
Abstract
Objective:
Acute respiratory distress syndrome (ARDS) is a severe pulmonary condition characterized by inflammation and lung damage, frequently resulting in poor clinical outcomes. Recent studies suggest that the gut-lung axis, mediated by gut microbiota, is critical in ARDS progression. This study investigates the therapeutic potential of fecal microbiota transplantation (FMT) in an ARDS rat model (n = 6).
Introduction:
The pathogenesis of ARDS involves complex interactions between the lungs and gut, with microbiota playing a key role. Understanding the effects of FMT on lung function and gut microbiota may provide new therapeutic strategies for ARDS management.
Methods:
Sprague-Dawley rats were pre-treated with a broad-spectrum antibiotic cocktail to create a germ-free state and subsequently exposed to intranasal lipopolysaccharide to induce ARDS. The rats then received FMT treatment. Lung samples were analyzed using histopathology and transcriptomics. Fecal samples were analyzed using 16S rRNA sequencing and metabolomics.
Results:
FMT treatment significantly reduced lung injury and improved pulmonary function, as evidenced by increased partial pressure of arterial oxygen (PaO2) and decreased partial pressure of arterial carbon dioxide (PaCO2). FMT also significantly altered in gut microbiota composition by regulating the gut microbiota composition of Akkermansia and Lactobacillus, restoring the abundance of genera such as Muribaculaceae, Clostridia_UCG-014, Prevotella, and Adlercreutzia, while reducing Romboutsia. FMT restored key metabolic pathways involved in lipid metabolism, amino acid biosynthesis, and immune regulation, including the modulation of immune pathways like mTOR signaling. These alterations contribute to reduced lung injury and improved pulmonary function.
Conclusion:
These findings indicate that FMT may exert its beneficial effects in ARDS by modulating the gut microbiota and enhancing metabolic and immune responses. However, given that this study remains in the preclinical stage, further validation in clinical studies is necessary before considering clinical application.
Keywords
Introduction
Acute respiratory distress syndrome (ARDS) is life-threatening condition characterized by acute-onset dyspnea, tachypnea, and hypoxemia, accompanied by bilateral opacities on chest imaging.1,2 The pathophysiology of ARDS involves a severe inflammatory response that leads to alveolar injury, increased alveolar-capillary permeability, and pulmonary edema. 3 ARDS presents clinical challenges due to its diverse etiologies and high mortality rates. Additionally, it has a 30-day readmission rate of 18.4%, further emphasizing the burden of the disease. 4 Current management focuses on supportive care strategies such as lung-protective ventilation, prone positioning, and extracorporeal membrane oxygenation (ECMO) in severe cases.5,6 However, the effectiveness of these interventions remains inconsistent. For instance, recent trials found no significant improvement in ECMO weaning time with prone positioning. Ongoing research into anti-inflammatory therapies has yielded inconsistent findings in clinical trials. 3 Given the complexity and variability in ARDS management, there is an urgent need to develop more effective and personalized therapeutic strategies that target both inflammatory pathways and individual patient profiles.
Recent studies have highlighted the gut-lung axis as a critical factor in ARDS development, particularly its role in gut microbiota-mediated modulation of inflammation and lung injury. 7 The gut-lung axis refers to the complex interplay between the gut microbiota and lung function, regulating metabolic and immune responses. 8 Gut microbiota, composed of bacteria, viruses, and fungi, plays a crucial role in immune regulation and inflammation, thereby contributing to ARDS onset and progression. 9 Additionally, changes in gut microbiota in response to ARDS can modulate immune and cellular functions, highlighting the bidirectional communication that affects disease progression. 10 Dysbiosis, characterized by an imbalance between beneficial and pathogenic gut bacteria, can compromise intestinal barrier integrity and alter immune cell populations, such as regulatory T cells (Tregs) and T helper 17 (Th17) cells, which are critical for lung inflammation regulation. 8 This disruption can trigger systemic inflammation, worsening pulmonary dysfunction and ultimately leading to multiple organ dysfunction. Lipopolysaccharide (LPS)-induced models are widely used in ARDS research. LPS, a component of Gram-negative bacteria, mimics ARDS-associated systemic inflammation and lung injury. LPS has been shown to induce significant inflammation in both the lungs and gut, providing valuable insights into ARDS-related inflammatory pathways.10,11 This model serves as a valuable tool for investigating the complex interactions between gut dysbiosis and lung injury in ARDS. Our previous research has demonstrated that gut microbiota and its metabolites are key regulators of immune responses and cellular functions during ARDS. These findings further reinforce the role of the gut-lung axis in ARDS pathophysiology. 12
Recent studies suggest that therapeutic strategies like FMT may help restore gut-lung axis balance and reduce ARDS complications. 13 FMT has been shown to effectively treat Clostridioides difficile infection in critically ill patients, as seen in a case where it outperformed antibiotics in a patient on ECMO support. 14 Additionally, FMT has demonstrated potential in addressing antibiotic-associated diarrhea in critically ill patients, improving gut microbiota composition and supporting faster recovery. 15 Although FMT may cause mild adverse effects like temporary diarrhea, most cases are transient. 16 A recent study demonstrated that direct stool testing for FMT donor screening effectively eliminates potential pathogen transmission, ensuring the safety of FMT procedures in clinical applications. 17 FMT alleviates acute lung injury model mice by reducing lung cell apoptosis, inhibiting inflammation and oxidative stress, and enhancing lung barrier integrity through tight junction proteins like ZO-1, claudin-1, and occludin. These effects are mediated by the Nrf2/HO-1 and TLR4/NF-κB pathways. 18 A comprehensive understanding of the mechanisms underlying FMT is crucial for the development of targeted therapeutic strategies aimed at improving outcomes in ARDS patients.
To investigate these interactions, advanced techniques such as 16S rDNA amplicon sequencing are employed to analyze gut microbiota composition and diversity. 19 In addition, metabolomics reveals biochemical processes and metabolic changes influenced by gut microbiota. 20 Transcriptomics enables the analysis of gene expression changes in response to microbiota alterations, further elucidating their role in ARDS pathophysiology. 21 In recent years, multi-omics technologies have advanced ARDS research significantly. However, current research primarily focuses on the host response mechanisms of ARDS22,23 and the identification of prognostic biomarkers,24,25 while studies on gut microbiota and its metabolic regulation in ARDS remain limited. In our previous research on ARDS, we found that FMT significantly improved LPS-induced lung injury in rats by reducing the lung wet/dry ratio, inflammatory markers, and histopathological damage, while increasing arterial oxygenation. 26 Additionally, FMT regulated key inflammatory signaling pathways, including TGF-β1/Smads/ERK and PI3K/AKT/NF-κB, to suppress inflammation and downregulate ICAM-1 expression, ultimately promoting recovery from LPS-induced acute lung injury. 27 This study integrates multi-omics approaches to explore the role of gut microbiota and its metabolites in ARDS. In contrast to previous studies, this study not only examines the impact of FMT on gut microbiota composition but also explores its effects on immune responses and metabolic pathways. By modulating the gut-lung axis, this study aims to uncover how FMT improves the pathophysiology of ARDS, providing new insights into its potential mechanisms.
Materials and methods
Animal housing and maintenance
Male specific pathogen-free (SPF) Sprague-Dawley rats (6–8 weeks, 180–250 g) were obtained from the Experimental Animal Center, Southwest Medical University. A total of 18 rats were randomly divided into three groups (n = 6 per group). The animals were acclimatized for 1 week under a 12 h light/dark cycle, with food and water provided ad libitum. The temperature was maintained at 23–25°C. All experimental procedures complied with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and adhered to the ARRIVE guidelines. This study was approved by the animal ethics committee of Liuzhou People’s Hospital (Ethics Approval Number: KY-2024-077).
Animal model and treatment
The rats in the LPS and FMT groups received a broad-spectrum antibiotic mixture via oral gavage for seven consecutive days. The antibiotic mixture contained neomycin (100 mg/kg, Sigma, N6386), ampicillin (100 mg/kg, Sigma, A0166), vancomycin (50 mg/kg, Sigma, 861987), and metronidazole (100 mg/kg, Sigma, M3761). 28 Following the antibiotic treatment, the rats were intranasally administered lipopolysaccharide (LPS, Sigma, L2880, 10 µg LPS dissolved in 50 µL PBS, 10 mg/kg) to induce the ARDS model. 29 All rats were anesthetized with 1% pentobarbital (intraperitoneal injection, 6 mL/kg) before LPS administration. The LPS solution was delivered with a 1-mL pipette inserted 0.5–1 cm into one nostril, while the other nostril was temporarily occluded for 1 min to ensure even pulmonary distribution. The control group received an equivalent volume of saline intranasally.
FMT procedure
Freshly collected feces from healthy donor rats were diluted in PBS (Gibco, 14190-144) at a 1:5 (g/mL) ratio. To ensure the preservation of anaerobic bacteria, L-cysteine (Sigma-Aldrich, C7352) was added to the suspension as a reducing agent. The fecal suspension was thoroughly mixed to achieve homogeneity, followed by filtering through a 0.8–1 mm mesh to remove particulate matter. To further purify the preparation, centrifugation was performed at 800 × g for 3 min to remove undissolved solids, as low-speed centrifugation helps clear away particulate matter without losing a significant number of bacteria. The suspension was stored at 4°C and used within 6 h to maintain bacterial viability. After the establishment of the ARDS model for 24 h, rats were designated for FMT received the fecal suspension via oral gavage (10 mL/kg) twice daily for 7 days, as previously validated in our studies26,27; other groups received an equivalent volume of sterile saline. Fresh feces were collected from the metabolic cages after the final FMT administration and immediately frozen in pre-chilled microcentrifuge tubes in liquid nitrogen, stored at −80°C for subsequent analysis. Rats were euthanized 24 h after the final FMT using sodium pentobarbital (40 mg/kg, intraperitoneal injection; Sigma-Aldrich, P3761) for humane euthanasia and sample collection. Following the intraperitoneal injection of sodium pentobarbital, the rats lost consciousness and all reflexes within a few minutes. If necessary, cervical dislocation was performed to confirm death. 30
Pulmonary function testing and lung wet/dry weight ratio
Arterial blood samples were collected to determine partial pressure of arterial carbon dioxide (PaCO2) levels with a blood gas analyzer, assessing respiratory function and CO2 elimination efficiency. Pulmonary function differences, particularly in the context of the ARDS model and FMT treatment, were assessed by comparing PaCO2 levels among groups. At the end of the study, arterial blood was sampled to measure partial pressure of arterial oxygen (PaO2) and calculate the Oxygenation Index (OI) as OI = PaO2/FiO2. After euthanasia, the left lung was immediately weighed to obtain its wet weight, then oven-dried at 70°C for 72 h to obtain dry weight. The wet/dry weight ratio was calculated to evaluate the severity of pulmonary edema.
Histopathological analysis
Freshly euthanized rats underwent thoracotomy to expose the lungs. Lung tissues were excised, snap-frozen in liquid nitrogen, and stored at −80°C. Samples were thawed, fixed in 4% paraformaldehyde (Sigma-Aldrich, P6148) for 24 h, then dehydrated, embedded in paraffin, and sectioned into 5-μm slices (Leica RM2235). Sections were deparaffinized, rehydrated, and stained with Hematoxylin (Sigma-Aldrich, H3136) for 5 min and Eosin (Sigma-Aldrich, E4009) for 2 min. Stained sections were examined under a light microscope (Olympus BX43) and images captured at ×400 magnification. Lung injury was scored on a scale of 0–8: 0 (no injury), 2 (mild, ⩽25% of the field), 4 (moderate, ⩽50% of the field), 6 (severe, ⩽75% of the field), 8 (extremely severe, diffuse injury). 31 Histological scoring was conducted by two blinded evaluators, both of whom were associate chief pathologists. In cases of discrepancy, a third blinded pathologist was consulted to reach a final consensus.
Masson’s trichrome staining
The sections were stained using a Masson’s trichrome staining kit (Sigma-Aldrich, HT15). The protocol involved staining with Weigert’s iron hematoxylin (Sigma-Aldrich, HT1079) for 10 min, followed by staining with Ponceau S-fuchsin (Sigma-Aldrich, P3504) for 5 min, and aniline blue (Sigma-Aldrich, 415049) for 5 min. After staining, the sections were dehydrated through a graded ethanol series, cleared in xylene, and mounted with Permount mounting medium (Fisher Scientific, SP15-100). Stained sections were examined under a light microscope (Olympus BX43), and images were captured at ×400 magnification to evaluate the degree of fibrosis in the lung tissues.
16S rRNA sequencing and analysis
Total DNA was extracted from rat fecal samples using the QIAamp Fast DNA Stool Mini Kit (Qiagen, 51604). The V3-V4 regions of the 16S rDNA were amplified using specific primers 341F (5'-CCTAYGGGRBGCASCAG-3') and 806R (5'-GGATACNNGGGTATCTAAT-3'). The PCR products were purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA), quantified using QuantiFluor™-ST (Promega), and checked for quality via 2% agarose gel electrophoresis. The purified PCR products were used to construct sequencing libraries with the TruSeq® DNA PCR-Free Library Prep Kit (Illumina, USA). Sequencing was performed on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) at Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). Raw sequencing reads were quality-filtered using QIIME software. Operational taxonomic unit (OTU) were clustered at a 97% similarity threshold using UPARSE and classified with the RDP classifier against the SILVA database. Further analysis of gut microbial diversity was conducted using QIIME and R.
Ultra-high performance liquid chromatography-mass spectrometry
Metabolites were extracted from 200 mg of rat fecal samples using the QIAamp Fast DNA Stool Mini Kit (Qiagen, 51604). Metabolites were extracted by adding 1 mL of pre-chilled 50% methanol solution (Sigma-Aldrich, M1775), followed by sonication and centrifugation. An extraction solution (400 µL, methanol = 4:1, containing 0.02 mg/mL L-2-chlorophenylalanine as internal standard) was added. The samples were then homogenized using a Wonbio-96c High Throughput Tissue Homogenizer for 6 min at −10°C and 50 Hz, followed by low-temperature sonication for 30 min at 5°C, 40 kHz. The samples were then left to stand at −20°C for 30 min before centrifugation at 13,000 × g for 15 min at 4°C. The supernatant was collected for analysis. Chromatographic separation was performed on an HSS T3 column (100 mm × 2.1 mm, 1.8 µm, Waters, Milford, MA, USA) using commercially available mobile phase A (95% water + 5% acetonitrile, containing 0.1% formic acid) and mobile phase B (47.5% acetonitrile + 47.5% isopropanol + 5% water, containing 0.1% formic acid), at a flow rate of 0.40 mL/min and a column temperature of 40°C. Mass spectrometry detection was conducted using a UHPLC-Q Exactive system (Thermo Fisher Scientific) under both positive and negative ion modes with a mass scan range of m/z 70–1050. Data were processed using Progenesis QI software, with metabolite identification performed against the HMDB and Metlin databases. Quality control (QC) samples were prepared by mixing equal volumes of all samples to assess analytical stability.
Differential metabolite analysis
Preprocessed data matrices were analyzed using the ropls package (Version 1.6.2) in R for principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA). Student’s t-test and fold change (FC) analysis were conducted for differential metabolite selection. Metabolites with VIP > 1 (from the PLS-DA model) and p < 0.05 (Student’s t-test) were considered significant. Volcano plots were generated based on log2FC and -log10 p-values, while heatmaps were used for hierarchical clustering visualization. Pearson correlation analysis was performed to assess relationships among differential metabolites. Predicted microbial functional analysis was conducted using PICRUSt2, and pathway abundance differences were calculated using the t-test with Benjamini-Hochberg correction for multiple comparisons. Metabolic pathway annotation was performed using the KEGG database (https://www.kegg.jp/kegg/pathway.html), and pathway enrichment analysis was conducted using the scipy.stats package in Python, with statistical significance assessed by Fisher’s exact test.
Transcriptomic analysis
Total RNA was extracted from lung tissue samples using TRIzol reagent (Invitrogen, 15596026) according to the manufacturer’s instructions. RNA purity and concentration were measured using a NanoDrop 2000 (Thermo Scientific), and integrity was assessed with an Agilent 5300 Bioanalyzer (Agilent Technologies). Library construction was performed using the Illumina® Stranded mRNA Prep, Ligation Kit (Illumina, USA), and sequencing was carried out on the NovaSeq X Plus system (Illumina, USA). Sequencing data underwent quality control using FastQC (v0.11.8), alignment with HISAT2, quantification with featureCounts, and differential expression analysis with DESeq2. Pathway enrichment analysis was performed using the Scipy.Stats package in Python and the KEGG database (https://www.kegg.jp/kegg/pathway.html), with the most relevant biological pathways identified via Fisher’s exact test. Gene Set Enrichment Analysis (GSEA) was performed to identify significantly enriched pathways associated with differentially expressed genes. The analysis was conducted using the GSEA software (v4.3.2, Broad Institute, USA) with gene sets obtained from the KEGG database. Genes were ranked based on their log2FC, and enrichment scores were calculated using 1000 permutations. Pathways with a nominal p-value < 0.05 and false discovery rate (FDR) <0.25 were considered significantly enriched. The results were visualized using the ggplot2 package in R.
Statistical analysis
All data are presented as mean ± standard deviation (SD). Statistical analyses were conducted using GraphPad Prism 9.0 (GraphPad Software, San Diego, CA, USA). Comparisons between two groups were performed using a two-tailed independent samples t-test, while comparisons among multiple groups were conducted using one-way ANOVA, with significance set at p < 0.05. For differential expression analysis, statistical testing was performed using DESeq2 (v1.24.0), with significant genes selected based on an adjusted p-value (p adj) <0.05. For comparisons of microbial community diversity, the Wilcoxon signed-rank test was used, and the ANOSIM test was applied to assess inter-group beta diversity differences. All statistical plots were generated using R (version 4.2.1) and the ggplot2 package (version 3.3.6). Additionally, correlations between microbes and metabolites were analyzed using Spearman correlation analysis, and relatedness networks were visualized using Cytoscape software, with significance thresholds for correlation analyses set at a p-value < 0.05 and an absolute correlation coefficient ⩾ 0.7.
Results
FMT reduces lung injury and improves pulmonary function in ARDS rats
Figure 1(a) and (b) illustrate the experimental design. H&E staining showed that the LPS group exhibited clear signs of lung injury, including inflammatory cell infiltration and structural damage, while these pathological features were visibly reduced in the FMT group (Figure 1(c)). Masson’s trichrome staining indicated increased fibrosis in the LPS group, with less fibrotic tissue observed in the FMT group (Figure 1(c)). Lung injury scores supported these observations, showing significantly higher scores in the LPS group compared to the control group and significantly reduced scores in the FMT group (Figure 1(d)). The wet/dry weight ratio of the left lung was significantly higher in the LPS group than in the control group, and significantly lower in the FMT group (Figure 1(e)). Regarding pulmonary function indices, PaO2 was significantly lower in the LPS group than in the control group but improved following FMT treatment (Figure 1(f)). Similarly, the partial pressure of PaCO2 was significantly higher in the LPS group compared to the control group, with a reduction observed in the FMT group (Figure 1(g)).

FMT reduces lung injury and improves pulmonary function in ARDS rats. (a–b) Experimental workflow diagram illustrating the ARDS model and FMT method; (c) Histopathological results of lung tissue stained with H&E and Masson’s trichrome. Scale bars = 100 µm; (d) Lung injury scores; (e) Wet/dry weight ratio of the left lung; (f) Partial pressure of arterial oxygen (PaO2). (g) Partial pressure of carbon dioxide (PaCO2). Data are represented as mean ± SD (n = 6). Compared with LPS group, ***p < 0.001.
FMT alters gut microbial diversity in ARDS rats
Microbial diversity analysis using primers 338F-806R was performed on 18 samples from control, ARDS, and FMT groups, followed by sequencing. Clustering at 97% similarity revealed OTU counts of 4426 for the control group with an average of 737 OTUs per sample, 3949 OTUs for the ARDS group averaging 658 OTUs per sample, and 3777 OTUs for the FMT group averaging 630 OTUs per sample. Analysis of α-diversity among the groups using Sobs and Chao indices showed no significant differences in community species richness. However Shannon and Simpson indices indicated no significant changes in α-diversity between the control and ARDS groups but significant differences between the control and FMT groups as well as between the ARDS and FMT groups, suggesting a substantial impact of FMT on the microbial community diversity (Figure 2(a)). β-diversity analysis based on Bray-Curtis distance at OTU and phylum levels using Principal Coordinate Analysis (PCoA) demonstrated distinct clustering of the groups, with the FMT group samples showing slight dispersion (Figure 2(b)). PCoA based on Bray-Curtis distance showed significant differences in β-diversity at the phylum level among the three groups (PERMANOVA, p = 0.009; Figure 2(c)), and pairwise comparisons between groups also exhibited significant differences (ANOSIM; Figure 2(d)).

FMT alters gut microbial diversity in ARDS rats. (a) Alpha diversity analysis at the OTU level (Sobs and Chao indices for species richness, Shannon and Simpson indices for diversity), analyzed using the Wilcoxon signed-rank test; (b) Beta diversity analysis (PCoA at OTU and phylum levels) showing group clustering; (c) Overall beta diversity comparison among the three groups, analyzed using PERMANOVA; (d) Pairwise comparisons of beta diversity between the groups using ANOSIM.
FMT modifies gut microbial composition in ARDS rats
Microbial annotation and comparative analysis were conducted on samples from the three groups. At the OTU level, a total of 854 OTUs were identified: the control group had the most unique OTUs (232), followed by the FMT group (205), while the ARDS group had the fewest (177; Figure 3(a)). In the ARDS group, the fecal microbiome was dominated by OTU419, OTU724, OTU425, and OTU373, whereas OTU391, OTU450, and OTU603 were predominant in the FMT group (Figure 3(b)). At the phylum level, the major taxa included Firmicutes, Bacteroidota, and Actinobacteriota. Notably, the proportion of Bacteroidota was lower in the ARDS group than in other groups, while Firmicutes were more abundant (Figure 3(c)). At the genus level, Lactobacillus was the most prevalent in the ARDS group, followed by the control group, with a reduced proportion post-FMT. Moreover, the proportion of Romboutsia was significantly higher in the ARDS group, possibly counteracting the growth of Clostridia_UCG-014 and Muribaculaceae due to its interaction with Lactobacillus (Figure 3(d)).

FMT modifies gut microbial composition in ARDS rats. (a) Venn diagram showing the number of shared and unique Operational Taxonomic Units (OTU) among the ARDS, Control, and FMT groups. The central region represents OTU common to all three groups, while the peripheral regions indicate OTU unique to each group; (b) Heatmap of community composition at the OTU level, displaying the relative abundance of OTU across different groups. Each row represents a specific OTU, and hierarchical clustering is applied to group similar OTU. The color gradient from blue (low abundance) to red (high abundance) indicates changes in microbial composition; (c) Phylum-level heatmap, showing the distribution of major bacterial phyla in each group. The color gradient corresponds to the relative abundance of each phylum, with red indicating higher abundance and blue indicating lower abundance; (d) Genus-level heatmap, illustrating the relative abundance of gut microbiota at the genus level. Key genera associated with microbial community changes across groups are labeled. The hierarchical clustering on the left organizes bacterial genera based on their similarity in distribution across groups.
FMT modulates gut microbial phenotypes and functional pathways in ARDS rats
Phenotypic predictions by BugBase indicated predominant phenotypes including Gram-positive, Gram-negative, biofilm-forming, pathogenic, mobile element-containing, oxygen-utilizing, and oxidative stress-tolerant categories. Phenotypic analysis revealed that microbial samples from all three groups were primarily associated with stress tolerance and Gram-positive phenotypes. Notably, ARDS-associated samples contained fewer potentially pathogenic phenotypes (Figure 4(a)). Differential abundance analysis among the groups showed that genera such as Romboutsia, Turicibacter, and Akkermansia were significantly more abundant in the ARDS group compared to the control group. Conversely, Prevotella and Treponema were less abundant (Figure 4(b)). Comparisons between the FMT and control groups revealed significantly higher abundance of Akkermansia, Adlercreutzia, Staphylococcus, and Jeotgalicoccus in the FMT group. Furthermore, Ruminococcaceae and Butyrivibrio showed significantly higher abundances in the control group’s feces, indicating significant changes in the butyrate metabolic pathways post-FMT (Figure 4(c)). Lactobacillus, Corynebacterium, and Prevotella levels were regulated in the FMT group relative to the ARDS group, suggesting their crucial role in FMT treatment of ARDS (Figure 4(d)). Functional pathway predictions using PICRUSt2 highlighted significant intergroup differences, particularly in metabolic pathways, biosynthesis of secondary metabolites, and microbial metabolism in diverse environments. Significant reductions were noted in fatty acid biosynthesis, the tricarboxylic acid (TCA) cycle, biosynthesis of pantothenic acid and coenzyme A, and thiamine metabolism in ARDS, profoundly impacting KEGG pathways (Figure 4(e)). After FMT treatment, compared to the ARDS group, the abundance of Romboutsia in the FMT group significantly decreased (p < 0.01, Figure 4(f)), while the abundances of Muribaculaceae, Clostridia_UCG-014, Prevotella, and Adlercreutzia were restored to normal levels (Figure 4(g)–(j)). These results, with statistically significant differences indicated by Kruskal-Wallis H tests, suggest that FMT may improve gut microbial function in ARDS by modulating these key genera.

FMT modulates gut microbial phenotypes and functional pathways in ARDS rats. (a) Microbial phenotype prediction, showing the relative abundance of predicted phenotypic categories across groups; (b–d) Differential analysis of microbial genera using the Wilcoxon rank-sum test with FDR correction: (b) ARDS versus Control, (c) FMT versus Control, (d) FMT versus ARDS. Bars indicate the proportion of genera in each group, with confidence intervals representing statistical differences; (e) Functional heatmap based on PICRUSt2 predictions, illustrating differences in bacterial metabolic pathways among groups. The color gradient indicates pathway abundance (red: high, blue: low); (f–j) Kruskal-Wallis H test for genus-level differences, highlighting significant variations in Romboutsia, Muribaculaceae, Clostridia_UCG-014, Prevotella, and Adlercreutzia across groups. *p < 0.5, **p < 0.01, ***p < 0.001.
FMT modulates metabolic pathways in ARDS rats revealed by metabolomics profiling of fecal samples
LC-MS metabolomics profiling was performed on rat fecal samples from control, ARDS, and FMT groups. In positive ion mode (POS), 1251 metabolites were identified, while in negative ion mode (NEG), 935 metabolites were identified. PCA and PLS-DA indicated distinct clustering of the groups, demonstrating separation in metabolic profiles (Figure 5(a)). The 95% confidence ellipses further revealed clear metabolic differences between the groups, with FMT treatment significantly altering the metabolic profile of ARDS rats, making it more similar to that of the control group. Differential metabolite analysis revealed 421 unique metabolites in the FMT group compared to the control group and 423 unique metabolites compared to the ARDS group (Figure 5(b) and (c)). The ARDS group exhibited 597 differential metabolites compared to the control group, predominantly showing a downward trend, suggesting that both ARDS and FMT induced significant changes in healthy rat metabolites (Figure 5(d)). Intersection analysis highlighted 20 common differential metabolites across the three groups, with a significant number of unique differential metabolites in each group (Figure 5(e)). Further analysis of differential metabolite KEGG pathways showed significant differences among the groups in amino acid metabolism, carbohydrate metabolism, and lipid metabolism (Figure 5(f)–(h)). The ARDS group showed an increase in metabolites involved in protein digestion and absorption, arginine and proline metabolism, and a decrease in purine metabolism, phenylalanine, tyrosine, and tryptophan biosynthesis compared to the control group (Figure 6(f)). Post-FMT treatment, a notable recovery was observed in lipid metabolism, glycolysis, and gluconeogenesis pathways (Figure 6(g)). Metabolite clustering heatmaps further illustrated that the FMT group’s metabolic profile closely resembled that of the control group, suggesting partial restoration of metabolic disturbances caused by ARDS (Figure 5(i)).

FMT modulates metabolic pathways in ARDS rats revealed by metabolomics profiling of fecal samples. (a) PCA and PLS-DA analysis in positive and negative ion modes, showing metabolic differences and group separation. Confidence ellipses represent the 95% confidence intervals of the data points, visually indicating the variability within each group and helping assess the statistical significance of clustering and group separation; (b) Volcano plot of differential metabolites comparing ARDS with control groups; (c) Volcano plot of differential metabolites comparing FMT with ARDS groups; (d) Volcano plot of differential metabolites comparing FMT with control groups; (e) Venn diagram showing common metabolites among the three groups; (f–h) KEGG pathway enrichment analysis for differential metabolites among the groups; (i) Heatmap of metabolite clustering illustrating group similarities and differences.
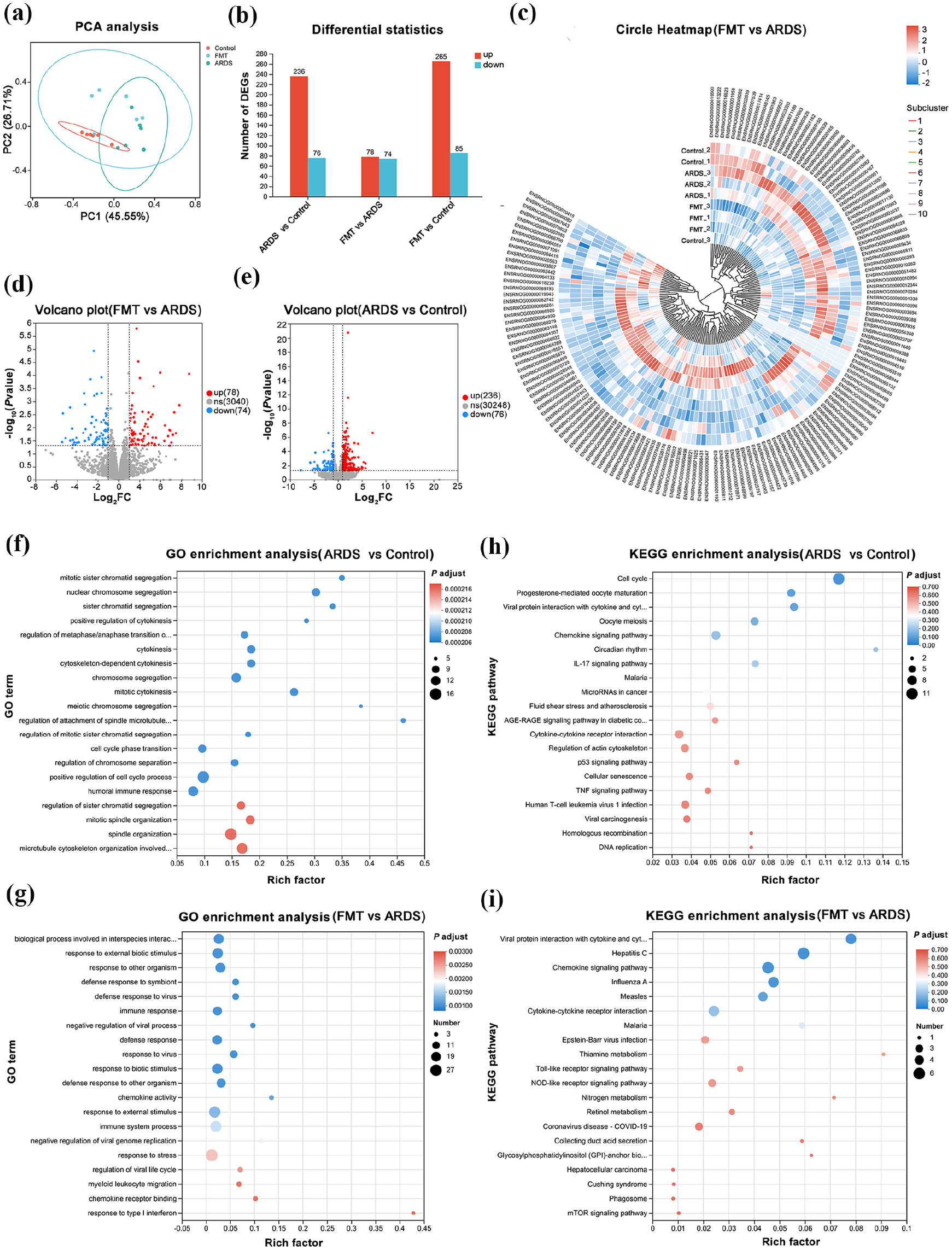
Differential gene expression and enrichment analysis between groups in lung tissue. (a) PCA analysis showing global gene expression patterns among the samples. The 95% confidence ellipses are shown around each group, representing the variability within each group and providing a visual indication of the statistical significance of the clustering; (b) Stacked bar chart depicting the number of differentially expressed genes; (c) Heatmap of clustered differential gene expression between the FMT and ARDS groups; (d) Volcano plot of differential gene expression between the FMT and ARDS groups; (e) Volcano plot of differential gene expression between the ARDS and control groups; (f) GO enrichment analysis of gene functions comparing ARDS and control groups; (g) GO pathway analysis of differential genes between FMT and ARDS groups; (h) KEGG enrichment analysis of gene functions comparing ARDS with control groups; (i) KEGG pathway analysis of differential genes between FMT and ARDS groups.
FMT regulates gene expression and signaling pathways in ARDS rats
Transcriptomic sequencing was conducted on nine lung tissue samples from each group, yielding an average of 48,249,809 raw reads per sample, with an average data output of 7.3 GB per sample. After data filtering, an average of 47,852,590 high-quality reads were obtained per sample. PCA revealed significant separation in global gene expression among the groups, indicating distinct gene expression patterns (Figure 6(a)). The 95% confidence ellipses highlighted the degree of group separation, with FMT treatment making the gene expression profile of ARDS rats more similar to that of the control group, suggesting a potential restorative effect of FMT on the gene expression profile in ARDS. Differential gene expression analysis showed a large number of significantly upregulated and downregulated genes in the comparisons of FMT versus ARDS and ARDS versus control groups (Figure 6(b)). Cluster analysis of selected gene sets among the samples showed significant differences in gene expression patterns between the ARDS and control groups and between the FMT and ARDS groups. The FMT group’s gene expression tended to align more closely with the control group, suggesting that FMT may partially restore gene expression patterns altered by ARDS (Figure 6(c)). Differential gene analysis identified 350 differential genes between the ARDS and control groups, and 152 between the FMT and ARDS groups (Figure 6(d) and (e)).
GO enrichment analysis revealed significant enrichment in biological processes related to chromosome segregation, mitotic cytokinesis, spindle organization, and cell cycle regulation in the ARDS group compared to the control group (Figure 6(f)). These findings suggest potential dysregulation in cell division and cytoskeletal organization in ARDS. KEGG pathway enrichment analysis indicated significant enrichment in pathways associated with cytokine-cytokine receptor interaction, IL-17 signaling, DNA replication, homologous recombination and cell cycle, suggesting the involvement of inflammatory responses, immune signaling, genomic instability, and dysregulated cell proliferation in ARDS pathogenesis (Figure 6(h)). Additionally, GO enrichment analysis revealed significant enrichment in biological processes related to immune response, antiviral defense, regulation of inflammation, and chemokine activity in the FMT group compared to the ARDS group (Figure 6(g)). These processes suggest that FMT treatment modulates host immune responses and viral defense mechanisms. KEGG pathway enrichment analysis indicated significant enrichment in pathways such as cytokine-cytokine receptor interaction, Toll-like receptor signaling, NOD-like receptor signaling, mTOR signaling and viral infection-related pathways (Figure 6(i)). These findings suggest that FMT treatment may regulate gene expression abnormalities induced by ARDS, involving multiple key immune and inflammatory signaling pathways.
GSEA analysis further validated part of the KEGG enrichment findings. In the comparison between the Control and ARDS groups, the DNA replication (NES = −1.600, p = 0.019, Figure 7(a)), Homologous recombination (NES = −1.523, p = 0.035, Figure 7(b)), and Cell cycle (NES = −1.504, p = 0.041, Figure 7(c)) pathways were significantly downregulated, suggesting that ARDS may be associated with the suppression of cell proliferation and genomic stability-related processes. In contrast, in the comparison between the ARDS and FMT groups, the mTOR signaling pathway (NES = 1.386, p = 0.017, Figure 7(d)), Coronavirus disease - COVID-19 pathway (NES = 1.281, p = 0.036, Figure 7(e)), and Viral protein interaction with cytokine and cytokine receptor pathway (NES = 1.365, p = 0.037, Figure 7(f)) were significantly upregulated. These findings suggest that FMT may influence ARDS progression by modulating immune and inflammatory signaling pathways.
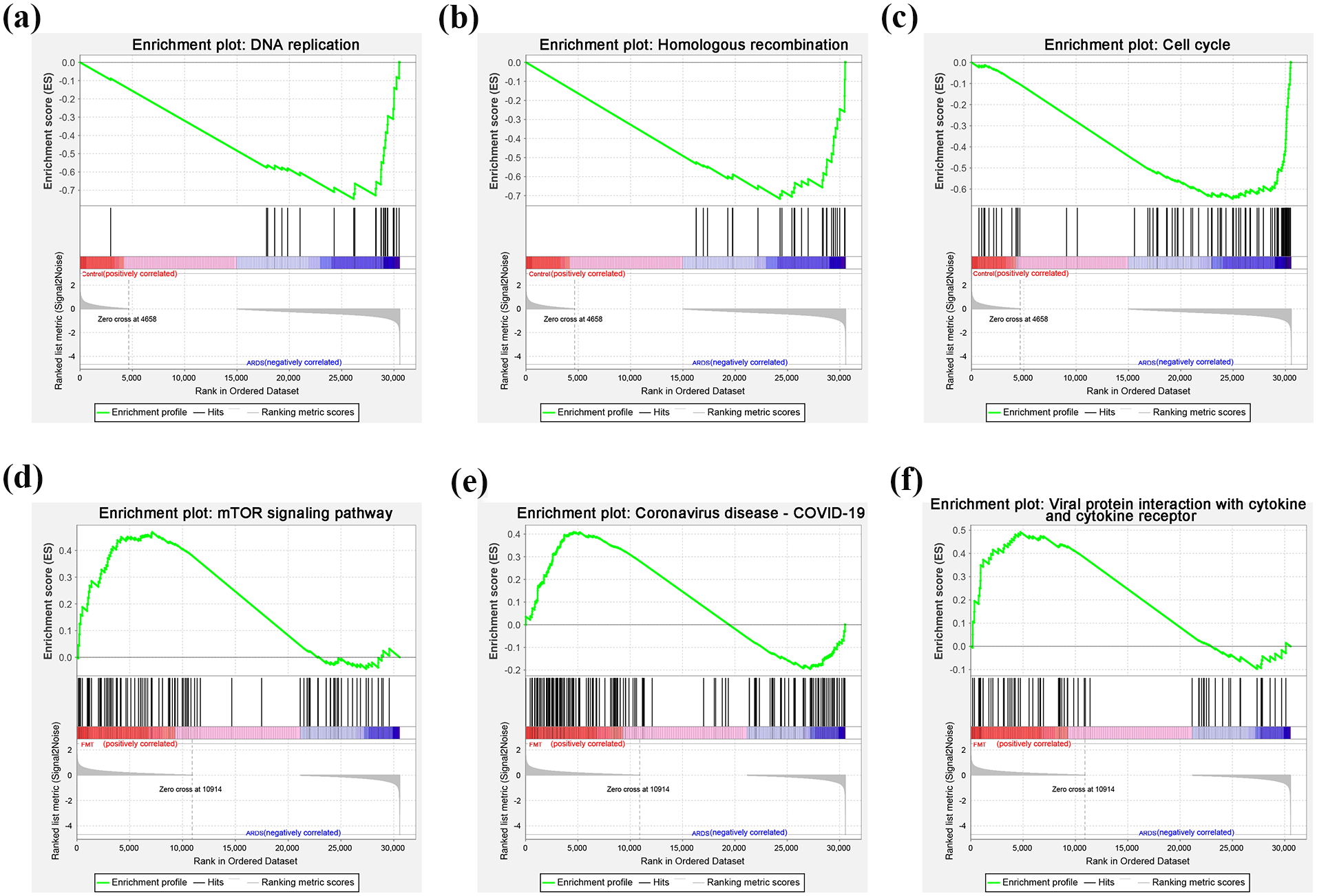
GSEA analysis further validated part of the KEGG enrichment findings. (a) DNA replication; (b) Homologous recombination; (c) Cell cycle; (d) mTOR signaling pathway; (e) Coronavirus disease - COVID-19; (f) Viral protein interaction with cytokine and cytokine receptor. Each enrichment plot illustrates the enrichment score (ES) profile (green line), ranked list metric (gray line), and the distribution of gene hits within the ranked gene set.
Integrated analysis reveals correlation between the microbiome and metabolome in the ARDS rats
Procrustes analysis and O2PLS highlighted the overall congruence and principal component distribution differences between the microbiome and metabolome datasets. Procrustes analysis showed that the microbial and metabolic datasets exhibited a high degree of similarity across the three experimental groups, with an M2 value of 0.817, and a Monte Carlo simulation p-value of 0.158, indicating no significant differences between these datasets (Figure 8(a)). O2PLS analysis further supported these findings, displaying a degree of separation among the groups, particularly between the control and ARDS groups on PCA (Figure 8(b)). To better understand the relationship between gut microbiota and pulmonary metabolites, we conducted a correlation analysis at the genus level between key differential metabolites and microbial taxa. The correlation heatmap illustrated the strength of these relationships, with Lactobacillus, Romboutsia, and others displaying significant positive or negative correlations with specific metabolites, providing molecular evidence of interactions between microbiota and host metabolism (Figure 8(c)).
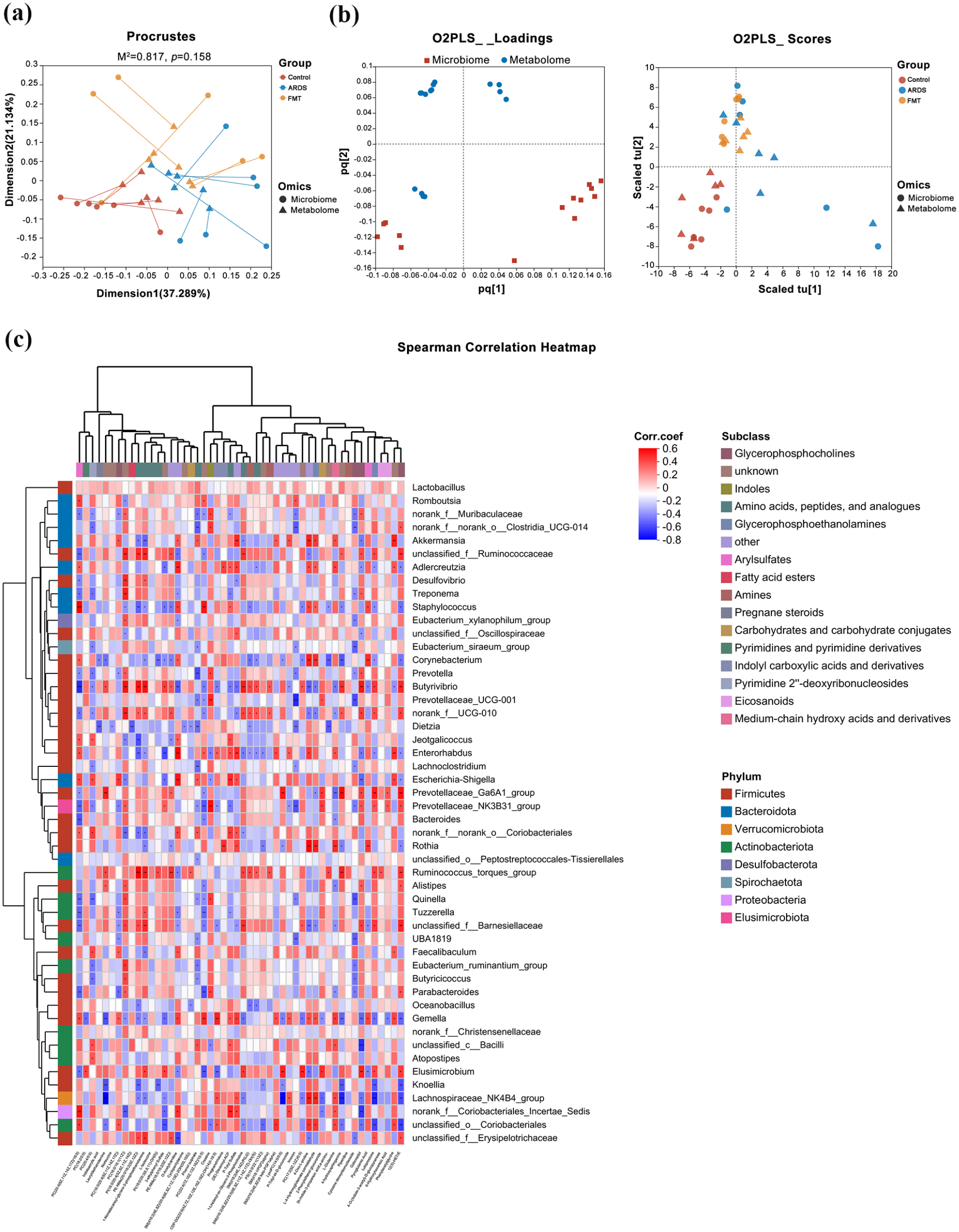
Correlation between the metabolites and microbiota. (a) Procrustes analysis showing shape similarity between microbial and metabolic datasets; (b) O2PLS analysis demonstrating group separation and alignment of principal components; (c) Correlation heatmap displaying relationships between the top 50 metabolites and key microbial genera.
Further analysis of key bacterial genera and critical metabolites involved aggregating differentially abundant taxa across the three microbiome groups, excluding unidentified taxa, yielding 24 distinct genera. These were correlated with key differential metabolites identified across the comparative groups, defining these metabolites as critical change agents. Cytoscape was used to create a network diagram illustrating metabolite-microbe interactions with p < 0.05 and absolute correlation coefficients ⩾ 0.7 (Figure 9(a)). Red lines indicate positive correlations, while blue lines indicate negative correlations; deeper colors signify stronger associations. The node size represents the number of connections, with Desulfovibrio showing the highest number of associations, followed by Escherichia-Shigella. Lactucin was the metabolite with the highest number of associations. Akkermansia had the fewest associations but was positively correlated with the metabolites Asn (asparagine), Phe (phenylalanine). A Sankey diagram illustrated these relationships between bacterial genera and metabolites (Figure 9(b)).

Network and flow associations between differential microbial taxa and metabolites. (a) Network diagram of differential microbial taxa and metabolites, indicating positive (red) and negative (blue) correlations; (b) Sankey diagram illustrating associations between bacterial genera and metabolites, with line width indicating strength of correlation.
Integrated analysis of transcriptomics and metabolomics in ARDS rats
Procrustes analysis and O2PLS analysis demonstrated that, although the FMT and ARDS groups exhibited a high degree of overall shape similarity in the transcriptomic and metabolomic datasets, they showed distinct separation in the PCA, indicating that FMT had a significant impact on metabolism and gene expression in ARDS (Figure 10(a) and (b)). Correlation heatmaps revealed significant correlations between specific metabolites and gene expression between the FMT and ARDS groups, with particularly pronounced interactions observed in lipid and peptide metabolites, suggesting that FMT modulates biomolecular changes induced by ARDS (Figure 10(c)). Moreover, compared to the control group, the FMT group exhibited distinct differences in peptide and lipid metabolites, particularly in pathways related to amino acid metabolism and central carbon metabolism (Figure 10(e)). The KEGG results from the integrated analysis of transcriptomics and metabolomics revealed that the control and ARDS groups exhibited significant enrichment in metabolism-related and cell process-related pathways (p < 0.01 and p < 0.05, Figure 10(d)), while the ARDS and FMT groups showed significant enrichment in immune regulation and inflammation-related pathways (p < 0.01 and p < 0.05, Figure 10(f)).

Integrated analysis of transcriptomics and metabolomics in ARDS rats. (a) Procrustes analysis showing shape similarity between transcriptomic and metabolomic datasets; (b) O2PLS analysis demonstrating group separation and alignment of principal components. Ltr: ARDS group (in lung tissue), Lfmt: ARDS group treated with FMT (in lung tissue); (c) Correlation heatmap comparing FMT and ARDS groups; (d) KEGG pathway enrichment analysis comparing FMT and ARDS groups; (e) Correlation heatmap comparing FMT and control groups; (f) KEGG pathway enrichment analysis comparing FMT and control groups.
Discussion
Recent studies indicate that the interaction between gut microbiota and the host immune system plays a pivotal role in ARDS progression, providing a basis for microbiota-targeted interventions to modulate disease outcomes.32,33 The gut microbiota, consisting of bacteria, archaea, fungi, and viruses, is essential for maintaining host health by regulating metabolism, immune function, and homeostasis.34,35 Clostridiales, Lactobacillales, and Bacteroidales constitute approximately 60% of the gut microbiota. 36 Beneficial bacteria, such as Lactobacillus and Bifidobacterium, produce short-chain fatty acids (SCFAs) that support gut integrity, modulate immune responses, and inhibit pathogenic microbes. 37 In contrast, harmful bacteria linked to dysbiosis can trigger inflammation and disrupt metabolic processes, leading to lung disease. 38 Alterations in microbial communities have been associated with ARDS severity and patient outcomes. Recent evidence suggests that microbial dysbiosis extends beyond the intestine and may contribute to respiratory dysfunction. A study on COVID-19-related ARDS found that changes in microbial composition were associated with respiratory mechanics and patient survival, highlighting the potential systemic effects of microbial alterations in ARDS. 39 In the context of ARDS, recent advances suggest that FMT can alleviate symptoms by restoring gut microbiota balance, which in turn reduces systemic inflammation and improves metabolic function. 13 According to existing literature, changes in the microbiome of ARDS patients exhibit specific trends. Prevotella has shown an increased abundance in ARDS patients, which is evident in both trauma and respiratory failure patients. 40 Akkermansia has been found to be elevated in some critically ill patients, potentially associated with the protective role of the gut barrier function. 41 In contrast, Clostridia generally exhibits a downregulation, particularly in the intestines of ARDS patients, where its reduced abundance may be linked to gut dysbiosis and abnormal immune responses. 42 Sanguinetti’s study highlights the altered pulmonary microbiota composition in Clostridioides difficile infection patients, showing an enrichment in Clostridium XI and a decrease in Faecalibacterium, which suggests a microbial imbalance. 43 This indirectly supports the hypothesis that FMT may influence ARDS through the gut-lung axis by modulating microbial communities. Our findings suggest that FMT may alleviate ARDS by regulating the gut microbiota composition of Akkermansia and Lactobacillus, restoring the abundance of genera such as Muribaculaceae, Clostridia_UCG-014, Prevotella, and Adlercreutzia, while reducing Romboutsia. These changes in microbiota composition are associated with the restoration of normal lipid, amino acid, and carbohydrate metabolism. These results provide new insights into how FMT can regulate the gut-lung axis in ARDS (Figure 11). The abundance of Prevotella is increased in ARDS patients, which is consistent with our findings in the FMT group. Akkermansia is elevated in critically ill patients, potentially associated with the protective role of the gut barrier, and similar expression patterns were observed in both the ARDS and FMT groups in our study. In contrast, Clostridia is downregulated in ARDS patients, likely linked to gut dysbiosis and abnormal immune responses, a trend that was also validated in our animal model, with Clostridia_UCG-014 abundance restored following FMT treatment. Furthermore, Romboutsia was reduced in the FMT group, suggesting that FMT may help restore the balance of the gut microbiota. These findings provide new preclinical evidence for microbiota-targeted interventions.

Proposed mechanism of FMT in alleviating ARDS-associated lung injury. This schematic diagram illustrates the potential mechanisms by which fecal microbiota transplantation (FMT) modulates gut microbiota, metabolic pathways, and transcriptional regulation, ultimately improving lung injury in acute respiratory distress syndrome (ARDS). The ARDS rat model was induced by lipopolysaccharide (LPS), leading to intestinal barrier disruption (dysbiosis), activation of inflammatory signaling, and exacerbation of lung injury. FMT intervention restored the gut microbiota composition by regulating the gut microbiota composition of Akkermansia and Lactobacillus, restoring Muribaculaceae, Clostridia_UCG-014, Prevotella, and Adlercreutzia, while reducing Romboutsia. These microbiota alterations were linked to metabolomic changes, including an increase in short-chain fatty acids (SCFAs, e.g. butyrate and propionate) and the restoration of amino acid and lipid metabolism, which contributed to immune modulation and inflammation reduction. At the transcriptomic level, FMT up-regulated cytokine signaling, mTOR signaling, and viral infection pathways, leading to upregulation of antiviral genes and enhancement of T cell receptor signaling, ultimately reducing lung inflammation and injury while improving pulmonary function. This study suggests that FMT exerts its protective effects on ARDS via the gut microbiota–metabolite–immune regulatory axis.
FMT has shown therapeutic potential in restoring gut microbiota balance and modulating systemic inflammation in conditions such as ARDS and Clostridium difficile infection.44–46 In our study, FMT significantly alleviated lung tissue damage and improved oxygenation in ARDS rats, likely by restructuring gut microbiota and reducing the production of inflammatory mediators. These effects appear to be linked to the restoration of gut-lung axis balance, promoting lung tissue repair. This aligns with previous findings that gut microbiota can mitigate LPS-induced lung injury by regulating pathways such as TLR4/NF-κB. 28 Studies have demonstrated that FMT can inhibit the TLR4/NF-κB pathway, thereby reducing the expression of pro-inflammatory cytokines and alleviating inflammation. 47 Additionally, FMT restores gut microbiota diversity and promotes the production of SCFAs, contributing to immune modulation. 48 Furthermore, FMT is closely associated with the mTOR signaling pathway, as it modulates gut microbiota to activate the PI3K/AKT/mTOR pathway, thereby improving metabolic homeostasis and promoting cellular repair. 49 Our study further demonstrates that FMT upregulates cytokine signaling, the mTOR pathway, and virus infection-related pathways, potentially regulating host immune responses to maintain inflammatory balance and immune homeostasis. Further microbiome analysis revealed that FMT regulates the gut microbiota composition of Akkermansia and Lactobacillus in ARDS, increasing the abundance of bacteria such as Muribaculaceae, Clostridia_UCG-014, Prevotella, and Adlercreutzia. This change likely contributes to gut microbial homeostasis by reducing the proportion of potentially pathogenic bacteria like Romboutsia. Significant correlations were observed between changes in Lactobacillus and Romboutsia with host metabolites, supporting the critical role of specific microbiota in modulating metabolism and immune responses. Lactobacillus rhamnosus modulates neutrophil phagocytosis and NETs formation, reducing pro-inflammatory cytokine levels and alleviating ARDS. This effect is primarily mediated by SCFAs, particularly butyrate, which decreases pulmonary neutrophil infiltration and inflammation. 50 SCFAs, including acetate, propionate, and butyrate, play a crucial role in maintaining metabolic homeostasis and regulating inflammation through G-protein-coupled receptors and histone deacetylases signaling pathways. 51 Akkermansia muciniphila enhances SCFA production, inhibits the TLR2/MyD88/NF-κB pathway, and reduces pro-inflammatory cytokine expression, thereby mitigating ALI pathology. 52 Although Akkermansia was less prominent in our study, its role in metabolic regulation aligns with findings by Zeng et al. 53 Furthermore, increasing Lactobacillus reuteri abundance through FMT has been shown to downregulate IFN-γ and IL-17 levels while upregulating IL-10 expression, promoting Treg cell expansion and reducing Th1/Th17 infiltration, ultimately ameliorating ARDS severity. 54 Our study’s increase in Lactobacillus might similarly regulate immune responses through metabolic products like butyrate, which exhibit anti-inflammatory effects. Importantly, our study identified correlations between specific microbial genera and host metabolites, suggesting that Akkermansia and Lactobacillus might help regulate immune responses and inflammation through their metabolic products, such as butyrate.50,53 This supports the hypothesis that restoring beneficial microbiota can improve energy metabolism and reduce inflammatory cytokine production, as seen in previous studies on microbiota-driven modulation of host metabolism.55,56 This hypothesis is reinforced by transcriptomic data showing that FMT significantly downregulated inflammation-related genes, underscoring the crucial role of probiotics in immune modulation.
Furthermore, our metabolomic and transcriptomic analyses indicated that FMT modulates key pathways, including lipid and amino acid metabolism, which are essential for energy balance and immune regulation. These findings align with existing literature that links metabolic dysregulation to impaired lung function in ARDS. 57 By enhancing metabolic processes like the TCA cycle, glycolysis, and amino acid biosynthesis, FMT may help restore energy homeostasis, thereby alleviating ARDS symptoms. Studies have demonstrated that butyrate-producing bacteria, such as those in the Firmicutes phylum, help maintain a stable intestinal environment and produce metabolites that inhibit inflammatory pathways like TLR4/MyD88/NF-κB, which are relevant to systemic inflammation seen in ARDS. 58 Our transcriptomic analysis also identified significant enrichment of pathways such as the cytokine-cytokine receptor interaction, mTOR signaling pathway and viral infection-related pathways, which are involved in immune modulation and inflammation, further supporting the role of FMT in regulating inflammation-related pathways in ARDS. The upregulation of the cytokine-cytokine receptor interaction pathway likely indicates that FMT modulates the immune system by enhancing cytokine-mediated immune responses and controlling inflammation, while the activation of viral infection-related pathways may further enhance host antiviral and anti-inflammatory defenses. 59 Furthermore, the increased activity of the mTOR signaling pathway suggests that FMT facilitates alveolar epithelial cell repair and regulates inflammatory factors through the modulation of cellular metabolism and autophagic processes. 60
Additionally, FMT has been shown to increase SCFA production, which not only contributes to restoring microbial diversity but also modulates immune function, as seen in experimental colitis and cognitive dysfunction models.58,61 Our study also suggests that FMT-induced changes in microbial composition may promote lung repair by modulating host gene expression, particularly by downregulating inflammation-related pathways such as TLR4/NF-κB. 57 The integrated analysis further identified potential therapeutic targets, such as Desulfovibrio and Akkermansia, which were highly associated with key metabolic changes. Desulfovibrio showed strong correlations with various metabolites, highlighting its potential role in ARDS pathogenesis and as a target for future microbiota-based therapies. Although our findings demonstrate the promise of FMT in managing ARDS, limitations such as variability in microbiota composition among individuals and the need for precise modulation strategies warrant further investigation. Future studies should focus on optimizing FMT protocols and exploring the role of key bacterial metabolites and genes to develop targeted, effective treatments for ARDS patients. Personalized FMT approaches, which tailor donor microbiota to the recipient’s unique microbial profile, have shown potential for enhancing therapeutic outcomes by improving strain engraftment and targeting specific dysbiotic features.62,63 Studies suggest that this individualized approach could optimize immune modulation and maintain long-term safety, supporting its future application in microbiome-targeted therapies for ARDS. 64
This study has some limitations that should be addressed in future research. Firstly, the relationship between gut microbiota and ARDS in this study remains largely correlational, and further causal experiments, such as using bacterial depletion strategies, are needed to validate the findings. While our animal model provides important insights into the mechanisms of ARDS, its translational relevance to human ARDS remains uncertain. Future studies should explore whether similar microbiota shifts are present in ARDS patients and assess how these findings may apply to clinical settings. Additionally, this study is the relatively small sample size (n = 6 per group), which results in a statistical power of 0.79, slightly below the conventional 0.80 threshold. While this power level is acceptable, a larger sample size could further enhance the robustness and generalizability of our findings. Future studies with larger cohorts would be beneficial to validate our results and provide more definitive conclusions.
Conclusions
This study demonstrates that FMT modulates the gut microbiota composition, including Akkermansia and Lactobacillus, restores the abundance of genera such as Muribaculaceae, Clostridia_UCG-014, Prevotella, and Adlercreutzia, and enhances the production of anti-inflammatory metabolites like butyrate. Additionally, FMT appears to restore key metabolic pathways related to lipid metabolism, amino acid biosynthesis, and immune regulation, including the modulation of immune pathways such as mTOR signaling. These changes contribute to improved lung tissue integrity and pulmonary function. However, the observed effects are associative rather than causal, and future research should focus on refining FMT protocols and exploring targeted microbial and metabolic interventions to develop more effective therapies for ARDS patients.
Footnotes
Author contributions
Dongwei Zhang: conceptualization, investigation, data curation, writing—original draft preparation, writing—review and editing; Zhenqiang Zhang: software, validation, formal analysis, investigation, data curation, visualization; Longxiong Liao: conceptualization, methodology, validation, investigation, writing—original draft preparation; Biying Dong: investigation, data curation, visualization; Xia Xiong: methodology, resources; Xuejun Qin: methodology, software, formal analysis, writing—review and editing, supervision; Xianming Fan: conceptualization, resources, writing—review and editing, supervision, project administration, funding acquisition.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Sichuan Science and Technology Program [grant number 2022YFS0631], Natural Science Foundation of Sichuan Province of China [grant number 2022NSFSC0046], Self-funded Research Project of Guangxi Autonomous Region [grant number Z-B20241275; GXZYB20240601], and Liuzhou Science and Technology Plan Project [grant number 2024YB0103B003].
Ethical considerations
Protocols for animal experiments were approved by the Animal Experimental Ethics Committee of Liuzhou People’s Hospital (Approval no. KY-2024-077) on May 5, 2024, in compliance with the National Institutes of Health guidelines for the care and use of laboratory animals.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Animal welfare
The present study followed national and institutional guidelines for humane animal treatment and complied with the National Institutes of Health guidelines for the care and use of laboratory animals.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors on request. The authors confirm that the data represented in this study are accurate, and the analysis was conducted following established standards to ensure reproducibility.