
Abstract
Background:
Oral mucosal diseases manifest primarily as inflammatory conditions. These diseases affect approximately half a billion people worldwide.
Objective:
Novel and effective strategies for treating inflammatory diseases of the oral mucosa have great potential for improving patient outcomes, and warrant study.
Methods:
The impact of melatonin on inflammation was investigated using RAW264.7 macrophages and HOEC and HSC-3 oral epithelial cells.
Results:
Melatonin decreased macrophage-induced inflammation by acting through the melatonin receptor MTNR1A. Additionally, melatonin mitigated macrophage-induced inflammation in oral epithelial cells. Importantly, the results demonstrated that the effects of melatonin on oral epithelial inflammation were mediated through the KEAP1/Nrf2 signaling pathway.
Conclusion:
These findings will contribute to the development of innovative therapies for inflammatory conditions affecting the oral epithelium.
Introduction
Oral mucosal diseases, which are predominantly inflammatory in nature, pose a global health concern affecting over half a billion individuals. 1 These conditions contribute to the deterioration of the periodontium, encompassing the soft tissue and alveolar bone surrounding the teeth. 1 Oral inflammation encompasses disorders, such as oral lichen planus and recurrent aphthous ulcers, which significantly impair the quality of life of affected individuals. 1 These diseases often exhibit prolonged courses or frequent relapses, further affecting the well-being of patients. 1 Although the precise pathogenesis of these conditions remains largely unknown, research has indicated that various pro-inflammatory cells and cytokines are involved in their initiation and persistence. 2 Oral mucosal inflammation is a complex pathological process regulated by oxidative stress, infection, injury, and underlying diseases. 3 Diverse molecular mechanisms contribute to this pathology, including abnormal gene expression, exposure to environmental chemicals, oxidative stress, and apoptosis. Accordingly, various models have been employed to enhance our understanding of the development of oral mucosal inflammation and its associated cellular events. 3 Innovative and effective strategies to address inflammatory diseases of the oral mucosa may improve patient outcomes and address the global public health burden of these conditions.
Macrophages play a vital role in the innate immune system and serve as essential regulators of both physiological homeostasis and pathological processes. 4 Macrophages, derived from hematopoietic progenitors, are instrumental in tissue remodeling during fetal development and contribute fundamentally to homeostasis in various organoids. 4 They are strategically distributed throughout the body and perform crucial homeostatic functions, including the removal of dead cells, debris, and lipoproteins from their microenvironment. 4 Moreover, beyond their role in homeostasis, macrophages are also involved in the repair of injured tissue. 5 They can polarize into distinct phenotypes in response to the surrounding microenvironment. 5 This polarization is critical for determining the fate of an organ during inflammation or injury. 5 In instances of infection or injury to an organ or tissue, macrophages undergo initial polarization to the pro-inflammatory M1 phenotype, releasing pro-inflammatory cytokines that facilitate the removal of antigens and necrotic cells. Subsequently, during the repair phase, M1 macrophages transition to the M2 phenotype, which secretes anti-inflammatory cytokines that suppress inflammation and promote tissue repair and remodeling. 5 Investigating the interplay between macrophage polarization and oral mucosal inflammation could clarify the effects of these processes on overall tissue health and resilience.
Melatonin is derived from tryptophan and is predominantly secreted by the pineal gland. It is an indolamine with high biological availability that readily crosses the blood–brain barrier and enters the brain parenchyma. 6 Extensive research has highlighted the diverse cellular activities of melatonin and has emphasized its significant antioxidant properties. Notably, melatonin scavenges reactive oxygen species and protects essential cellular components and molecules, such as mitochondrial oxidoreductase, superoxide dismutase (SOD), and other critical proteins and enzymes, thereby alleviating DNA oxidative damage. 6 In addition to its antioxidant capabilities, melatonin has been increasingly recognized for its anti-inflammatory properties in both acute and chronic inflammatory processes. 6 Studies using animal models have revealed that exogenous melatonin administration can suppress inflammatory stimuli by reducing pro-inflammatory cytokines, such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), while concurrently elevating levels of the anti-inflammatory cytokine IL-4 in the serum. 6 Given these findings, exploring the effects of melatonin on oral mucosal inflammation and elucidating the underlying mechanisms are promising avenues for research.
Previous studies have shown that concivia nuclear factor erythroid 2-related factor 2 (Nrf2), which is considered to be a sensor of oxidative stress. The main function of this signaling pathway is to maintain physiological conditions by inducing redox balance.7,8 Kelch-like ECH-associated protein 1 (KEAP1) is a classic protein upstream of Nrf2. KEAP1/Nrf2 signaling is important in regulating inflammation.8,9 Thus, it is important to explore whether the effect of melatonin on inflammation occurs via KEPA1/Nrf2 signaling.
In this study, we examined the effect of melatonin on inflammation in macrophages and oral epithelial cells, specifically focusing on a human oral epithelial cell line (HOEC) and human oral squamous carcinoma cell line (HSC-3). The goal was to understand the effects of melatonin on macrophage inflammation and delineate the underlying mechanisms. Additionally, we explored the effect of melatonin on inflammation in oral epithelial cells, induced by a macrophage conditioned medium (CM), to elucidate the detailed mechanisms by which melatonin modulates inflammation in the oral mucosa. Our findings provide valuable insights that will inform the development of therapeutic strategies for oral mucosal inflammation.
Materials and methods
Cell culture
RAW264.7 macrophages obtained from the Cell Bank of The Fourth Hospital of Hebei Medical University were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific; catalog number: 11960044), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific; catalog number: 16140071), and 1% penicillin-streptomycin (Thermo Fisher Scientific; catalog#15140148). RAW264.7 cells, with a 70%–80% confluency, were seeded in 96-well plates and maintained in an incubator at 37°C in an atmosphere of 5% CO2.
HOEC and HSC-3 oral epithelial cell lines were used in this study. HOEC cells were purchased from the American Type Culture Collection (catalog number AC34021, passage 4), while HSC-3 cells were purchased from Mingzhou Bio (catalog number MZ-0878, https://www.mingzhoubio.com/goods-2286.html, passage 10). Both cell lines were cultured in high-glucose DMEM (Thermo Fisher Scientific; catalog number: 10569010) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific; catalog number: 16140071) and 1% penicillin-streptomycin (Thermo Fisher Scientific; catalog number: 15140148). All cells were maintained at 37°C in a humidified chamber in an atmosphere of 5% CO2.
The macrophages and two oral epithelial cell lines were treated with different doses (0, 10, and 50 μM) of melatonin and lipopolysaccharide (LPS, 100 ng/mL) for 24 h at 37°C in a humidified chamber in the 5% CO2 atmosphere.
Preparation of macrophage CM
Cells were cultured in high-glucose DMEM (Thermo Fisher Scientific; catalog#11960044) supplemented with the aforementioned 10% FBS and 1% penicillin-streptomycin for 48 h. The culture was harvested and filtered through a 0.22-μm filter to remove cell debris. The resulting cell-free conditioned medium served as a control (CM-). Murine RAW macrophages were activated by incubating them overnight with 100 ng/mL LPS. The cells were washed twice with PBS and cultured with DMEM supplemented with 10% FBS for 48 h. Subsequently, they were harvested and filtered through a 0.22-μm filter. This cell-free medium represented the CM.
ELISA detection of inflammatory and anti-inflammatory factors
RAW264.7, HOEC, and HSC-3 cells were subjected to various stimuli including treatment with LPS or melatonin and transfection with small interfering RNA (siRNA) in low-glucose DMEM (Thermo Fisher Scientific; catalog number: 11885084) for 24 h. Culture supernatants were collected to quantify the levels of TNF-α, IL-6, and IL-11 cytokines using specific ELISA kits (Shanghai ExCell Biotechnology) in accordance with the manufacturer’s instructions.
RNA isolation and reverse transcription-quantitative PCR (RT-qPCR)
The total RNA was isolated from RAW264.7, HOEC, and HSC-3 cells seeded at a density of 6,000 cells per well in 96-well plates using TRIzol reagent (Beyotime Institute of Biotechnology) following the manufacturer’s protocol. Subsequently, the BeyoRT First Strand cDNA Synthesis kit (Beyotime Institute of Biotechnology; catalog number: D7166) was used to synthesize cDNA from total RNA. RT-qPCR was performed using the BeyoFast SYBR Green qPCR Mix (2×, Beyotime Institute of Biotechnology; catalog Number: D7260-25 mL) using an Applied Biosystems 7500 Fast Real-Time PCR System (Thermo Fisher Scientific). The qPCR protocol included an initial step at 50°C for 2 min and 95°C for 2 min, followed by 40 cycles at 95°C for 15 s, 60°C for 1 min, and an extension at 72°C for 1 min, with a final extension step at 72°C for 10 min. Glyceraldehyde 3-phosphate dehydrogenase served as the internal control. The relative gene expression levels were determined using the 2−ΔΔCq method. 10 The primers used in this study are listed in Table 1. The experiment was repeated thrice.
Primers used in the present study.

Knockdown using siRNA
Gene knockdown was performed using siRNAs. The empty vector siRNA‑negative control (NC) was purchased from Synbio Technologies. siRNA sequences against melatonin receptor 1A (MTNR1A), KEAP1, and Nrf2 were designed using siRNA-Target-Finder (GeneScript, https://www.genscript.com/tools/sirna-target-finder) according to the cDNA backbone of MTNR1A (ACCESSION number: NM_005958), KEAP1 (ACCESSION number: NM_012289), and Nrf2 (ACCESSION number: NM_001145412), followed by synthesis, and were purchased from Synbio Technologies. The siRNA sequences were:
NC, 5’-AAGAAGCTCAGGAACGCAGGA-3’; KEAP1, 5’-AAGTGCGAGATCCTGCAGTCC-3’; high mobility group A1 (HMGA1), 5’-AAGTAGGTAACTGTAGTCCAC-3’; and NRF2, 5’ AAGTAGGTAACTGTAGTCCAC-3’.
Non-targeting control siRNA and target siRNA were transiently transfected into RAW264.7, HOEC, and HSC-3 cells using the FuGENE HD Transfection Reagent (Promega Corporation; catalog number: E2311), according to the manufacturer’s instructions, and placed in a cell culture incubator (37°C, 5% CO2) for 24 h to facilitate siRNA transfection prior to the subsequent experiments. Knockdown efficiency was evaluated using RT-qPCR and western blot assays, following the protocols described in this paper.
Western blot
Cell samples were collected using RIPA lysis buffer (Beyotime Biotechnology; catalog number: P0013B) containing protease and phosphatase inhibitors to ensure protein preservation. Protein concentrations were determined using Bradford or BCA assays. Equal amounts of protein (typically 20–50 µg) were denatured by boiling in sodium dodecyl sulfate sample buffer and then separated by polyacrylamide gel electrophoresis. The resolved proteins were transferred to polyvinylidene difluoride or nitrocellulose membranes using a semi-dry or wet transfer system. To prevent nonspecific binding, the membrane was blocked with 5% nonfat dry milk or bovine serum albumin in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h at room temperature. The membrane was then treated with primary antibodies:
MTNR1A (Abcam, catalog number: ab87639), KEAP1 (EPR22664-26, Abcam; catalog number: ab227828), Nrf2 (Abcam; catalog number: ab137550), light chain 3A/3B (LC3A/B, Cell Signaling Technology; catalog number: #4108), P62 (Abcam; catalog number: ab91526), IL-11 (EPR15253, Abcam; catalog number: ab187167), and rabbit monoclonal antibody to TNF-α (D1G2, Cell Signaling Technology; catalog number 8184), diluted in block buffer, and incubated overnight at 4°C. After extensive washing with TBST, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Abcam; catalog number: ab2761) for 1 h at room temperature. After a subsequent wash with TBST, protein bands were visualized using enhanced chemiluminescence substrate and imaged using a chemiluminescence detection system. Quantitative analysis of band intensity was performed using ImageJ software (NIH).
Statistical analyses
All data are presented as means ± standard errors of the mean. Statistical analysis of continuous variables was performed using a one-way ANOVA and Tukey’s post-hoc test. Categorical variables were analyzed using Fisher’s exact test. Correlation analyses, specifically Pearson’s correlation, and overall statistical analyses were conducted using GraphPad Prism software (version 5.0; GraphPad Software, Inc.). Statistical significance was set at p < 0.05.
Results
Melatonin inhibits macrophage inflammation
RAW264.7 macrophages were treated with 0, 10, and 50 μM of melatonin and LPS (100 ng/mL) for 24 h at 37°C in a humidified chamber in an atmosphere of 5% CO2. LPS significantly induced inflammation in the macrophages, as evidenced by an increased expression of TNF-α, IL-6, and IL-11 inflammatory cytokines (Figure 1(a–c), respectively). Interestingly, melatonin significantly attenuated macrophage inflammation in a dose-dependent manner, reducing the expression of TNF-α, IL-6, and IL-11 in RAW264.7 cells (Figure 1(a–c), respectively). Furthermore, melatonin produced a significant dose-dependent reduction in the protein levels of inflammatory cytokines, including TNF-α, IL-6, and IL-11 (Figure 1(d–f), respectively), in the supernatants of RAW264.7 cells. LPS significantly decreased the expression of CAT mRNA. This decrease was attenuated by melatonin (Figure 1(g)). LPS also significantly decreased the expression of SOD1 mRNA. Melatonin also significantly attenuated this decrease in RAW264.7 cells (Figure 1(h)). To further explore the effect of melatonin on inflammation, cells treated with LPS and melatonin for 48 h were analyzed by flow cytometry to measure CD68. The results indicated that LPS increased M1 macrophages and melatonin reversed this increase (Figure 1(i)). Overall, these findings demonstrate that in macrophages LPS induces inflammation and melatonin effectively inhibits it.

Melatonin inhibits macrophage inflammation. (a) LPS significantly mRNA expression of TNFɑ, and Mel significantly attenuated increased mRNA expression of TNFɑ induced by LPS on RAW264.7 cells (*p < 0.05, **p < 0.01, ***p < 0.001). (b) LPS significantly mRNA expression of IL11, and Mel significantly attenuated increased mRNA expression of IL11 induced by LPS on RAW264.7 cells (**p < 0.01, ***p < 0.001). (c) LPS significantly mRNA expression of IL6, and Mel significantly attenuated increased mRNA expression of IL6 induced by LPS on RAW264.7 cells (*p < 0.05, **p < 0.01, ***p < 0.001). (d) LPS significantly TNFɑ protein level, and Mel significantly attenuated increased TNFɑ protein level induced by LPS on RAW264.7 cells (*p < 0.05, **p < 0.01, ***p < 0.001). (e) LPS significantly TIL11 protein level, and Mel significantly attenuated increased IL11 protein level induced by LPS on RAW264.7 cells (*p < 0.05, **p < 0.01, ***p < 0.001). (f) LPS significantly IL6 protein level, and Mel significantly attenuated increased IL11 protein level induced by LPS on RAW264.7 cells (**p < 0.01, ***p < 0.001). (g) LPS significantly decreased the expression level of CAT mRNA, and Mel significantly attenuated decreased expression level of CAT mRNA induced by LPS on RAW264.7 cells (*p < 0.05, **p < 0.01). (h) LPS significantly decreased expression level of SOD1 mRNA, and Mel significantly attenuated decreased expression level of SOD1 mRNA induced by LPS on RAW264.7 cells (*p < 0.05, **p < 0.01). (i) Cell were treated with LPS and melatonin, and after 48 h cells were analyzed by flow cytometry to measure CD68.
Melatonin inhibits macrophage inflammation via MTNR1A
MTNR1A is an important receptor for melatonin. 11 Accordingly, we investigated whether the effects of melatonin on macrophage inflammation could be mediated by targeting MTNR1A. MTNR1A expression in RAW264.7 cells treated with LPS and melatonin was knocked down using MTNR1A-targeted siRNA and assessed through western blotting (Figure 2(a and b)). MTNR1A knockdown attenuated the inhibitory effects of melatonin on the LPS-induced increase in mRNA and protein levels of inflammation-related genes, including TNF-α (Figure 2(c and f)), IL-11 (Figure 2(d and g)), and IL-6 (Figure 2(e and h)). To further demonstrate the effects of MTNR1A on inflammation, western blot analysis also assessed the influence of MTNR1A knockdown on IL-6, TNF-ɑ, and IL-11 protein levels. MTNR1A

Melatonin inhibits macrophage inflammation via its receptor, MTNR1A. (a) MTNR1A was knockdown by siNRA detected using western blot. (b) Quantification of western blot of (a) (***p < 0.001). (c) MTNR1A knockdown attenuated inhibitory effects of Mel on LPS induced increased mRNA expression level of TNFɑ on RAW264.7 cells (*p < 0.05, ***p < 0.001). (d) MTNR1A knockdown attenuated inhibitory effects of Mel on LPS induced increased mRNA expression level of IL11 on RAW264.7 cells (*p < 0.05, ***p < 0.001). (e) MTNR1A knockdown attenuated inhibitory effects of Mel on LPS induced increased mRNA expression level of IL6 on RAW264.7 cells (*p < 0.05, **p < 0.01, ***p < 0.001). (f) MTNR1A knockdown attenuated inhibitory effects of Mel on LPS induced increased protein level of TNFɑ on RAW264.7 cells (*p < 0.05, **p < 0.01, ***p < 0.001). (g) MTNR1A knockdown attenuated inhibitory effects of Mel on LPS induced increased protein level of IL11 on RAW264.7 cells (**p < 0.01, ***p < 0.001). (h) MTNR1A knockdown attenuated inhibitory effects of Mel on LPS induced increased protein level of IL6 on RAW264.7 cells (*p < 0.05, **p < 0.01, ***p < 0.001). (i) MTNR1A knockdown attenuated inhibitory effects of Mel on LPS induced increased protein level of IL6 on RAW264.7 cells detected by Western Blot. (j) Quantification of western blot of (i). (k) MTNR1A knockdown attenuated inhibitory effects of Mel on LPS induced increased protein level of IL6 on RAW264.7 cells detected by Western Blot. (l) Quantification of western blot of (k) (*p < 0.05, **p < 0.01, ***p < 0.001). (m) MTNR1A knockdown attenuated inhibitory effects of Mel on LPS induced increased protein level of IL6 on RAW264.7 cells detected by Western Blot. (n) Quantification of western blot of (m) (*p < 0.05, **p < 0.01, ***p < 0.001).
knockdown mitigated the inhibitory effects of melatonin on LPS-induced increases in mRNA expression and protein levels of inflammatory genes, including IL-6 (Figure 2(i and j)), TNF-α (Figure 2(k and l)), and IL-11 (Figure 2(m and n)). THP-1 human macrophage cells were used to explore the role of MTNR1A in inflammation. MTNR1A knockdown attenuated the inhibitory effects of melatonin on LPS-induced increases in mRNA expression of inflammatory genes, including TNF-α (Supplemental Figure 2(A)) and IL-6 (Supplemental Figure 2(B)). Collectively, these results demonstrate that melatonin inhibits macrophage inflammation through the MTNR1A receptor in both mouse and human macrophages.
Melatonin inhibits inflammation of oral epithelial cells induced by macrophage CM
Given the pivotal role of macrophages in oral epithelial inflammation, 12 we investigated whether activated macrophages induce inflammation in HOEC and HSC-3 oral epithelial cells. MTNR1A was knocked down using specific siRNA (Figure 3(a)). CM was collected from RAW264.7 cells treated with 100 ng/mL LPS and was used to treat HOEC and HSC-3 cells. Macrophage CM significantly induced inflammation in both HOEC (Figure 3(b–d)) and HSC-3 cells (Figure 3(d–f)). Importantly, melatonin concentrations of 10 and 50 μM significantly attenuated the increased protein levels of cytokines detected by ELISA, including TNF-α (Figure 3(e)), IL-11 (Figure 3(f)), and IL-6 (Figure 3(g)), induced by macrophage CM in HOEC cells. These concentrations of melatonin also significantly attenuated the macrophage CM-induced increase in protein levels of IL-6 (Figure 3(h)) in HSC cells. Collectively, these findings demonstrate the inhibition of inflammation induced by macrophage CM in HOEC and HSC-3 cells treated with melatonin.
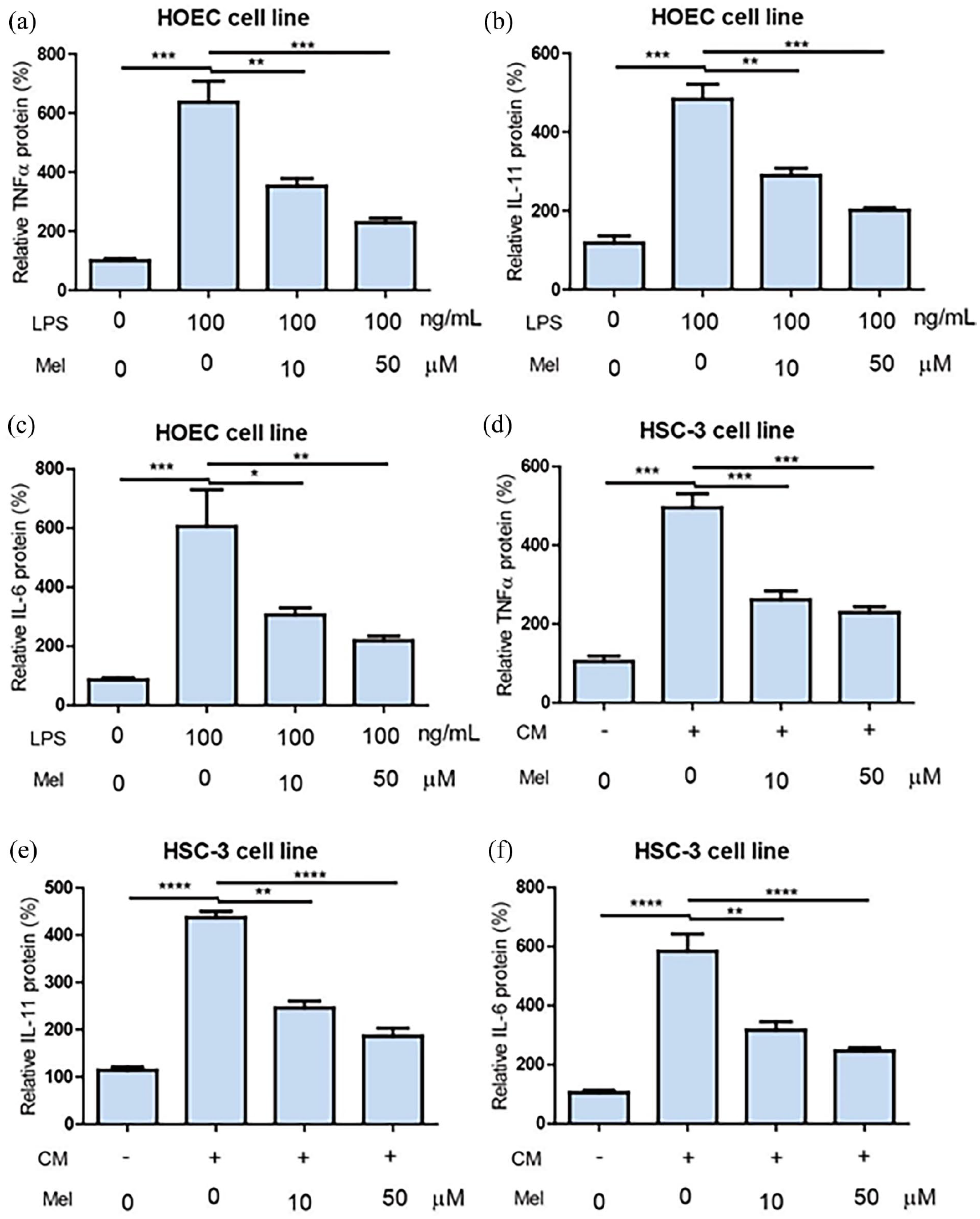
Melatonin inhibits inflammation of oral epithelial cells induced by macrophage condition medium. (a) Macrophage condition medium (CM) significantly induced increase TNFɑ protein level, while melatonin (10 and 50 μM) significantly attenuated macrophage CM induced increase of TNFɑ protein level of on HOEC cells (**p < 0.01, ***p < 0.001). (b) Macrophage condition medium (CM) significantly induced increase IL11 protein level, while melatonin (10 and 50 μM) significantly attenuated macrophage CM induced increase of IL11 protein level of on HOEC cells (**p < 0.01, ***p < 0.001). (c) Macrophage condition medium (CM) significantly induced increase IL6 protein level, while melatonin (10 and 50 μM) significantly attenuated macrophage CM induced increase of IL6 protein level of on HOEC cells (*p < 0.05, **p < 0.01, ***p < 0.001). (d) Macrophage condition medium (CM) significantly induced increase TNFɑ protein level, while melatonin (10 and 50 μM) significantly attenuated macrophage CM induced increase of TNFɑ protein level of on HSC-3 cells (***p < 0.001). (e) Macrophage condition medium (CM) significantly induced increase IL11 protein level, while melatonin (10 and 50 μM) significantly attenuated macrophage CM induced increase of IL11 protein level of on HSC-3 cells (**p < 0.01, ****p < 0.0081). (f) Macrophage condition medium (CM) significantly induced increase IL6 protein level, while melatonin (10 and 50 μM) significantly attenuated macrophage CM induced increase of HSC-3 protein level of on HOEC cells (**p < 0.01, ****p < 0.0001).
KEAP1 is closely involved in inflammation of oral epithelial cells induced by macrophage CM
KEAP1 is closely involved in the regulatory effects of melatonin on cellular activities. 13 To investigate the involvement of KEAP1 in the inhibitory effects of melatonin on inflammation induced by macrophage CM in oral epithelial cells, MTNR1A was knocked down in HOEC cells (Figure 4(a)). The knockdown significantly reduced the mRNA expression of KEAP1 in the cells (Figure 4(b)). To explore the effects of KEAP1 on inflammation in HOEC cells, KEAP1 was knocked down, as detected by qPCR (Figure 4(c)) and confirmed by western blotting (Figure 4(d and e)). KEAP1 knockdown significantly attenuated the increase in TNF-α (Figure 4(f)) and IL-6 (Figure 4(g)) cytokines induced by macrophage CM in HOEC cells. To further assess the effects of melatonin on inflammation induced by macrophage CM, MTNR1A was knocked down in HSC-3 cells (Figure 4(h)). MTNR1A knockdown significantly reduced the mRNA expression of KEAP1 in these cells (Figure 4(i)). KEAP1 knock down was evident by qPCR (Figure 4(j)), and western blotting confirmed the knockdown effect in HOEC cells (4k and 4l). KEAP1 knockdown significantly attenuated the increase in cytokines, including TNF-α (Figure 4(m)) and IL-6 (Figure 4(n)), induced by macrophage CM in HSC-3 cells. Taken together, these findings demonstrate that melatonin inhibits macrophage CM-induced inflammation in oral epithelial cells via KEAP1.

KEAP1 is closely involved in oral inflammation induced by macrophage. (a) MTNR1A was knockdown on HOEC cells (**p < 0.01). (b) MTNR1A knockdown significantly mRNA expression of KEAP1 on HOEC cells (**p < 0.01). (c) KEAP1 was knockdown on HOEC cells detected by qPCR (***p < 0.001). (d) KEAP1 was knockdown on HOEC cells detected by western blot. (e) Quantification of western blot of (d) (***p < 0.001). (f) KEAP1 knockdown significantly attenuated macrophage CM induced increase of cytokines including TNFɑ on HOEC cells (**p < 0.01, ***p < 0.001). (g) KEAP1 knockdown significantly attenuated macrophage CM induced increase of cytokines including IL6 on HOEC cells (**p < 0.01, ***p < 0.001). (h) MTNR1A was knockdown on HSC-3 cells (***p < 0.001). (i) MTNR1A knockdown significantly mRNA expression of KEAP1 on HSC-3 cells (*p < 0.05, **p < 0.01, ***p < 0.001). (j) KEAP1 was knockdown on HSC-3 cells detected by qPCR (***p < 0.001). (k) KEAP1 was knockdown on HSC-3 cells detected by western blot. (l) Quantification of western blot of (k) (***p < 0.001). (m) KEAP1 knockdown significantly attenuated macrophage CM induced increase of cytokines including TNFɑ on HSC-3 cells (**p < 0.01, ****p < 0.0001). (n) KEAP1 knockdown significantly attenuated macrophage CM induced increase of cytokines including IL6 on HSC-3 cells (***p < 0.001, ****p < 0.0001).
KEAP1/Nrf2 pathway is closely involved in the inflammation of oral epithelial cells induced by macrophage CM
Nrf2 is a downstream protein of KEAP1.
13
Accordingly, we assessed the interaction between Nrf2 and KEAP1 (Figure 5(a)) using STRING analysis (https://cn.string-db.org/). KEAP1 knockdown decreased Nrf2 mRNA expression in both HOEC (Figure 5(b)) and HSC-3 cells (Figure 5(c)). siRNA-mediated knockdown was validated through western blotting (Figure 5(e and f)). Nrf2 knockdown in
HOEC cells significantly attenuated the CM-induced increase in cytokines, including TNF-ɑ (Figure 5(g)) and IL-6 (Figure 5(h)). Similarly, Nrf2 knockdown in HSC-3 cells (Figure 5(i, g, and k)) demonstrated that the macrophage CM-induced increase in cytokines, including TNF-ɑ and IL-6 was significantly attenuated (Figure 5(l and m), respectively). Taken together, these results demonstrate that melatonin inhibits macrophage CM-induced inflammation in oral epithelial cells via the KEAP1/Nrf2 pathway.

KEAP1/Nrf2 pathway is closely involved in inflammation of oral epithelial cells induced by macrophage. (a) Nrf2 interacts with KEAP1 (Figure 5(a)) analyzed using String (https://cn.string-db.org/). (b) KEAP1 knockdown was found to decrease mRNA expression of Nrf2 on HOEC cells (*p < 0.05). (c) KEAP1 knockdown was found to decrease mRNA expression of Nrf2 on HSC-3 cells (**p < 0.01). (d) Nrf2 was knockdown on HOEC cells detected by qPCR. (e) Nrf2 was knockdown on HOEC cells detected by Western Blot. (f) Quantification of western blot of (e) (***p < 0.001). (g) Nrf2 knockdown significantly attenuate macrophage CM induced increase of TNFɑ protein level on HOEC cells (*p < 0.05, **p < 0.01, ***p < 0.001). (h) Nrf2 knockdown significantly attenuate macrophage CM induced increase of IL6 protein level on HOEC cells (**p < 0.01, ***p < 0.001). (i) Nrf2 was knockdown on HSC-3 cells detected by qPCR (**p < 0.01). (j) Nrf2 was knockdown on HSC-3 cells detected by Western Blot. (k) Quantification of western blot of (j) (***p < 0.001). (l) Nrf2 knockdown significantly attenuate macrophage CM induced increase of TNFɑ protein level on HSC-3 cells (***p < 0.001, ****p < 0.0001). (m) Nrf2 knockdown significantly attenuate macrophage CM induced increase of IL6 protein level on HSC-3 cells (***p < 0.001, ****p < 0.0001).
Discussion
Oral mucosal inflammatory diseases are a significant global health challenge.
14
The incidence of oral mucosal inflammatory diseases is closely associated with various systemic conditions.
14
To elucidate new mechanisms and support the development of innovative therapies for oral mucosal inflammation, we investigated the effects of melatonin on macrophages and inflammation in the oral epithelium. Melatonin effectively suppressed macrophage inflammation via the MTNR1A receptor. Melatonin also significantly curtailed macrophage-induced inflammation in
HOEC and HSC-3 oral epithelial cells, which was attributed to the activation of the KEAP1/Nrf2 signaling pathway. The study findings offer valuable insights for a deeper understanding of the underlying mechanisms involved in oral mucosal inflammation.
Accumulating evidence has highlighted the multifaceted role of melatonin in various physiological processes, including circadian rhythm regulation, immune system modulation, cancer, and energy metabolism. Melatonin is a potent regulator of inflammation. In atherosclerotic mice, melatonin suppresses Galectin-3, thereby reducing nuclear factor-kappa B pathway activity. This in turn enhances Galectin-3 secretion, activates the phosphatidylinositol 3-kinase/protein kinase B pathway, and impairs autophagy, ultimately suppressing inflammation. 15 In a previous study, melatonin effectively mitigated intervertebral disc degeneration and inflammation in rats by modulating IL-1β-induced extracellular matrix remodeling in human nucleus pulposus cells. 16 In addition, melatonin protects against autophagy-induced injury by regulating FOXO3a, thereby attenuating neuroinflammation and depression in mice. 17 The results of the present study corroborate these findings by demonstrating the potent inhibition of macrophage inflammation and oxidative stress by melatonin (Figure 1), consistent with a previous study. 18 This previous study also indicated that cell morphology is altered by inflammation. 18 However, another study found that inflammation does not have a significant impact on cellular morphology. A possible reason for this dichotomy could be that the inflammation in the current study was not intense enough to trigger a change in cellular morphology. Notably, our investigation revealed that the regulatory effects of melatonin on macrophage inflammation are mediated through the MTNR1A receptor in both mouse and human macrophages (Figure 2). Melatonin typically exerts its physiological functions by interacting with the MTNR1A or MT2 receptors. Consistent with this, a previous study demonstrated that LPS treatment leads to decreases in melatonin content, melatonin receptor content (MTNR1A), and antioxidant potential (catalase and SOD), while simultaneously increasing nitro-oxidative stress markers (C-reactive protein, nitrate, and TNF-α). 19 Additionally, another study reported that melatonin can inhibit LPS-stimulated phosphorylation of extracellular signal-regulated kinase 1/2, c-Jun N-terminal kinase, and nuclear factor-kappa B p65 through its receptors. 20
Macrophages play a critical role in regulating inflammation in the oral epithelium through various mechanisms. 21 Upon encountering pathogens or tissue damage, macrophages are activated and release a range of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6. 21 These cytokines act on oral epithelial cells, inducing the expression of adhesion molecules and other inflammatory mediators that facilitate the recruitment and activation of additional immune cells at the site of inflammation. 22
In the present study, LPS significantly increased the expression of TNF-α, IL-6, and IL-11 pro-inflammatory cytokines (Figures 1 and 2). Thus, understanding the precise molecular pathways by which macrophages regulate inflammation in the oral epithelium could provide insights into new therapeutic strategies for oral inflammatory diseases.
The pathogenesis of various oral mucosal inflammatory diseases remains elusive and requires further investigation. Macrophage inflammation has been identified as a driver of epithelial inflammation in various tissues. In the present study, macrophage inflammation induced inflammation in the oral epithelium (Figure 3). Macrophages are the major inducers of inflammation in different organs. 23 Oral inflammatory diseases, such as oral lichen planus and recurrent aphthous ulcers, significantly diminish the quality of life of those who are affected. 24 Several factors influencing anti-inflammatory properties have been identified in oral inflammation. For example, antimicrobial peptides counteract inflammatory cytokines, thus suppressing oral inflammation and serving as potential targets for the prevention or treatment of oral inflammatory conditions. 25 In the present study, we confirmed that melatonin significantly inhibited the macrophage-induced inflammation of the oral epithelium by inhibiting TNF-ɑ, IL-11, and IL-6 (Figure 3). IL-11 plays an important role in promoting inflammation and fibrosis in various cell types, 26 as well as cell in proliferation. 27 Therefore, it is reasonable to conclude that melatonin plays an important role in reducing inflammation and inhibiting cancer cell growth by suppressing the expression of IL-11. Consistent with our findings, melatonin suppresses neuroinflammation and improves axonal hypomyelination by regulating microglial polarization in postnatal rats exposed to LPS. 28 Another study demonstrated that melatonin ameliorated ochratoxin A-induced liver inflammation, oxidative stress, and mitophagy in mice. 29 Increased levels of melatonin and MTNR1A expression in the inflamed oral mucosal tissue of patients with oral lichen planus have been described. 30 Additionally, topical melatonin niosome gels are reportedly beneficial in treating oral mucositis induced by 5-fluorouracil in mice. 31
Nrf2 and its endogenous inhibitor, Keap1, constitute a ubiquitous and evolutionarily conserved intracellular defense mechanism that counteracts oxidative stress. 32 Under normal conditions, KEAP1 functions as an E3 ubiquitin ligase that tightly regulates the activity of the transcription factor NRF2 by promoting its ubiquitination and subsequent proteasome-dependent degradation. 33 The Keap1/Nrf2 pathway is crucial in the regulation of inflammation. For example, polydatin has been shown to monitor the Keap1/Nrf2 signaling pathway, attenuating fructose-induced liver inflammation and lipid deposition. 34 Additionally, panaxydol attenuates ferroptosis in mice with LPS-induced acute liver injury ALI via the Keap1-Nrf2/Heme oxygenase-1 pathway. 35 In the present study, MTNR1A knockdown significantly reduced the mRNA expression of KEAP1 in oral epithelial cells (Figure 4), indicating that MTNR1A may regulate KEAP1 by influencing its expression. Furthermore, KEAP1 regulated the expression of NRF2 (Figure 5). KEAP1 and NRF2 are potent regulators of oral epithelial inflammation (Figures 4 and 5). Thus, MTNR1A may regulate oral epithelial inflammation via the KEAP1/NRF2 pathway. Consistent with our findings, melatonin reportedly regulates endoplasmic reticular stress in rats with chronic hepatotoxicity by activating the Keap1/Nrf2 pathway. 13 Similarly, the activation of melatonin receptors protects the brain from traumatic injury by attenuating oxidative stress and inflammation, which are mediated by the Nrf2 signaling pathway. 36 Although solid evidence is still lacking regarding the mechanism underlying how Keap1 knockdown reduces Nrf2 transcription, the literature suggests several potential explanations. In the absence of Keap1, cells may upregulate other compensatory pathways, such as Hrd1 (an E3 ubiquitin ligase), which targets Nrf2 for degradation or represses its transcriptional activity. 37 Additionally, the absence of Keap1 may increase the activity of MafK, which can form heterodimers with Bach1, enhancing the latter’s repressive effects on Nrf2 target genes. 38 Thus, the Keap1/Nrf2 signaling pathway appears to be crucial for the regulatory role of melatonin in inflammation.
Although the results of our study clearly demonstrate that macrophages play a crucial role in regulating inflammation in the oral epithelium, there are a few limitations that should be noted. Although human macrophages have been used to explore their effects on oral epithelial inflammation, a comprehensive analysis of these effects is lacking. Therefore, future studies should focus on thoroughly investigating the effects of human macrophages on oral epithelial inflammation. This would provide a more detailed understanding of the interactions between macrophages and oral epithelium, potentially leading to improved therapeutic strategies for managing oral inflammatory conditions. Moreover, oral mucosal inflammation is complicated and often coupled with oxidative stress. 18 Thus, future studies should explore the effects of melatonin on oxidative stress in oral epithelial cells.
Conclusion
Melatonin exerts anti-inflammatory effects on macrophage-induced inflammation through its receptor, MTNR1A. Additionally, melatonin attenuates macrophage-induced inflammation in oral epithelial cells. Importantly, these findings show that the regulatory effects of melatonin on oral epithelial inflammation involve the KEAP1/Nrf2 signaling pathway. Taken together, the results of our study provide valuable insights for the development of novel therapeutic strategies to treat oral epithelial inflammation.
Supplemental Material
sj-docx-1-iji-10.1177_03946320251318147 – Supplemental material for Melatonin alleviates oral epithelial cell inflammation via Keap1/Nrf2 signaling
Supplemental material, sj-docx-1-iji-10.1177_03946320251318147 for Melatonin alleviates oral epithelial cell inflammation via Keap1/Nrf2 signaling by Nan Zhang, Wenjing Wang, Rongxia Zhang, Yaxuan Liu, Yamei Wang, Yang Bai and Chencong Li in International Journal of Immunopathology and Pharmacology
Footnotes
Acknowledgements
None.
Authors’ contributions
N.Z., W.W., R.Z., Y.L., Y.W., Y.B., and C.L. performed the experiments. N.Z., W.W., and C.L. designed the study. N.Z., W.W., and C.L. wrote the manuscript and C.L. supervised the project. N.Z., W.W., and C.L. confirmed the authenticity of the raw data. All the authors have read and approved the final version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Medical Science Research Project of Hebei (20241083).
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Availability of data and materials
The datasets used and/or analyzed in the current study are available from the corresponding author upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.