
Abstract
Introduction
Type 2 diabetes is one of the risk factors for digestive diseases and is a chronic disease caused by hyperglycemia and metabolic disorders, which may result in multi-organ complications. The gastrointestinal tract is one of the pathogenic targets of type 2 diabetes.1–3 Diabetic enteropathy is a common complication of diabetes. Gastroparesis, diarrhea and constipation, and changes in gastrointestinal function in diabetic patients have been widely studied.4–6 Studies have found that there are hyperproliferation and abnormal differentiation of intestinal epithelial cells (IECs) in diabetes.7,8 It has been confirmed that multiple modulation mechanisms are involved in the proliferation and differentiation of IECs. Signal pathways and epigenetic regulation are important regulators in maintaining intestinal homeostasis and disease progression. 9 It is reported that the proliferation of small IECs in diabetic rats was significantly enhanced. 8 Previous study has found that inhibiting the cellular Notch/Hes1 signaling pathway resulted in increased proliferation and abnormal differentiation of IECs in streptozotocin-induced diabetes in mice, as well as intestinal barrier dysfunction. 7 However, the molecular mechanism involved in this process remains unclear.
Studies have shown that high blood glucose levels can inhibit autophagy in mammalian cells.10,11 The accumulation of endogenous p62 is observed in C17.2 cells cultured under high glucose.12,13 It has been demonstrated that high glucose played a dual role in autophagy in GMCs, which could activate the autophagy signaling pathway in short time periods, but inhibit autophagy over 12 h. 14 Continuous and chronic hyperglycemia promoted increased expression of ROS, caspase 1 and IL-1β by inhibiting autophagy levels. 16 However, there is no definite evidence whether the effect of hyperglycemia on intestinal function is related to the inhibition of autophagy.
Multiple lines of evidence demonstrate the critical link between intestinal function and autophagy. This relationship is complex because autophagy can dramatically regulate multiple aspects of intestinal physiology, from maintenance of the epithelial architecture to metabolic regulation, function of specific intestinal epithelial subsets, regulation of inflammatory pathways, and defense against infection. 15 It is shown that autophagy plays an important role in maintaining intestinal homeostasis, participates in regulating the interaction between the gut microbiota and innate and adaptive immunity, and enhances the host’s defense against intestinal pathogens. 16 It has been reported that autophagy defect is related to various intestinal pathological changes such as inflammatory bowel disease (IBD). 17 Intestinal epithelial dysfunction, intestinal dysbiosis, enterocyte endoplasmic reticulum stress response are closely related to autophagy deficiency, and abnormal intestinal immune response to pathogenic bacteria is a hallmark of IBD pathogenesis. 18 In 2015, the study reported for the first time that autophagy could regulate intestinal barrier function by reducing intestinal epithelial permeability, because autophagy could induce the lysosomal degradation pathway of the tight junction protein claudin 2. 19 The study found that DRAM1 derived from IECs was involved in the pathogenesis of IBD, and the possible mechanism was that DRAM1 played a positive role in regulating autophagy and apoptosis of IECs. 20
The effect of diabetes on intestinal function is likely to be related to the down-regulation of autophagy. It has been verified that inflammatory response can be relieved by autophagy. However, whether diabetes induced intestinal dysfunction is related to impaired autophagy, and, whether the damage of autophagy can lead to the aggravation of inflammation (such as enterocolitis and colitis) has not been studied. In this study, we observed the effect of hyperglycemia on autophagy, and then explored the effect of autophagy on the inflammation level of IECs. Different concentrations of glucose were used to culture IECs. The AMPK (AMP-activated protein kinase) agonists and antagonists were also used.
Materials and methods
Chemicals and reagents
The mouse IEC-6 cells were purchased from Cobioer (CBP60938, Nanjing, China). The Dulbecco’s modified Eagle’s medium (DMEM). Fetal bovine serum, penicillin-streptomycin, and 0.25% trypsin were purchased from Hyclone (Logan, USA). The rabbit anti-mouse P62, anti-mouse LC3 and anti-mouse AMPK antibodies were purchased from R&D (San Diego, USA). ULK1 antibody was purchased from Novus (Colorado, USA). Cytokine detection kits for nitric oxide synthase 2 (NOS2), tumor necrosis factor-α (TNF-α), interferon-γ (INF-γ) and interleukin 22 (IL-22) were purchased from Cloud-clone (Wuhan, China). HRP-conjugated secondary antibodies and ECL detection system were purchased from Santa Cruz Biotechnology (Santa Cruz, USA). Lipofectamine 2000 was purchased from Invitrogen (Waltham, USA). Goat anti-mouse LC3 was purchased from Merck (Woodbridge, USA). GSK621, Dorsomorphin and dihydrochloride were purchased from Merck (Woodbridge, USA). Rat anti-mouse ULK1 was purchased from Abcam (USA). Rat anti-mouse p-AMPK antibody was purchased from Sigma (St Louis, USA).
Cell culture and treatments
IEC-6 cells were cultured in DMEM containing 10% fetal bovine serum and penicillin-streptomycin (100 U/ml; 0.1 mg/ml) in a 5% CO2-humidified incubator at 37°C. After trypsin digestion, cells were subsequently seeded into 6-well plates, with 100 μl of 106 cells per well. To explore suitable diabetic mellitus (DM) conditions, IEC-6 cells were cultured in different concentrations of D-glucose (5 mM–50 mM). The control group was exposed to L-glucose (LG) in normal medium with D-glucose to account for medium hyperosmolarity. Cells were collected for subsequent experiments after 48 h of stimulation.
Cell viability assay
The cytotoxic effect of high glucose on IEC-6 cells was determined using a CCK-8 kit (Wuhan, China). Cells were seed into 96-well plates at a density of 106 cells per well and incubated at 37°C overnight. After the treatment, the IEC-6 cells were incubated for another 4 h with 100 µl of serum-free DMEM and 10 µl of CCK-8 at 37°C. Then, we used a microplate reader to measure the absorbance at 450 nm wavelength.
Western blot analysis
IEC-6 cells were collected and subjected to lysis with RIPA lysis buffer (Sigma, USA) containing protease inhibitors. The proteins were obtained after centrifugation. Subsequently, the BCA (Beyotime, shanghai, China) was used to determine the protein concentration. Equal amounts of proteins (40 μg) were subjected to 10% SDS-PAGE and then transferred onto nitrocellulose membranes (Millipore, MA, USA). Subsequently, the membranes were then blocked with 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 2 h at room temperature, and then incubated separately with anti-mouse LC3, rabbit anti-mouse P62 and anti-mouse GAPDH primary antibodies (R&D, San Diego, USA) at 4°C overnight. Then, the membranes were incubated with HRP–conjugated goat anti-rabbit or goat anti-mouse secondary antibodies (Santa Cruz, USA) for 1 h at room temperature. After washing for three times with TBS-T, the membranes were detected using the ECL detection system (Merck, USA) and analyzed using ImageJ software.
Immunofluorescence
IEC-6 cells after various treatments were fixed with 4% paraformaldehyde for 10 min at room temperature, washed twice with PBS, and then incubated in 5% normal goat serum. To reduce nonspecific binding, we incubated the slides with 0.05% Tween 20 for 1 h. Subsequently, incubation with anti-LC3b primary antibody (R&D, San Diego, USA) for 1 h at room temperature was performed. After washing the slides three times with PBS, we then incubated the slides with Alexa Fluor 488 secondary antibody (Merck, USA) for 1 h at 37°C. In addition, we also set up negative controls. All images were acquired at 488 nm wavelength with a fluorescence microscope (Zeiss, Germany).
Cytokine assay
Amounts of NOS2, TNF-α, INF-γ and IL-22 in the culture medium were detected. Cells were treated as previously described. Levels of cytokines in cell culture supernatants were examined using corresponding ELISA kits (Cloud-clone, Wuhan, China), according to the manufacturer’s instructions. Absorbance values at 450 nm were measured using a microplate reader (Bio-Rad, USA).
Statistical analysis
SPSS version 17.0 software (IBM, Chicago, USA) was used for Statistical analysis of the data. Data obtained from three independent experiments were shown as the mean ± SEM (standard error of mean). T test was used for comparisons between two groups, and one-way ANOVA was used for multiple comparisons among groups. A value of p < .05 was considered significant.
Results
High glucose is not conducive to the survival of IEC-6 cells in vitro
The effect of different concentrations (5–50 mM) of glucose on cell growth status was observed. IEC-6 cells were cultured with medium containing different concentrations of glucose for 48 h. The cells treated with 5 mM glucose were used as normal control. The images of cells in each group under ×40 magnification were shown (Figure 1(a)). Next, we analyzed the effects of glucose levels on cell morphology by counting the number of shrunken and necrotic cells (Figure 1(b)). In addition, we evaluated cell viability by CCK-8 assay. The results indicated that cells exposed to high glucose showed a profound increase in the number of necrotic cells, and CCK-8 assay confirmed a significant decrease in cell viability, especially in the 50 mM group (**p < .01). These findings demonstrate that high glucose is not conducive to the survival of IEC-6 cells in vitro. Growth status of IEC-6 cells under different glucose concentrations. (a) IEC-6 cells were divided into four groups and cultured with glucose of different concentrations (5 mM, 10 mM, 30 mM, and 50 mM) for 48 h. (b) The number of shrunken and necrotic cells per high power field (40x). (c) CCK-8 assay was performed to assess cell viability of IEC-6 cells. Data are presented as the mean ± SD; n = 3; *p < .05; **p < .01 vs. control.
High glucose inhibits autophagy and increases cytokines in IEC-6
To investigate whether high glucose affects autophagy in IECs, we detected the levels of proteins after autophagy in IEC cells on Day 2. The results showed that in the high glucose group, there was downregulated IC3II but upregulated P62, which indicates that the autophagy level is decreased compared with the LG group (Figure 2(a)). Furthermore, it was also shown that autophagy level was positively correlated with glucose concentration. The effect of high glucose on cytokines of IL-22, INF-γ, NOS2 and TNF-α was detected with ELISA (Figure 2(b)). The results indicated that their levels were significantly increased in the high glucose group (30–50 mM) compared with the LG group (*p < .05; **p < .01). These results suggest that high glucose could not only inhibit the expression of autophagy proteins but also increase the secretion of pro-inflammatory factors. Autophagy and cytokine levels under different glucose concentrations. (a) Western blot image and semi-quantitative profile of LC3I/II and P62 protein levels in IEC-6 cells cultured with high glucose for 48 h. (b) ELISA analysis of TNF-α, NOS2, IL-22, and INF-γ levels. Data are presented as the mean ± SD; n = 3; **p < .01; ***p < .001; #p > .05 vs. control.
Activation of AMPK pathway favor the survival of IEC-6 cells in high glucose
To investigate the effect of AMPK signaling pathway on the growth of IEC-6 cells cultured in high glucose, we used different AMPK modulators, including Dorsomorphin (an AMPK inhibitor), GSK621 (an AMPK activator), and rapamycin (an autophagy activator), to interfere with the level of AMPK. The group treated with 30 mM glucose served as blank control. After 48 h of treatment, IEC-6 cells of each group were observed under 40x magnification (Figure 3(a)). Next, we verified the effects of AMPK modulators on cell morphology in high glucose medium by counting the number of shrunken and necrotic cells (Fig3(b)). We also evaluated the cell viability by CCK-8 assay. The results indicated that Dorsomorphin group showed a profound increase in the number of necrotic cells but significant decrease in cell viability compared with control group (**p < .01). However, GSK621 group and Rapamycin group showed a low necrosis rate and a high cell viability compared with Dorsomorphin group (***p < .001). These results demonstrated that activating AMPK pathway or enhancing autophagy can reduce cellular damage caused by high glucose and promote cell survival. Growth status of IEC-6 cells co-cultured with Dorsomorphin, GSK621, and Rapamycin under high glucose (30 mM). (a) IEC-6 cells were cultured for 48 h in the presence of 10 µm Dorsomorphin, 10 µm GSK621 and 10 µm Rapamycin. (b) The number of shrunken and necrotic cells per high power field. (c) CCK-8 assay was performed to assess cell viability of IEC-6 cells under different treatments. Data are presented as the mean ± SD; n = 3; *p < .05; **p < .01 vs. control.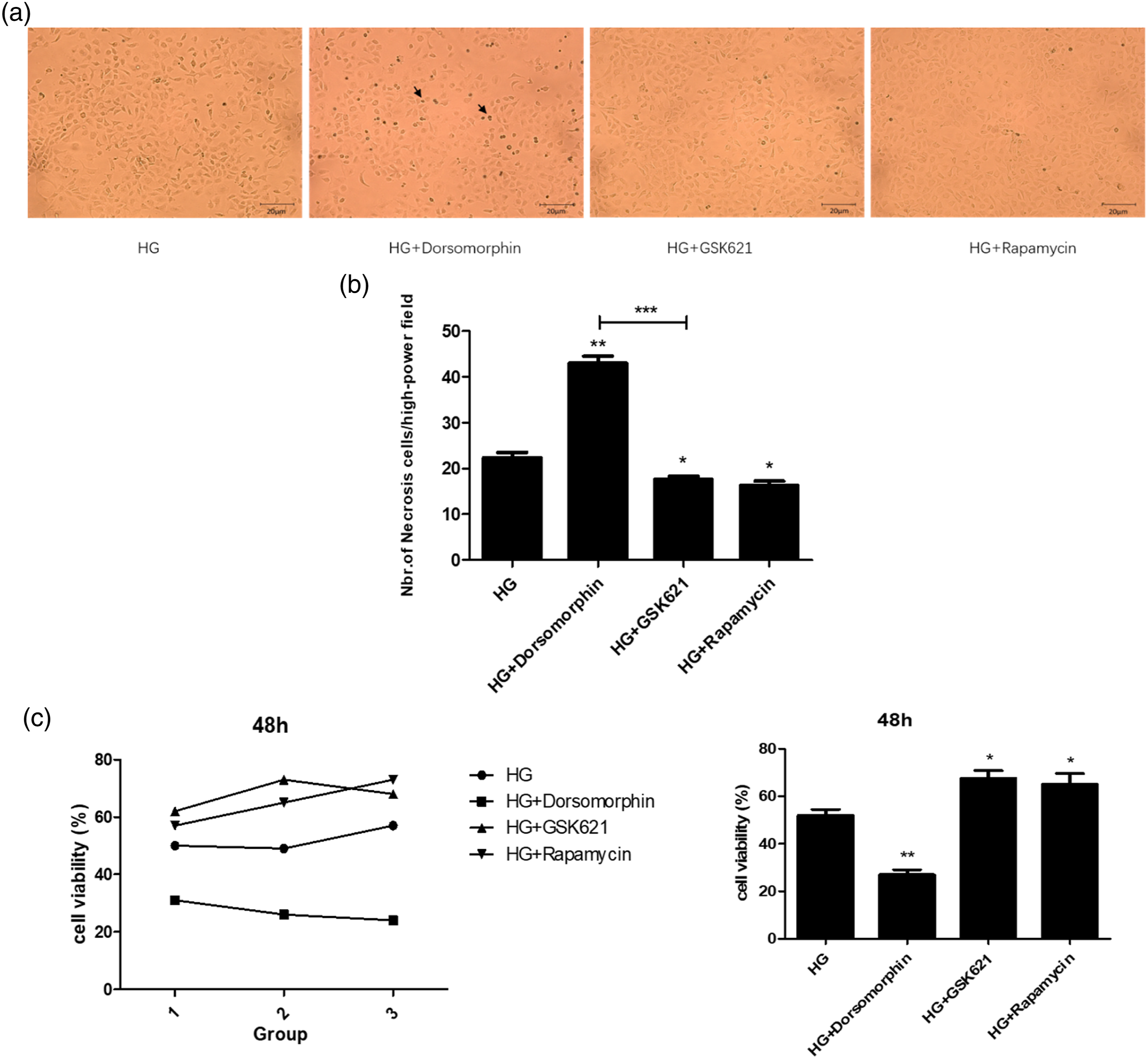
AMPK-mediated autophagy is involved in regulating cytokine secretion
We next investigated the relationship between AMPK pathway and autophagy under high glucose. We used AMPK antagonist Dorsomorphin and agonist GSK621 to analyze the effect on the level of autophagy. Furthermore, we analyzed the expression of LC3II, P62, AMPK, and ULK1 in IEC-6 cells on day 2. Western blot results suggested that exposure to Dorsomorphin reduced the levels of LC3II, phosphorylated AMPK (p-AMPK) and phosphorylated ULK1 (p-ULK1), while P62 was increased compared with the control group (**p < .01). However, GSK621 and rapamycin increased the levels of LC3II, p-AMPK and p-ULK1 compared with the control group (**p < .01), indicating that autophagy is enhanced (Figure 4(a)). Consistently, cellular immunofluorescence showed that the fluorescence intensity of autophagy protein LC3B in the GSK621 group and the Rapamycin group was increased, while that in the Dorsomorphin group was significantly reduced compared with the control group (Figure 4(b)). In addition, ELISA was used to detect the expression of IL-22, INF-γ, NOS2 and TNF-α (Figure 4(c)). The results indicated that the levels of pro-inflammatory factors NOS2 and TNF-α were significantly increased in Dorsomorphin group compared with the control group (*p < .05). The expressions of NOS2 and TNF-α in GSK621 group and Rapamycin group were significantly lower than those in Dorsomorphin group (*p < .05; **p < .01). Furthermore, the anti-inflammatory factors IL-22 and INF-γ in the GSK621 group were higher than those in the Dorsomorphin group (Figure 4(e)). Our results indicate that GSK621 and Rapamycin can up-regulate autophagy and reduce the expression of pro-inflammatory factors, which is conducive to cell survival. AMPK-mediated autophagy modulates the inflammatory cytokine expression in IECs. (a–b) Western blot images and semi-quantitative profiles of LC3II/I, P62, p-AMPK, and p-ULK1 protein levels in IEC-6 cells cultured for 48 h in the presence of 10 µm Dorsomorphin, 10 µm GSK621, and 10µm Rapamycin. (c–d) Immunofluorescence images showing LC3B (green) expression in IEC-6 cells at 48 h of treatment. The histogram shows the average number of single LC3+ autophagosomes from three independent experiments. (e) ELISA analysis of TNF-α, NOS2, IL-22, and INF-γ. All data are presented as the mean ± SD; n = 3; *p < .05; **p < .01; ***p < .001; #p > .05 vs. control.
Discussion
The effects of high glucose on IEC proliferation and cytokine secretion are rarely studied, while the results remain controversial. Previous study reported that there were over proliferation and abnormal differentiation of IECs in the intestinal epithelium of DM. 21 Elinav 22 et al. have indicated that hyperglycemia markedly interfered with homeostatic epithelial integrity, causing retrograde movement of glucose into IECs via GLUT2 and resulting in altered intracellular glucose metabolism. However, the mechanism of intestinal mucosal barrier damage caused by hyperglycemia has not been thoroughly studied, especially the inflammatory signaling mechanism in IECs. Our study found that autophagy was inhibited by high glucose. In addition, high glucose induced the secretion of inflammatory cytokines. Interestingly, AMPK agonists could increase the level of autophagy and down-regulate the secretion of pro-inflammatory factors compared with AMPK antagonists. Therefore, our results demonstrate that AMPK mediated autophagy may regulate levels of inflammatory cytokines and improve cell survival under high glucose.
Our results showed that high glucose had an adverse effect on the survival of IECs in vitro. The results indicated that the high glucose significantly inhibited the growth of IECs and induced more shrinkage and necrosis of cells. Furthermore, the expression levels of cytokines IL-22, INF-γ, NOS2, and TNF-α were significantly increased, which were positively correlated with glucose concentration. In particular, the levels of pro-inflammatory factors NOS2 and TNF-α were significantly increased compared with the control group. It has been found that high glucose induces oxidative stress in cells, and autophagy induction helps remove damaged proteins and organelles. 23 Regulation of cellular stress by autophagy and protection from stress–induced apoptosis is one aspect of how autophagy plays a role in IEC death.24,25 Consistently, this study demonstrated for the first time the effects of high glucose on autophagy and inflammatory cytokines of IECs. High glucose had a negative effect on autophagy, as revealed by the decrease in LC3II expression and the increase in P62 expression. Furthermore, high glucose induced the secretion of pro-inflammatory factors, which in turn led to more cell damage.
The AMPK (AMP-activated protein kinase) signaling pathway is activated in the absence of glucose. AMPK regulates autophagy level in response to energy stress by directly suppressing mTOR activity and phosphorylating ULK1.26,27 Interestingly, it is found that high glucose decreased the autophagy level of osteoclasts, which was demonstrated by decreased expression of Beclin-1 and LC3II and increased expression of P62. 28 Several studies have found that high glucose has an inhibitory effect on autophagy. However, Kobayashi reported that high glucose toxicity was alleviated by inhibiting autophagy in cardiomyocytes, playing a role in protecting cardiomyocytes. 29 Consistently, our study found that high glucose has an inhibitory effect on IEC autophagy. Additionally, we used AMPK antagonist Dorsomorphin and AMPK agonist GSK621 to observe the effect of AMPK signaling pathway on autophagy and IECs survival under high glucose. It has been confirmed that Dorsomorphin down-regulates the expression of p-AMPK and autophagy protein compared with GSK621. Consistently, cellular immunofluorescence also detected low autophagy protein expression. However, GSK621 and Rapamycin up-regulated the level of autophagy. Therefore, our study confirms that AMPK agonists are beneficial to cell survival by up-regulating the level of autophagy and reducing the secretion of inflammatory factors.
It has been reported 30 that the activation of various inflammation-related signaling pathways such as MAPK and NF-κB is related to the expression of inflammatory factors, such as NO, TNF-α, PGE2 and IL-1β. Autophagy is also involved in regulating the secretion of these inflammatory factors. Previous studies31,32 have shown that excessive activation of the inflammasome elevates CASP1 activity, increases IL1B and IL18 production, and finally leads to an increased susceptibility to experimental intestinal inflammation in mice. Interestingly, abnormal activation of the inflammasome is closely related to the loss of autophagy function. 32 Our study also showed that high glucose inhibited autophagy and induced the increase of pro-inflammatory factors. Interestingly, AMPK agonist or autophagy activation significantly down-regulated the secretion of cytokines, among which the levels of cytokines IL-22, NOS2, and TNF-α were significantly lower than those in the AMPK antagonist group. It has been shown that AMPK-mediated autophagy is not only effective in regulating cytokines but also has a cytoprotective function. However, inhibiting AMPK signaling pathway can inhibit autophagy and induce cell apoptosis. In recent years, it has been reported that autophagy is closely related to innate and adaptive immune responses, and is involved in antigen presentation, cytokine secretion, and production of antimicrobial peptides.33,34 Similarly, autophagy plays an important role in intestinal immune function. Under high glucose, IEC dysfunction is manifested by autophagy inhibition and increased expression of inflammatory factors. Chronic intestinal inflammation and intestinal functional disorders may occur in diabetic individuals. However, precious study has demonstrated a role for autophagy in regulating intestinal inflammation and restoring intestinal homeostasis. 35 Intestinal epithelial susceptibility to inflammation is associated with defective autophagy, which leads to breakdown of the intestinal epithelial barrier and cell death.36,37
Therefore, the present study showed that high glucose induced the secretion of inflammatory cytokines and down-regulated autophagy, Furthermore, AMPK agonists were found to be beneficial to cell survival by up-regulating the level of autophagy and reducing the secretion of inflammatory factors. However, there are some limitations in our study. For example, the mechanism underlying the regulation of AMPK pathway on autophagy was not investigated. Animal models will be used in our follow-up research.
Conclusion
High glucose had an adverse effect on cellular homeostasis and function of IECs in vitro. We demonstrated that autophagy was inhibited by high glucose and participated in the regulation of cytokine secretion. In addition, the AMPK/ULK1 pathway was involved in glucose-induced autophagy activity in IECs. The induction of autophagy might be a default mechanism to prevent high glucose-induced IECs injury. Our study provides new insights into the effects of AMPK-mediated autophagy in regulating cell survival under high glucose and may help identify novel therapeutic strategies for the protection of diabetes-related enteritis.
Footnotes
Acknowledgements
This work was supported by Central Hospital Affiliated to Shandong First Medical University.
Author contributions
Kun Ma: experiments and manuscript writing; Jingjing Guo: study conception and design; Yun Li: material preparation and data collection; Xiaolin Dong: data collection and analysis; All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was Supported by the Youth Training Program for High-level Projects of Jinan Central Hospital (Project no.202,007,020) and Shandong Provincial Medical Health Technology Development Project (Project no.2021BJ000018).
Ethical approval
The experiments complied with the Regulations of the Administration of Laboratory Animals of the Ministry of Health of China (Document No.55 of 2001) and were approved by the animal Ethics Committee of Central Hospital Affiliated to Shandong First Medical University, China (JNCH2021-5).