
Abstract
Introduction
Bladder cancer represents a significant health challenge globally, ranking among the most common malignancies of the urinary system.1,2 It exhibits diverse clinical manifestations, ranging from non-invasive, low-risk tumors to aggressive, muscle-invasive forms with metastatic potential.3,4 The complexity of bladder cancer arises from the intricate interplay of genetic, environmental, and lifestyle factors in its etiology.1,5,6 The incidence of bladder cancer demonstrates marked geographic variation, with higher rates observed in regions with greater exposure to these risk factors.
Copper, a trace element, acts as a cofactor for over 30 enzymes involved in a wide array of physiological processes. 7 Recent studies have revealed that cells tightly regulate copper levels, with elevated cumulative copper concentrations leading to cytotoxicity and potential cell death.8,9 Moreover, the disruption of copper homeostasis is implicated in the facilitation of tumor metastasis. A novel form of cell death termed “cuproptosis” has been proposed by Tsvetkov, suggesting its contribution to cancer progression and metastasis. 10 This concept identifies 13 genes associated with cuproptosis, including FDX1, SLC31A1 (solute carrier family 31 member 1), LIPT1, LIAS, DLD, DBT, GCSH, DLST, DLAT, PDHA1, PDHB, ATP7A, and ATP7B. 11 Notably, SLC31A1 plays a role in the regulation of dietary copper uptake through copper transporters in the cell membrane. 12 Intriguingly, an increasing body of research links disrupted copper levels with various cancer types, encompassing lung, breast, kidney, and colorectal cancers. Given the pivotal role of SLC31A1 in copper homeostasis, a comprehensive exploration of its specific functions in Chinese bladder cancer patients’ contexts becomes imperative.
This study focuses on bladder cancer within the Chinese population, a region where the disease presents unique challenges and features. Investigating the molecular alterations in the SLC31A1 gene and their implications in Chinese bladder cancer patients is a critical step towards unraveling the complexity of this malignancy and developing tailored approaches for its management. We conducted molecular experiments to assess SLC31A1 gene expression, evaluate promoter methylation status, and identify genetic mutations in bladder cancer patients from China. To corroborate our findings, we further validated these aspects of SLC31A1 using datasets from The Cancer Genome Atlas (TCGA). Additionally, we explored the association between SLC31A1 expression and patient survival through online databases. To gain deeper insights into the underlying molecular mechanisms and potential therapeutic implications, we conducted gene enrichment analyses and examined patterns of immune cell infiltration.
Methodology
Sample collection
In our study, we meticulously collected a total of 50 tissue samples, comprising 25 specimens of bladder cancer tissue and 25 adjacent control samples. These tissues were diligently procured from The First Affiliated Hospital of Fujian Medical University, China. Our sampling strategy aimed to provide a comprehensive and representative set of specimens, allowing for a rigorous analysis of bladder cancer and its adjacent normal tissue. The current study received approval from the Ethics Committee of Fujian Medical University (FU# 2341-11), China, and was conducted in strict adherence to the Helsinki guidelines. 13 Furthermore, prior to the collection of samples, written informed consent was obtained from all patients.
Nucleic acid extraction
DNA extraction from tissue samples was done following organic method 14 while RNA was extracted using TRIzole method. 15
RT-qPCR-based expression analysis of SLC31A1
The quality and purity of the extracted RNA were evaluated using an Agilent Bioanalyzer (Santa Clara, CA, USA). Subsequently, the RNA was subjected to reverse transcription to generate complementary DNA (cDNA) using a Reverse Transcription kit (TOYOBO, Shanghai, China). Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) was carried out with SYBR Green PCR mix (Thermo Fisher Scientific, Waltham, USA) on an ABI 7900HT FAST Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). PCR amplification was performed at 95 °C for 10 minutes. Subsequently, the amplification process consisted of denaturation at 95°C for 15 seconds followed by annealing and extension at 60°C for 60 seconds, which were repeated for a total of 40 cycles. To ensure standardization, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was utilized as an internal control. The relative mRNA expression levels were determined using the widely accepted method known as the 2−ΔΔCT method. In this particular study, GAPDH and SLC31A1 genes were amplified employing the following primers. GAPDH-F 5′-ACCCACTCCTCCACCTTTGAC-3′, GAPDH-R 5′-CTGTTGCTGTAGCCAAATTCG-3′ SLC31A1-F: 5′-GGGGATGAGCTATATGGACTCC-3′ SLC31A1-R: 5′-TCACCAAACCGGAAAACAGTAG-3′
Receiver operating characteristic (ROC) curve
The Receiver Operating Characteristic (ROC) curve provides a holistic assessment, incorporating the continuous variables of sensitivity and specificity (Reference). The Area under the ROC curve (AUC) serves as an indicator of the diagnostic efficacy of the test. Typically, an AUC exceeding 0.9 is indicative of a highly accurate diagnostic test. The ROC curve analysis was carried out using Graph Pad Prism 7.0 with data derived from RT-qPCR.
Promoter methylation analysis
Library preparation for targeted bisulfite sequencing analysis
The first step involved the fragmentation of 1 µg of total DNA into fragments of approximately 200–300 bp using the Covarias sonication system (Covarias, Woburn, MA, USA). Subsequently, the DNA fragments underwent a series of processes, including repair and phosphorylation of blunt ends, facilitated by a mixture of enzymes such as T4 DNA polymerase, Klenow Fragment, and T4 polynucleotide kinase. The repaired fragments were then subject to 3′ adenylation using Klenow Fragment (3′-5′ exo-), followed by ligation with adapters. These adapters contained 5′-methylcytosine in place of 5′-cytosine and index sequences, and the ligation was executed using T4 DNA Ligase. After the construction of libraries, quantification was performed utilizing a Qubit fluorometer with the Quant-iT dsDNA HS Assay Kit (Invitrogen, Carlsbad, CA, USA). The prepared libraries were subsequently dispatched to Beijing Genomic Institute (BGI), China, for the purpose of targeted bisulfite sequencing. Once the sequencing was completed, the methylation data was subjected to a normalization process, resulting in the generation of beta values.
Mutational analysis of SLC31A1 gene
The genomic DNA from a subset of 10 bladder cancer samples was subjected to fragmentation, resulting in fragments of 150–200 bp in size. This fragmentation process was carried out using the Covaris M220 Focused-ultrasonicator™ Instrument (Covaris, Massachusetts, USA). Subsequently, libraries for the fragmented DNA and were prepared using the KAPA HTP Library Preparation Kit (Illumina platforms) from KAPA Biosystems (Massachusetts, USA), following the manufacturer’s instructions. For the targeted sequencing of the SLC31A1 gene, a custom Genescope panel (Genecast, Beijing, China) was employed. The prepared DNA libraries were then subjected to a capture process using this panel. The captured samples were subsequently subjected to paired-end sequencing on an Illumina HiSeq X-Ten platform. Finally, the observed genetic mutations were interpreted according to the American College of Medical Genetics and Genomic (ACMG) guidelines 16 and annotated by utilizing the ClinVar database. 17
Immunohistochemical (IHC) analysis
Deparaffinization and rehydration were carried out on paraffin-embedded sections of two bladder cancer tissues and one adjacent control tissue. Antigen retrieval was then performed according to standard protocols. The tissue samples were subsequently prepared for immunohistochemical (IHC) staining using the primary anti-SLC31A1 antibody (abcam, ab133385). Following this, slides were subjected to incubation with an HRP-conjugated anti-rabbit secondary antibody, followed by the application of DAB and counterstaining with hematoxylin. The stained slides were observed under a light microscope for further analysis.
Validation of SLC31A1 expression using The Cancer Genome Atlas (TCGA) datasets and Human Protein Atlas (HPA) database
UALCAN 18 and GEPIA 19 are powerful web-based platforms that have revolutionized the field of cancer genomics and gene expression analysis. UALCAN offers an intuitive interface for researchers to explore and interpret cancer-related data, providing comprehensive insights into gene expression, clinical outcomes, and molecular subtypes. It is particularly valuable for in-depth investigations of TCGA data. GEPIA, on the other hand, is a user-friendly tool for gene expression profiling and interactive visualization. It enables the comparison of gene expression in normal and tumor tissues across TCGA datasets, allowing users to explore potential biomarkers and their relevance in various cancers. Both UALCAN and GEPIA have become indispensable resources for researchers and clinicians seeking to understand the intricacies of cancer biology, aiding in the discovery of diagnostic biomarkers. In the present study, we used both these databases for the validation of SLC31A1 gene expression at the mRNA level across bladder cancer TCGA datasets.
The HPA is a comprehensive and freely accessible resource that provides extensive insights into the human proteome. 20 This database offers detailed information on the expression patterns and localization of proteins in various tissues and cells. With its wealth of immunohistochemistry and immunofluorescence data, the HPA aids researchers in understanding the roles of specific proteins in health and disease. In the present study, the HPA database was used to validate SLC31A1 expression at the protein level across bladder cancer samples.
Validation of SLC31A1 promoter methylation level and mutational analysis across the Cancer Genome Atlas (TCGA) datasets
OncoDB and UALCAN are valuable databases for researchers and clinicians involved in oncology. 21 These databases offer a repository of cancer-specific information, encompassing promoter methylation data. In the current study, we used OncoDB and UALCAN databases to validate the promoter methylation level of SLC31A1 across TCGA bladder cancer patients.
cBioPortal is a powerful web-based platform that facilitates the exploration of complex cancer genomics data. 22 It offers an intuitive interface for researchers to interactively analyze and visualize multidimensional cancer datasets, encompassing genetic mutations and clinical information. Researchers and clinicians rely on cBioPortal to gain insights into the molecular intricacies of cancer, enabling the discovery of potential therapeutic targets, biomarkers, and personalized treatment strategies. In our study, the cBioPortal database was utilized for the mutational analysis of SLC31A1 across the TCGA bladder cancer patients.
Survival analysis of SLC31A1
The KM plotter tool is an invaluable resource for survival analysis in cancer research. 23 It provides researchers with a user-friendly platform to assess the impact of specific genes on patient survival outcomes. By analyzing large-scale clinical datasets, the KM plotter helps identify potential prognostic markers and therapeutic targets. In the present research, along with UALCAN, the “pan-cancer” survival analysis feature of the KM plotter tool was utilized to perform survival analysis of SLC31A1 in bladder cancer patients.
Gene enrichment analysis
UALCAN was used to identify positively correlated genes with SLC31A1 in bladder cancer patients. After identification, all genes were then subjected to gene enrichment analysis employing the DAVID tool. 22 DAVID is a bioinformatics tool that facilitates the functional analysis of large gene lists. Researchers can gain insights into gene function, pathways, and biological processes, aiding in the interpretation of high-throughput genomics data.
Exploration of SLC31A1 expression regulatory drugs
DrugBank is a prominent resource for comprehensive information on drugs, including their interactions, mechanisms of action, and therapeutic applications. 24 In our study, we harnessed the capabilities of DrugBank to investigate drugs that may regulate the expression of SLC31A1 in the treatment of bladder cancer.
Cell culture
The human bladder cancer cell line (RT4) was sourced from the Institute of Cell Research, Chinese Academy of Sciences in Shanghai, China. The RT4 cells were cultivated in McCoy’s 5A Medium (Gibco, Life Technologies, Carlsbad, CA, USA) as per the manufacturer’s guidelines. RT4 cells were maintained in culture media supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin (Gibco) under standard conditions of 5% CO2 at 37°C.
Knockdown of SLC31A1 in bladder cancer cell line
The siRNA designed to target SLC31A1 was procured from OBiO Company, with the following sequences: Si126 Forward: 5′-GAUGCCUAUGACCUUCUACUUTT-3′ and Reverse: 5′-AAGUAGAAGGUCAUAGGCAUCTT-3′. The RT4 bladder cancer cell line was evenly seeded in 6-well plates (1 × 105 cells/well) using McCoy’s 5A Medium (Gibco, Life Technologies, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS; Gibco) and 100 μL/mL penicillin and streptomycin. Cells were maintained at 37°C in a 5% CO2 environment. Transfection of 110 pmol of siRNA was carried out using PolyPlus’s jetPRIME® transfection reagent, following the provided instructions. Transfection efficiency was assessed through RT-qPCR and western blotting. RT-qPCR was performed according to the previously mentioned protocol while for western blotting, the cells were collected and the protein was blotted onto polyvinylidene fluoride (PVDF) after using 10% SDS-PAGE separation. After that, the membrane was blocked with 5% bovine serum albumin (BSA, Sigma, CAS, NO: 9048-46-8) in Tris-buffered saline with Tween 20 (TBST) before incubation with specific antibodies, including Anti-SLC31A (abcam, ab129067) and Anti-beta actin (abcam, ab8227) at 4°C overnight. Then, the membranes were incubated with the secondary antibody for 2 h at room temperature (15∼30°C) before the blot samples were imprinted using an Easysee Western Blot Kit (Transgene, Alsace, France).
Cell counting kit-8 (CCK-8) assays
After the transfection process, RT4 cells were plated in 96-well plates at a concentration of 1 × 105 cells/mL and allowed to proliferate for 48 h. To assess cell viability, we employed a CCK-8 kit (provided by Meilunbio, China), following the manufacturer’s instructions. Absorbance measurements at 450 nm were taken using a Bio-Rad model 550 microplate reader.
Colony-forming assays
The RT4 cells were distributed into 6-well plates, with each well receiving 500 cells, and were then cultured for 48 h. Subsequently, the cells were exposed to the correct doses of ATO (2 μM for RT4 cells). Following one-week incubation, the cells were immobilized using 4% paraformaldehyde sourced from Thermo Fischer (USA). Afterward, they were subjected to staining with 2% crystal violet from Thermo Fischer (USA). Colonies that were clearly visible and consisted of at least 50 cells per clone were enumerated under a microscope.
Statistics
In this study, for GO and KEGG enrichment analysis, we employed the Fisher’s Exact test to compute the differences. 25 Correlational analyses were carried out using the Pearson method. A Student t test was employed for making comparisons. All the analyses were conducted using R version 3.6.3 software.
Results
Expression and promoter methylation analyses of SLC31A1 across clinical samples of bladder cancer
RT-qPCR analysis was conducted to assess SLC31A1 expression in 25 bladder cancer samples alongside their paired control samples. The findings demonstrated a significant increase in SLC31A1 expression within bladder cancer tissues compared to adjacent control tissues (Figure 1(a)). Immunohistochemical analysis of two bladder cancer tissue samples and one control tissue sample revealed that SLC31A1 protein expression was also high (staining: medium) in bladder cancer samples as compared to the control sample (Figure 1(c)). Subsequently, we performed targeted bisulfite sequencing analysis of the SLC31A1 gene in both the bladder cancer samples (n = 25) and their corresponding control samples (n = 25). Following bisulfite sequencing and the exclusion of CpG sites with coverage of less than five reads, a minimum of 4 CpG sites for SLC31A1 per sample were obtained. To ensure reliability, only the CpG sites detected in all 25 bladder cancer and paired control samples were retained. Ultimately, three CpG sites were identified for further analysis, with most of these sites being either fully methylated or unmethylated. The results unveiled a striking disparity in the promoter methylation levels of SLC31A1. Specifically, the bladder cancer samples exhibited a significantly lower promoter methylation level compared to their paired control samples (Figure 1(d)). RT-qPCR, immunohistochemistry, targeted bisulfite sequencing, and ROC analysis of SLC31A1 gene across clinical samples sourced from the local bladder cancer patients. (a)–(b) RT-qPCR analysis and expression-based ROC analysis of the SLC31A1 gene across bladder cancer samples paired with adjacent controls, (c) immunohistochemistry-based analysis of SLC31A1 protein expression in two bladder cancer sample and one adjacent control samples, and (d)–(e) Targeted bisulfite sequencing and promoter methylation-based ROC analysis of the SLC31A1 gene across bladder cancer samples paired with adjacent controls. A p-value < .05 was considered significant.
Furthermore, ROC curves were generated for SLC31A1 based on both expression and promoter methylation levels, revealing robust AUC values of 0.946 and 0.900, each with a p-value below 0.05 (Figure 5(b)–(e)). These ROC curves underscore the significant diagnostic potential and exceptional discriminatory capacity of SLC31A1.
Mutational analysis of the SLC31A1 gene across clinical samples of bladder cancer
It is well known that the accumulation of genetic mutations is the cause of expression dysregulation,26–28 so we analyzed genetic mutations in SLC31A1 across bladder cancer samples. The mutational analysis of the SLC31A1 gene was conducted following the described methodology, which included DNA fragmentation, library preparation, targeted sequencing, and adherence to ACMG guidelines for interpretation. The analysis outcomes for SLC31A1 mutation detection in the 10 bladder cancer samples indicated the presence of two benign mutations: NM_001859.4 (SLC31A1): c.308A>G (p.Tyr103Cys) and NM_001859.4 (SLC31A1): c.505G>A (p.Gly169Ser) (Figure 2). Depiction of amino acid alterations in the encoded protein as a result of SLC31A1 mutations detected via Next Generation Sequencing (NGS) in cancer samples sourced by the local bladder cancer patients.
Validation of the SLC31A1 expression using The Cancer Genome Atlas (TCGA) datasets and Human Protein Atlas (HPA) database
To confirm the elevated expression of the SLC31A1 gene in bladder cancer patients from the TCGA dataset, we conducted an analysis using various databases. Initially, we assessed SLC31A1 expression by comparing bladder cancer samples with their corresponding normal controls, utilizing data from the UALCAN and GEPIA databases (Figure 3(a)–(b)) (N = 19, T = 408). Figure 1(a)–(b) illustrates the up-regulation of SLC31A1 gene expression at the mRNA level in bladder cancer patients compared to the normal control samples. To further corroborate these findings, we employed the GEPIA database to examine SLC31A1 mRNA expression in bladder cancer patients of different cancer stages. Results of this analysis showed that SLC31A1 expression was more aggressive in stages III and IV bladder cancer patients as compared to the stage I bladder cancer patients (Figure 3(c)). We extended our investigation to assess the SLC31A1 protein expression level using data sourced from the HPA database. The results revealed an up-regulation of SLC31A1 protein levels in bladder cancer tissue samples, with a medium level of staining. In contrast, no SLC31A1 expression was detected in the normal control tissue sample (Figure 3(d)). SLC31A1 expression validation at both mRNA and protein levels utilizing The Cancer Genome Atlas (TCGA) datasets and the Human Protein Atlas (HPA) database. (a) SLC31A1 mRNA expression level validation results by utilizing bladder cancer TCGA datasets from the UALCAN, (b) SLC31A1 mRNA expression level validation results by utilizing bladder cancer TCGA datasets from the GEPIA, (c) SLC31A1 mRNA expression level validation results across bladder cancer patients of different stages using GEPIA, and (d) SLC31A1 protein expression level validation results by utilizing HPA. A p-value < .05 was considered significant.
Validation of the SLC31A1 promoter methylation level and survival analysis across The Cancer Genome Atlas (TCGA) datasets
Next, we proceeded to validate the promoter methylation level of the SLC31A1 gene using the UALCAN and GEPIA databases in TCGA bladder cancer samples (n = 832) alongside their corresponding normal control samples (n = 38). The analysis results provided compelling evidence of SLC31A1 hypomethylation in bladder cancer tissue samples when compared to the control samples, as demonstrated in Figure 4(a). This observation emphasizes a significant epigenetic alteration associated with SLC31A1 in bladder cancer, potentially contributing to its dysregulation in this disease. SLC31A1 promoter methylation level validation and survival analysis utilizing The Cancer Genome Atlas (TCGA) datasets. (a) SLC31A1 promoter methylation level validation in bladder cancer TCGA dataset from the UALCAN, (b) SLC31A1 promoter methylation level validation in bladder cancer TCGA dataset from the OncoDB, (c) SLC31A1 survival analysis outcomes using UALCAN, and (d) SLC31A1 survival analysis outcomes using KM plotter. A p-value < .05 was considered significant.
To explore whether elevated SLC31A1 gene expression could serve as a prognostic indicator for bladder cancer, we divided bladder cancer cases obtained from the TCGA database via UALCAN and KM plotter databases into high- and low-expression groups based on their SLC31A1 expression levels. Subsequently, we assessed the association between SLC31A1 expression and overall survival (OS) using Kaplan–Meier survival curves. The results indicated a significant (p-value < .05) reduction in the survival rate among bladder cancer patients with high SLC31A1 expression compared to those with low SLC31A1 expression (Figure 4(b)).
Mutational analysis of SLC31A1 across The Cancer Genome Atlas (TCGA) dataset
To scrutinize mutations within the SLC31A1 gene across TCGA bladder cancer patients, we utilized the rich data resource of the TCGA dataset via the cBioPortal database. Our findings, visually depicted in Figure 5(a)–(b), unveiled that SLC31A1 mutations were discernible in a limited subset, specifically, only five out of the 986 samples subjected to analysis. What’s noteworthy is that all the mutations identified within SLC31A1 were of the missense mutation type. Upon closer examination, we discovered that the C>T missense mutations dominated in terms of frequency among the bladder cancer samples featuring mutations (Figure 5(b)). SLC31A1 mutational analysis outcomes across bladder cancer TCGA datasets sourced from the cBioPortal. (a) This panel shows the frequency and types of the observed mutations and (b) This panel shows the summery of the observed mutations.
Gene enrichment analysis
Initially, we employed the UALCAN resource to identify the top 10 genes that exhibited a positive correlation with SLC31A in bladder cancer. This analysis revealed RAD23A, NAA35A, C90rf80, GPR107, SURF4, GAPVD1, HIATL1, PPP6C, NCBP1, and FUBP1 as the prominent candidates which positively correlated with SLC31A1 in bladder cancer (Figure 6(a)). Subsequently, we subjected SLC31A1 and these positively correlated genes to a comprehensive analysis using the DAVID tool to unveil their associated GO and KEGG terms within the context of bladder cancer. In the CC, “NatC complex, XPC complex, N-terminal acetyltransferase complex, Nucleotide-excision repair, and DNA repair complex, etc.” were significantly associated with the analyzed set of genes (Figure 6(b)). Concerning MF, the “Copper ion transmembrane transporter activity, proteasome binding, damaged DNA binding, and ubiquitin binding, etc.” were closely associated with the analyzed set of genes (Figure 6(c)). In BP, some vital functions including “Copper ion import, cellular response to cisplatin, copper ion transmembrane transport, response to cisplatin, and cellular copper ion homeostasis, etc.” were significantly associated with the analyzed set of genes (Figure 6(d)). The analyzed genes associated KEGG pathways include “Nucleotide excision repair, mineral absorption, platinum drug resistance, and protein processing in endoplasmic reticulum” (Figure 6(e)). Correlation and gene enrichment analysis outcomes of the SLC31A1. (a) A list of the top 10 positively correlated genes with SLC31A1, obtained via UALCAN, (b) SLC31A1 and its correlated gene’s associated CC terms, (c) SLC31A1 and its correlated gene’s associated MF terms, (d) SLC31A1 and its correlated gene’s associated BP terms, (e) SLC31A1 and its correlated gene’s associated KEGG terms. A p-value < .05 was considered significant.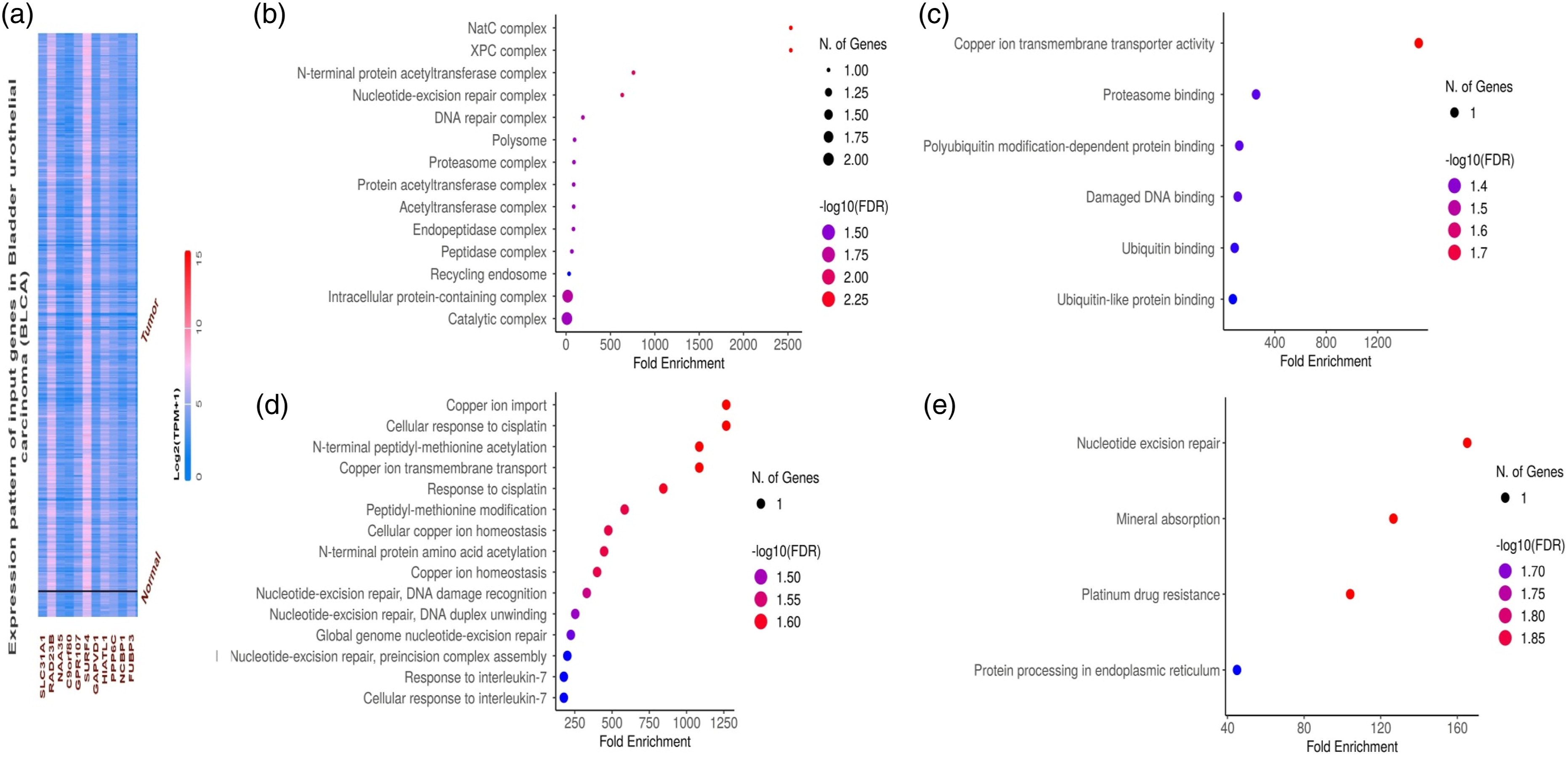
Functional verification of the in vitro and in vivo roles of SLC31A1 in bladder cancer
Following this, we proceeded with the silencing of SLC31A1 in RT4 cells using specific siRNA (Si126), and subsequently assessed the efficiency of this silencing approach at both mRNA and protein level through RT-qPCR and western blot analyses. The results, depicted in Figure 7(a)–(b), clearly illustrated a significant decrease in the mRNA and protein expression of SLC31A1 in the transfected RT4 cells in comparison to the control RT4 cells. Knockdown of SLC31A1 impairs the growth and metastatic potential of bladder cancer cells (RT4). (a)–(b) The transfection efficiency of si126-SLC31A1 was checked at both mRNA and protein levels with the help of RT-qPCR and western blot analyses, (c) RT4 control and transfected cells were analyzed proliferation, (d)–(e) Colony formation.
We further assessed the impact of SLC31A1 knockdown on cell proliferation, and CCK-8 assay results demonstrated a significant reduction in the proliferative ability of the transfected RT4 cells upon SLC31A1 knockdown (Figure 7(c)). Additionally, findings from the colony formation assay revealed a substantial decrease in the number of colonies formed by the transfected RT4 cells compared to control RT4 cells (Figure 7(d)–(e)).
Exploration of SLC31A1 expression regulatory drugs
DrugBank-based SLC31A1-associated drugs.

Discussion
Bladder cancer represents a significant medical challenge, with variable outcomes and limited therapeutic options. 29 The recently reported pathophysiological role of cuproptosis may provide new insight into anticancer treatments. SLC31A1, a widely expressed protein, serves as the central regulator for copper uptake in most cells.30,31 Notably, cuproptosis, a recently discovered cell death mechanism hinging on mitochondrial respiration and the tricarboxylic acid cycle, has been emerging in the spotlight. 32 SLC31A1 is currently under the spotlight as a potential biomarker in cancer therapy and may influence chemoresistance in a few cancer types.33,34 The high-affinity copper uptake protein 1 (CTR1), a product of the SLC31A1 gene, plays a fundamental role in cellular copper uptake. 35 Recent research unveiled a fascinating facet, indicating that CTR1 can act as a redox sensor, driving neovascularization. 36 Additionally, a compelling connection has been observed between CTR1 and Programmed death-ligand 1, prompting clinical trials evaluating copper chelators as potential immune checkpoint inhibitors.37,38 Our study analyzed the involvement of SLC31A1 in bladder cancer to enhance our understanding of its role in this context. The findings provide critical insights into the role of SLC31A1 in bladder cancer progression, with implications for diagnosis, prognosis, and potential therapeutic interventions.
In our study, we observed a notable overexpression of SLC31A1 in bladder cancer tissues compared to normal controls. The implications of elevated SLC31A1 expression in bladder cancer are multifaceted. Copper is a cofactor for numerous enzymes involved in processes like angiogenesis, antioxidant defense, and cell proliferation. 39 Therefore, the overexpression of SLC31A1 might facilitate copper availability for these processes, potentially promoting tumor growth.
Increasingly, a mounting body of evidence suggests that genomic mutations play a pivotal role in shaping tumor progression and determining responses to chemotherapy.40–43 For instance, there is substantiated evidence indicating that genetic polymorphisms within SLC31A1 have been linked to resistance to chemotherapy and have been shown to influence clinical outcomes in cancer patients. 44 In our mutational analysis, we identified genetic mutations in SLC31A1 in a very small subset of bladder cancer samples. These mutations were primarily missense and benign mutations. Functional studies are needed to determine whether these mutations affect copper transport and other cellular processes.
Our study also investigated the promoter methylation of SLC31A1 in bladder cancer. We found that SLC31A1 gene promoter regions were hypomethylated in bladder cancer tissues compared to adjacent normal tissues. DNA methylation patterns are essential for regulating gene expression, and hypomethylation can lead to increased gene expression.45,46 The observed hypomethylation of SLC31A1 in bladder cancer has implications for its overexpression. Hypomethylation-induced overexpression may contribute to increased copper transport and subsequently impact tumor progression. These findings highlight the significance of epigenetic alterations in bladder cancer.
The diagnostic and prognostic value of SLC31A1 in bladder cancer is evident. We observed a significant up-regulation of SLC31A1 in bladder cancer samples. Elevated SLC31A1 expression could serve as a diagnostic biomarker, aiding in the early detection of bladder cancer. Our ROC analysis demonstrated the remarkable diagnostic potential of SLC31A1, with a substantial AUC. The clinical utility of SLC31A1 extends to prognosis. Bladder cancer patients with high SLC31A1 expression levels had significantly lower survival rates. These results emphasize SLC31A1’s potential as a prognostic marker. Integrating SLC31A1 expression into clinical practice could enhance bladder cancer patient care by identifying high-risk individuals. Moreover, our gene enrichment analysis unveiled a robust association between genes co-expressed with SLC31A1 and metabolic pathways within the endoplasmic reticulum. These findings indicate that SLC31A1 might exert a pivotal influence on cancer by modulating processes related to metabolism and copper homeostasis. Previous studies also highlighted the involvement of the SLC31A1 gene in similar pathways among other cancers.47–49
The identification of drugs in the DrugBank database that can modulate SLC31A1 expression presents a promising avenue for therapeutic intervention. Six drugs were identified as potential candidates for reducing SLC31A1 expression. Targeting SLC31A1 may offer a novel approach to bladder cancer therapy. However, the journey from drug discovery to clinical application is complex. Further preclinical studies are required to validate the efficacy of these drugs in bladder cancer models. Additionally, their safety and potential side effects need careful evaluation.
While our study provides valuable insights, it has limitations. The sample size is relatively small, and larger cohorts are necessary for robust conclusions. Future research should explore how SLC31A1’s regulation affects tumor microenvironment, immune responses, and therapy responses.
Conclusion
In conclusion, our study sheds light on the pivotal role of SLC31A1 in bladder cancer. Elevated expression, genetic mutations, and hypomethylation of SLC31A1 are all associated with bladder cancer. The clinical implications of SLC31A1 extend to diagnosis, prognosis, and the potential for targeted therapy. Future research endeavors are needed to unravel the mechanistic intricacies and translate these findings into improved patient care and therapeutic strategies in the fight against bladder cancer.
Footnotes
Author contributions
The idea was conceived by Xue-Yi Xue. Yun-Zhi Lin, Wei-hui Liu, Yu-Peng, Hai Cai, and Qing-Shui Zheng performed all the Boinformatics and wet lab experimental work. Yong Wei and Ning Xu wrote first draft of the manuscript which was finalized by Xue-Yi Xue. Lin, YZ, and Liu, WH conceived and designed the research; Wu YP., Cai H, and Zheng QS analyzed and interpreted the data; Wei Y and Xu N wrote first draft of the manuscript which was finalized by Xue XY. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the Natural Science Foundation of Fujian Province, (No.2021J01219).