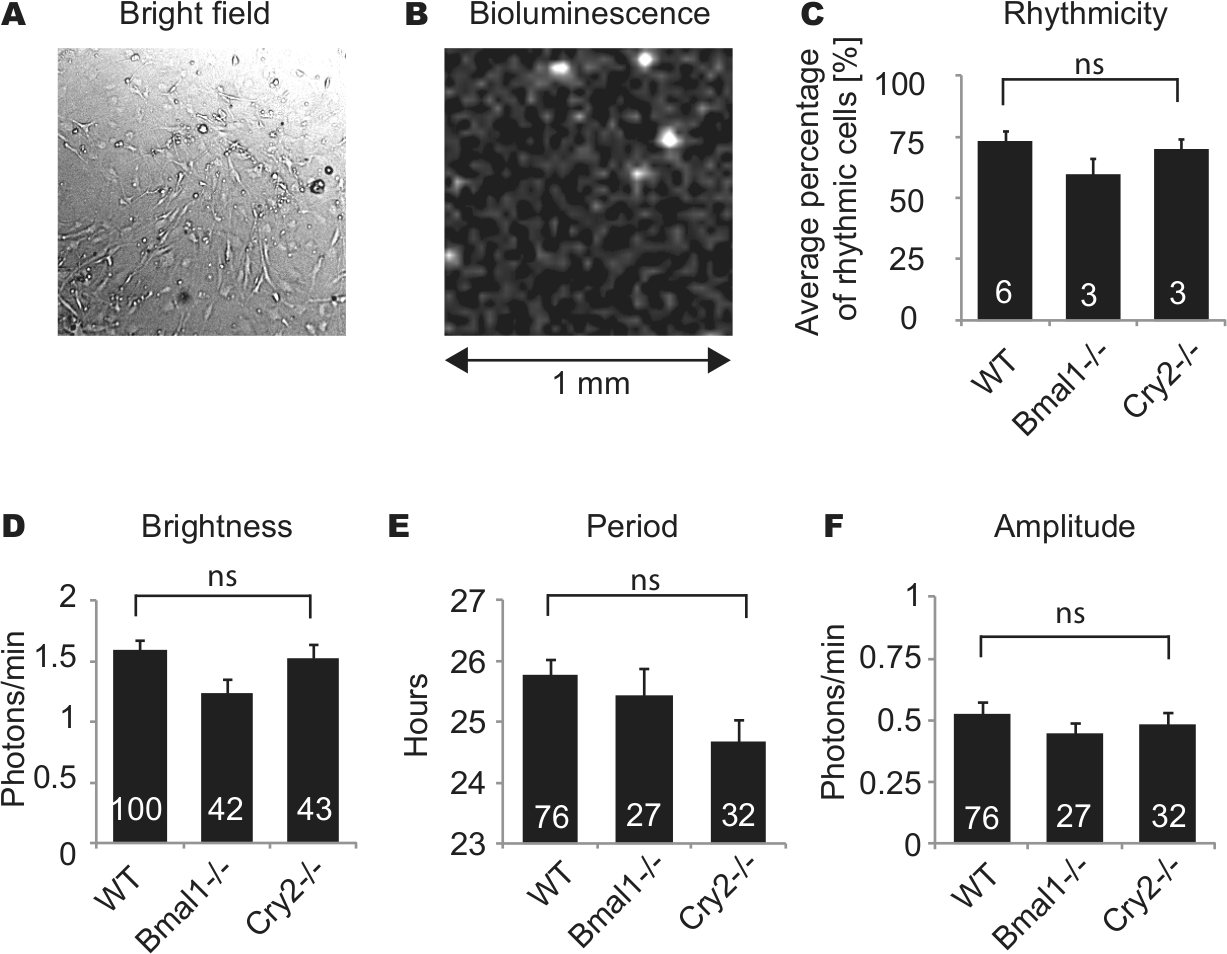
PER2::LUC fibroblasts were mixed with a 20- to 100-fold excess of nonluminescent fibroblasts from WT,
Conditioned Medium Rescues Rhythmicity of Fibroblasts in Low-Density Cultures
To determine whether paracrine signals that support rhythmicity of fibroblasts are diffusible factors released in medium, we cultured low-density fibroblasts with conditioned medium obtained from high-density fibroblast cultures. Rhythmicity of fibroblasts in low-density cultures was rescued by addition of conditioned medium (50% conditioned medium, 50% fresh explant medium) (Fig. 4, A and E). Average percentage of rhythmic cells was 66.6% with conditioned medium, which was not significantly different from high-density cultures (n = 3 cultures with conditioned medium, n = 8 high-density cultures; p > 0.05, ANOVA). In previous work, we found that low K+ suppressed fibroblast rhythmicity and that either low K+ or low Ca2+ suppressed SCN rhythmicity (Lundkvist et al., 2005; Noguchi et al., 2012). Therefore, to test whether high K+ or Ca2+ can rescue rhythmicity of fibroblasts in low-density culture, we tested medium containing 21 mM K+ or 3.6 mM Ca2+ (vs. 5.3 mM K+ and 1.8 mM Ca2+ in regular medium). High Ca2+ produced highly variable results ranging from 20% to 77.8% rhythmic cells in a culture. On average, high Ca2+ rescued rhythmicity partially (48.4% of cells rhythmic). This percentage was significantly higher than for low-density cultures without any treatment (Fig. 4A) (n = 3 cultures with high Ca2+, n = 4 low-density cultures; p < 0.05, ANOVA followed by Dunnett’s test). High K+ treatment produced slightly higher percentages of cell rhythmicity in low-density cultures (29.7%), but this effect was not significant (n = 3 cultures with high K+, n = 4 low-density cultures; p > 0.05, ANOVA). Brightness of cells was not significantly different across conditions (n = 54, 27, 34, 42, 120 cells in nontreated low-density culture, high K+, high Ca2+, conditioned medium, high-density culture, respectively; p > 0.05, ANOVA) (Fig. 4B). For cells categorized as rhythmic, periods and amplitudes were analyzed. Fibroblasts showed a 1.8- or 1.4-h longer period with high K+ or high Ca2+, respectively, and a 1-h shorter period with conditioned medium, although these effects were not statistically significant (n = 10, 8, 16, 28, 92 cells in untreated low-density culture, high K+, high Ca2+, conditioned medium, high-density culture, respectively; p > 0.05, ANOVA followed by Dunnett’s test) (Fig. 4C). There was no statistical difference in amplitudes (p > 0.05, ANOVA (Fig. 4D).
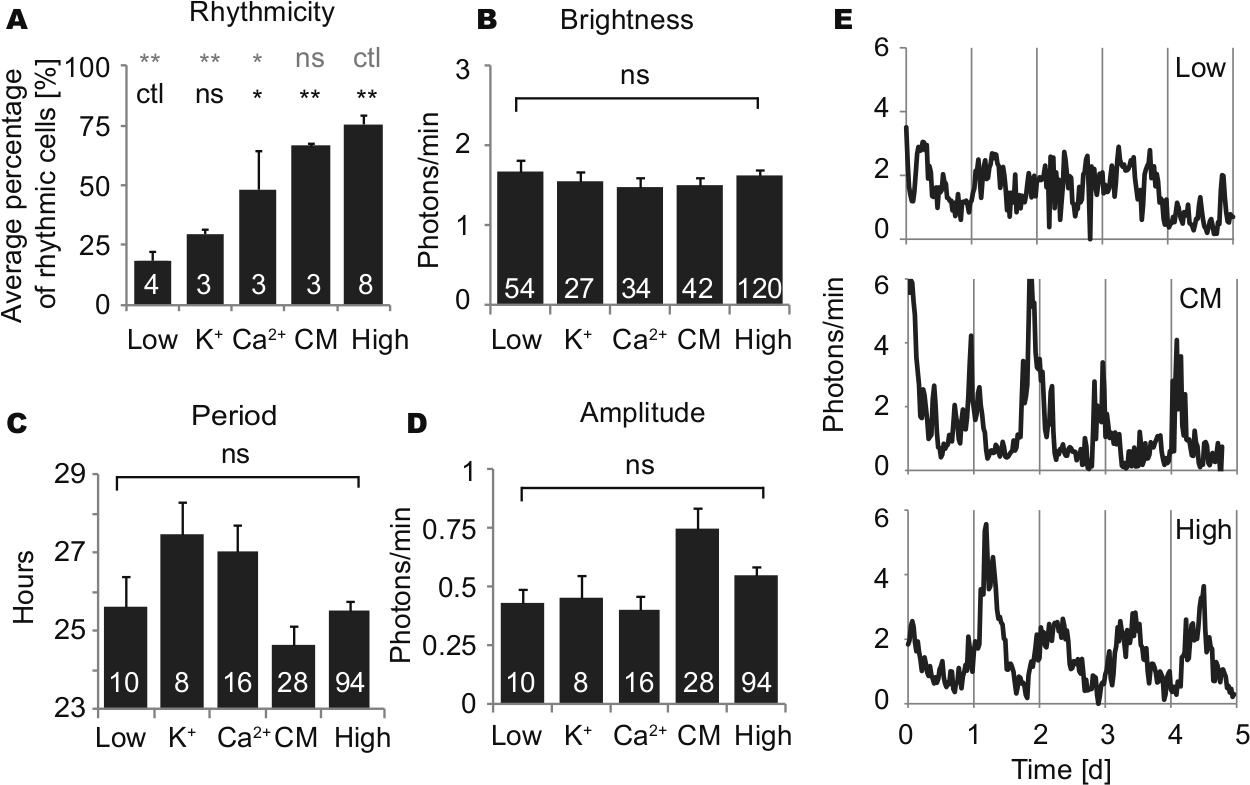
Circadian rhythm parameters in low- and high-density cultures without supplement and in low-density cultures with high K+ (final 21 mM), high Ca2+ (final 3.6 mM), or 50% conditioned medium: percentage of rhythmic cells (A), brightness of cells (B), and (for rhythmic cells) period (C) and amplitude (D). Columns show average values ± SEM, and numerals within columns indicate number of cultures (A) or number of cells (B-D). Low = low-density culture; K+ = high K+; Ca2+ = high Ca2+; CM = conditioned medium; ctl = control; ns = not significant; *
Discussion
In the SCN, the importance of intercellular signaling for supporting circadian rhythmicity is well established. Neuron rhythmicity in the SCN is impaired by blockade of neuronal firing with tetrodotoxin (TTX) (Yamaguchi et al., 2003) or genetic disruption of vasoactive intestinal polypeptide (VIP) signaling (Aton et al., 2005; Maywood et al., 2006). When SCN cells are dissociated and cultured at very low density, the percentage of rhythmic cells is reduced (Webb et al., 2009), suggesting that many SCN neurons require neighboring cells to maintain rhythmicity. It is unknown whether the intercellular signals supporting SCN rhythmicity are synaptic transmitters or diffusible paracrine signals. It is also unknown whether these signals are effective when present at a tonic level or whether they must vary in a circadian manner. However, diffusible synchronizing factors from SCN have been demonstrated by experiments in which an encapsulated SCN graft rescued circadian behavioral rhythms with the donor’s period (Silver et al., 1996). Diffusible SCN factors have also been shown to induce circadian rhythms in co-cultured nonrhythmic SCN slices (Maywood et al., 2011) and to prolong rhythmicity in populations of astrocytes (Prolo et al., 2005) and fibroblasts (Allen et al., 2001; Farnell et al., 2011).
In this paper, we show that, like SCN neurons, most fibroblasts lose PER2::LUC circadian rhythmicity in low-density cultures (Figs. 1 and 2), suggesting that signals from neighboring cells are necessary to generate circadian rhythms in cultured fibroblasts. Co-culture with nonrhythmic Bmal1–/– (Liu et al., 2008) or longer period Cry2–/– (Liu et al., 2007) fibroblasts rescues rhythmicity of fibroblasts in low-density cultures but does not affect the periods of recovered rhythms (Fig. 3). These results suggest that intercellular signals help to generate rhythmicity but do not work as circadian synchronizers among fibroblasts, consistent with previous studies that showed independently phased circadian rhythms of individual fibroblasts (Nagoshi et al., 2004; Welsh et al., 2004). Our finding that conditioned medium rescues rhythmicity of fibroblasts in low-density culture (Fig. 4), where most cells do not have physical contact with other cells, suggests that the important intercellular factors are diffusible paracrine signals rather than direct contacts with neighboring cells, such as gap junctions. Fibroblasts do express connexins and are connected through gap junctions in high-density cultures (Goodenough et al., 1996), but even in such high-density cultures there is no evidence that disruption of gap junctions affects circadian rhythms of fibroblasts (O’Neill and Hastings, 2008). We found that in some low-density cultures, addition of Ca2+ or K+ partially restores rhythmicity of fibroblasts (Fig. 4A), possibly by stimulating the release of paracrine signals or activating downstream signaling pathways.
The nature of the intercellular signal supporting rhythmicity in fibroblasts is unknown, but fibroblasts secrete many diffusible factors, including growth factors, cytokines, and extracellular matrix proteins. For example, fibroblast growth factor (FGF), epidermal growth factor (EGF), insulin-like growth factor (IGF), transforming growth factor (TGF) α and β (Wang et al., 2008), interleukins (Kumar et al., 1992), collagen, and fibronectin are secreted from dermal fibroblasts (Bhowmick and Moses, 2005). Fibroblast conditioned medium also contains metabolites such as amino acids, nucleotides, and intracellular proteins possibly released via cell death (Lim and Bodnar, 2002). Factors in conditioned medium affect proliferation, differentiation, and cell death of neighboring cells (Bhowmick and Moses, 2005; Millis et al., 1992; Schuldiner et al., 2000). Fibroblast conditioned medium also supports cell growth and attachment in low-density culture (Millis et al., 1992). Interestingly, TGFβ resets circadian rhythms of Rat-1 fibroblasts (Kon et al., 2008), TGFα is expressed rhythmically in the SCN and inhibits locomotor activity through hypothalamic EGF receptors (Kramer et al., 2001), and such growth factors as FGF, EGF, and IGF are rhythmic in human serum (Haus et al., 2001). Thus, many diffusible molecules are reasonable candidates for paracrine signals supporting rhythmicity of fibroblasts; as an initial approach, boiling or fractionating conditioned medium by molecular size would help to determine whether the paracrine signals are proteins.
At high-density, fibroblast cultures could be maintained for 8 to 12 weeks, with medium replenished once per week. At low density, fibroblasts could survive for only a few weeks (data not shown). Although low-density culture of fibroblasts may compromise long-term survival, raising the question of whether the paracrine signals supporting rhythmicity work as trophic factors, the fibroblasts we analyzed survived throughout 5- to 7-day experiments, producing PER2::LUC bioluminescence as bright as in high-density culture (Fig. 2C). Bioluminescence is produced by an ATP-requiring enzymatic reaction and for this reason is an excellent test of cell viability. Cells that did not maintain bioluminescence for the entire duration of the experiment were excluded from analysis. Therefore, all cells analyzed were viable. Furthermore, individual cells cultured at low density produced the same average level of PER2::LUC bioluminescence as cells cultured at high density (Fig. 2C), suggesting that they were equally healthy, although stress signaling pathways may increase PER2 stability (Uchida et al., 2012), possibly confounding PER2::LUC as a quantitative measure of cell health.
We performed 2 additional independent tests of cell viability in low-density fibroblast cultures. We confirmed membrane integrity by exclusion of 20% trypan blue (Sigma; data not shown). We also used fluo4-AM calcium imaging to measure response to thapsigargin, an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPase, and found a 50% increase in Ca2+ (mean of 4 cells), which returned to baseline levels after drug washout (see Fig. S1). Furthermore, addition of conditioned medium to fibroblasts in low-density culture induced cell migration and proliferation (data not shown), consistent with a previous study (Millis et al., 1992). These observations confirm that nonrhythmic fibroblasts in low-density culture are viable and maintain metabolic activities, so the paracrine signals supporting circadian rhythmicity in fibroblasts cannot work merely as trophic factors promoting cell survival.
Previous studies suggested that cell proliferation perturbs cellular timekeeping (Nagoshi et al., 2004; O’Neill and Hastings, 2008), raising the question of whether higher rates of cell proliferation in low-density cultures may account for reduced rhythmicity. However, in our culture conditions using serum-free medium, we rarely observed cell division in high- or low-density cultures, and the number of cells did not increase substantially during imaging. Thus, it is unlikely that differences in cell proliferation rates were responsible for differences in rhythmicity.
In this study, the percentage of rhythmic cells in high-density cultures was 77.5% and did not reach 100% as reported in previous studies (Leise et al., 2012; Liu et al., 2007; Welsh et al., 2004). There are several possible explanations for this. First, as suggested by Leise et al. (2012), cells may appear to be nonrhythmic in shorter recordings due to stochastic variations in amplitude. Second, since we mixed PER2::LUC fibroblasts with excess nonluminescent WT fibroblasts to facilitate tracking, dimmer fibroblasts were more evident than in previous studies, and dimmer cells are more likely to appear nonrhythmic due to a decreased signal-to-noise ratio. Finally, because of the low resolution of bioluminescence images, we cannot exclude the possibility that morphologically unusual (damaged) cells or clusters of multiple cells were included in our analysis in some cases. Improved spatial resolution of bioluminescence images, or interleaving bioluminescence images with higher resolution bright field or fluorescence images, may clarify this issue in future studies.
In summary, here we demonstrate the existence of paracrine signals necessary to maintain circadian rhythmicity in fibroblasts. Further investigation is needed to identify these paracrine signals released into medium by high-density fibroblast cultures. Studying the effects of various paracrine signals on circadian rhythms will provide further insight into how the mechanism of circadian rhythm generation may involve not only clock genes but also extracellular signals.
Footnotes
Acknowledgements
Supported by NIH (R01 MH082945 to D.K.W.) and a V.A. Career Development Award (to D.K.W.).
Conflict of Interest Statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.