
Abstract
Objective: Evidence suggests that aldehyde dehydrogenase 2 (ALDH2) offers protection against damage caused by oxidative stress in diverse rodent models. Nonetheless, the effect of Alda-1, a compound that activates ALDH2, on acute lung injury (ALI) induced by air embolism (AE) remains unclear. The objective of this study was to explore the protective effects of Alda-1 in ALI induced by AE. Methods: A rat model of in situ isolated perfused lung was established to investigate AE-induced ALI. Air was infused into the pulmonary artery at 0.25 mL/min for 1 minute. Before inducing AE, different doses (10, 20, or 30 mg/kg) of Alda-1 were given through intraperitoneal injection. Pathological changes in lung tissue were assessed using hematoxylin-eosin staining. We performed Western blot analysis to assess the protein levels of ALDH2,4-hydroxy-trans-2-nonenal (4-HNE), Bcl-2, caspase-3, phosphatidylinositol 3-kinase (PI3K), Akt, IκB-α, and nuclear NF-κB. Results: Notably, AE results were demonstrated as harmful to the lungs, which is evidenced by intensified lung edema and disruption of lung tissue structure. Furthermore, AE caused a decrease in ALDH2 expression, increased accumulation of 4-HNE and MDA, infiltration of neutrophils, increased production of inflammatory cytokines, apoptosis, and upregulation of the PI3K/Akt and NF-κB signaling pathways within the lungs. Administration of a 20 mg/kg dose of Alda-1 alleviated the detrimental effects induced by AE. Conclusion: Alda-1 shows promise in mitigating AE-induced ALI, possibly through the upregulation of ALDH2 expression and suppression of the PI3K/Akt and NF-κB signaling pathways. Further research is warranted to validate these findings and to explore their translational potential in human subjects.
Introduction
Air embolism (AE) can occur due to various activities, such as diving, barotrauma, and clinically invasive procedures, particularly those involving vascular access.1,2 The most frequent site of AE accumulation is the pulmonary artery. 1 Under normal circumstances, the pulmonary vasculature is tasked with air absorption. Nonetheless, when the volume of incoming air exceeds the lung's capacity for absorption, it can result in acute lung injury (ALI). When pulmonary AE interacts with the pulmonary endothelium, the release of inflammatory mediators such as reactive oxygen species (ROS) and cytokines is triggered. Furthermore, AE obstructs the pulmonary microvasculature, resulting in tissue ischemia.3,4 These factors contribute to oxidative stress, lipid peroxidation, neutrophil infiltration, endothelial dysfunction and the subsequent development of lung edema and ALI.4,5 In addition to supportive care, there is currently no effective therapy available for AE-induced ALI. Hence, it is essential to explore novel treatment strategies for ALI induced by AE.
Notably, the longevity and harmfulness of the aldehydes produced during lipid peroxidation at the site of oxidative damage surpassed those of ROS. 6 4-hydroxy-trans-2-nonenal (4-HNE) and malondialdehyde (MDA) are highly reactive endogenous aldehydes commonly generated in tissues experiencing ischemic injury.7-9 The buildup of 4-HNE and MDA culminates in the formation of a cellular “aldehydic load” that results in irreversible tissue damage.7,10,11 Aldehyde dehydrogenase 2 (ALDH2), with abundant expression in the lung, heart, brain, and liver, possesses the capability to metabolize 4-HNE and MDA into non-harmful carboxylic acid. 10 By increasing the expression of ALDH2 through the administration of alpha-lipoic acid, there is a significant reduction in the levels of 4-HNE and MDA, which leads to protective mechanisms enforced against heart ischemia/reperfusion (IR) injury. 12 Furthermore, mice with a deleted ALDH2 gene exhibit heightened 4-HNE accumulation and aggravated oxidative stress in the brain and lung tissues.8,13,14
Alda-1, a small molecule chemical compound, acts as an agonist for ALDH2. It exhibits selective binding to ALDH2, leading to a remarkable 11-fold increase in the catalytic activity of the enzyme. 15 Alda-1 reduced ischemic damage in the heart by inhibiting cytotoxic aldehyde formation. 15 A previous study found that Alda-1 decreased oxidative stress levels and markers of lung endothelial dysfunction induced by heatstroke. 16 Alda-1 reduced immune cell infiltration into alveoli, alveolar epithelial tissue permeability, and oxidative stress responses in hemorrhagic shock-, hyperoxia-, acrolein- and lung IR-induced ALI by activating ALDH2 activity and decreasing the level of accumulation of 4-HNE.11,17-19
Nevertheless, the effect of Alda-1 on AE-induced ALI has yet to be determined. Our hypothesis indicates that Alda-1, by activating ALDH2, has a positive impact on AE-induced ALI. Therefore, this study aimed to examine this hypothesis through the utilization of a rat model for ALI induced by AE in an in situ isolated perfused lung.
Methods
Animals
Male Sprague‒Dawley rats weighing approximately 350 g (±20 g) were used for this investigation, and the animals were treated with compassion in accordance with the regulations laid out by the National Institutes of Health guidelines (National Academy Press, 1996). The research received approval from the Institutional Animal Care and Use Committee of the National Defense Medical Center, with the approval number IACUC-15-179. The rats were maintained in an environment with standard temperature and humidity, where they experienced a 12-h light and dark cycle and had continuous access to both food and water.
Air embolism model in rats
To induce AE, we applied the method outlined in our earlier investigation using an isolated perfused lung in situ model in rats. 9 Briefly, all rats were anesthetized using Zoletil (50 mg/kg) via intraperitoneal injection and were subjected to tracheostomy. Throughout the procedure, the rats were exposed to humidified air with 5% CO2 and maintained with a positive end-expiratory pressure set at 1 cm H2O. The mechanical ventilator was configured to deliver a 3 mL tidal volume with a respiration rate of 60 breaths per minute. After a median sternotomy, we administered heparin at a dosage of 1 U/g of body weight into the right ventricle, followed by performing a cardiac puncture to collect 10 mL of blood. We inserted a polyethylene (PE)-10 catheter into the pulmonary artery, and simultaneously, advanced a PE-30 catheter into the left atrium. The side arm of the cannula allowed continuous recording of pulmonary venous pressure (PVP) and pulmonary arterial pressure. For perfusing the isolated lung, we utilized a physiological saline solution composed of the following constituents: 119 mM NaCl, 4.7 mM KCl, 1.17 mM MgSO4, 22.6 mM NaHCO3, 1.18 mM KH2PO4, 1.6 mM CaCl2, 5.5 mM glucose, and 50 mM sucrose, along with 4% bovine serum albumin. The obtained 10 mL blood sample was subsequently introduced into the perfusate to create a “half-blood” solution prior to initiating recirculation. We utilized a roller pump to ensure a constant flow rate of 8 mL/min. Following the surgical procedure, the rats were positioned on an electronic balance, enabling continuous monitoring of changes in lung weight (LW). We introduced air into the lung through the pulmonary artery catheter at a rate of 0.25 mL/min for 1 min using an infusion pump, which led to the induction of AE injury.3,4
Experimental design
The rats were assigned in a random manner to different groups: a control + vehicle group receiving dimethyl sulfoxide (DMSO) (n = 6), a group receiving AE alone, and groups receiving AE with varying doses of Alda-1 (10 mg, 20 mg, or 30 mg/kg BW) (n = 6 in each group). We administered Alda-1 (Sigma Aldrich Company, St. Louis, MO, USA) dissolved in DMSO via intraperitoneal injection to each animal 30 min before the surgical procedure. The dosages of Alda-1 employed in this investigation were established in accordance with a prior study. 17
Lung weight/body weight and wet/dry weight ratios
The LW/BW and W/D weight ratios were evaluated following previously described methods. 9 In brief, we promptly collected and weighed the right middle lung after the surgery, representing the wet LW. The LW/BW ratios were calculated by dividing the wet LW by the BW. Next, the right middle lung was positioned in a consistently heated incubator at 60°C for 48 h to acquire the dry LW. The W/D weight ratios were calculated by dividing the wet LW by the dry LW.
Vascular filtration coefficient
We determined the Kf value by assessing the change in LW induced by an elevation in venous pressure, following the established methodology. 9 Kf was defined as the y-intercept of the plot (in g·min−1) divided by the PVP (10 cm H2O) and LW and expressed in whole units of g·min−1 cm H2O−1 × 100 g.
Assessment of CINC-1 and TNF-α levels in bronchoalveolar lavage fluid
Commercial mouse ELISA kits (R&D Systems Inc., Minneapolis, MN, USA) were used to measure the levels of CINC-1 and TNF-α in BALF. Quantification was performed according to the protocols provided by the manufacturer.
Lung myeloperoxidase (MPO) activity (Immunohistochemical analyses)
MPO plays a vital role as an inflammatory enzyme capable of inducing both oxidative stress and inflammation. The identification of MPO through immunohistochemical staining was conducted following the previously described methods. 20 In summary, formalin-fixed paraffin lung tissue sections were prepared through the processes of deparaffinization and antigen retrieval pretreatment. We inhibited endogenous peroxidase activity by immersing the sections in a solution of 3% H2O2 and 100% methanol for a duration of 15 min. Subsequently, the lung sections underwent immunostaining with a rabbit polyclonal antibody against MPO (diluted at 1:100, Cell Signaling Technology). After two rinses with phosphate-buffered saline (PBS), the slides were exposed to a rat-specific horseradish peroxidase-conjugated secondary antibody (supplied by Nichirei Corporation, Tokyo, Japan) for a duration of 30 min. We detected the presence of horseradish peroxidase through a chromogenic reaction using diaminobenzidine for 3 min, and subsequently, the lung tissue sections were counterstained with hematoxylin.
Malondialdehyde level in lung tissue
Lung tissue was homogenized in a 1.15% KCl aqueous solution. Following this, 100 μL of the resulting homogenized lung tissue was mixed with a solution containing 200 μL of 8.1% thiobarbituric acid and 700 μL of distilled water. The mixture was heated at 100°C for 30 min and then centrifuged at 3000 × g for 10 min. The malondialdehyde level in the supernatant was quantified by measuring its absorbance at 532 nm and reported in units of nmol/mg protein.
Western blotting
We isolated the cytoplasmic and nuclear fractions from the lung tissue and determined the protein content using a Bradford assay. Subsequently, lung protein lysates (30 μg per lane) were separated via 10%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Immunoblots were then performed in accordance with the method described previously. 9 The blots were subjected to probing with specific antibodies, including anti-ALDH2, anti-4-HNE (diluted at 1:1000, Abcam, Waltham, MA, USA), anti-PI3K, anti-AKT, anti-pAKT, anti-cleaved caspase 3, anti-NF-κB p65, anti-phospho-NF-κB p65, anti-IκB-α (diluted at 1:1000, Cell Signaling Technology, Danvers, MA, USA), anti-Bcl-2 (diluted at 1:1000, Bioss Inc., Woburn, Massachusetts, USA), anti-lamin B1 (diluted at 1:200, Santa Cruz Biotechnology, Dallas, TX, USA), or β-actin (diluted at 1:10,000, Sigma Chemical Company, St. Louis, MO, USA). The data were expressed by determining the relative ratio of the target protein's content to that of the reference protein. The relative ratio of the target protein's content in the control group was established as 1.
Histopathological analysis
We followed previously established methods for the analysis of neutrophil counts and lung injury scoring in the lung tissue, as detailed in reference.2 21 In brief, lung tissues were fixed, sectioned, and subjected to hematoxylin and eosin (HE) staining. Morphological assessments were conducted using light microscopy. At least 10 fields were randomly selected for the evaluation of neutrophil infiltration in the airspace or vessel wall, as well as alveolar wall thickening. Two pathologists, blinded to the experimental groups, independently scored lung damage on a four-point scale: none (0), mild (1), moderate (2), or severe (3). The scores from each pathologist were combined to determine the overall lung injury score. Histological changes were assessed in randomly chosen fields under a 200× magnification.
Statistical analysis
Data analysis was performed using GraphPad Prism 9 statistical software (GraphPad Software, San Diego, CA, USA). The data is displayed in the form of the means ± standard deviations (SD). Based on the findings from our previous experiment, 4 the Cohen's d assumption is calculated as 4.25, which corresponds to a large effect size. We established a two-tailed significance level of 0.05 and an expected power of 0.8. The sample size for each group was determined to be three, which is enough for detecting differences between the two groups, as calculated using GPower version 3.1.9.7. For the comparison between the treatment and control groups, we performed a one-way analysis of variance (ANOVA) followed by post hoc Bonferroni correction. Furthermore, we compared the lung weight gain (LWG) between groups through a two-way ANOVA with repeated measurements, and subsequent post hoc Bonferroni testing. Statistical significance was established with a threshold of p < .05.
Results
Alda-1 administration attenuates AE-induced acute pulmonary edema
The increased levels of inflammation markers in rats with AE-induced lung injury led to enhanced microvascular permeability and the accumulation of fluid in the alveoli. This was evident through higher values of LWG, LW/BW, W/D ratio, Kf, and protein levels in BALF after AE-induced lung injury. However, rats treated with Alda-1 at a dose of 20 mg/kg demonstrated significantly lower LWG, LW/BW, W/D ratio, Kf, and BALF protein levels than rats with AE-induced injury (Figure 1). Alda-1 effectively mitigated acute lung edema induced by AE, as evidenced by the significant improvements in several parameters: (a) LWG, (b) Kf, (c) LW/BW, (d) W/D weight ratios, and (e) protein content in BALF, which were significantly elevated in the AE group. Alda-1 significantly attenuated these increases. Among the different doses tested, 20 mg/kg Alda-1 demonstrated superior efficacy in reducing lung edema. The data, presented as the mean ± SD (n = 6 in each group), indicated statistical significance (*p < .05, **p < .01, ***p < .001) compared to the control + vehicle group and (#p < .05, ##p < .01, ###p < .001) compared to the AE group.
Alda-1 administration decreased AE-induced lung injury scores and neutrophil accumulation
In comparison to the control + vehicle group, the AE group showed significant infiltration of neutrophils in both the intra-alveolar and interstitial areas, as well as significantly thickened alveolar walls. Rats treated with 20 mg/kg Alda-1 before AE challenge displayed reduced infiltration of inflammatory cells in the alveolar space, primarily attributed to a decrease in neutrophil recruitment. Histological examination of lung tissue sections confirmed significantly higher lung injury scores in rats exposed to AE than in control rats. However, the elevated lung scores observed in AE-exposed rats were significantly attenuated by treatment with 20 mg/kg Alda-1 (Figure 2). Alda-1 attenuated AE-induced ALI as observed through various assessments: (a) HE staining of lung tissue exhibited increased alveolar septal thickness and neutrophil infiltration in the lung interstitium and alveolar space in the AE group when compared to the control + vehicle group. However, treatment with Alda-1 at a dosage of 20 mg/kg ameliorated alveolar damage and reduced neutrophil recruitment; (b) Alda-1 administration at 20 mg/kg significantly reduced AE-induced lung injury scores; (c) The number of neutrophils per high-power field was elevated in the AE group, while the administration of Alda-1 at a dosage of 20 mg/kg significantly reduced the count of infiltrated neutrophils in the context of AE-induced injury. The data, presented as the mean ± SD (n = 6 in each group), demonstrated statistical significance (***p < .001) compared to the control + vehicle group and (###p < .001) compared to the AE group.
Alda-1 administration increased ALDH2 expression and diminished 4-HNE expression
Western blot analysis revealed a notable decrease in ALDH2 protein expression and an increase in 4-HNE protein expression in lung tissue after AE induction. However, administration of 20 mg/kg Alda-1 notably enhanced ALDH2 protein expression while decreasing the expression of 4-HNE protein after AE-induced injury (Figure 3). Alda-1 treatment enhances ALDH2 activity and diminishes 4-HNE expression in the context of AE-induced injury, as confirmed through Western blot analysis of (a) ALDH2 and (b) 4-HNE protein levels in lung tissue, with β-actin used as a loading control. AE significantly suppressed ALDH2 expression and induced upregulation of 4-HNE protein expression in lung tissue. Nonetheless, the administration of Alda-1 produced a substantial reversal of these effects induced by AE. The data, presented as the mean ± SD (n = 6 in each group), demonstrated statistical significance (***p < .001) compared to the control + vehicle group and (#p < .05 and ###p < .001) compared to the AE group.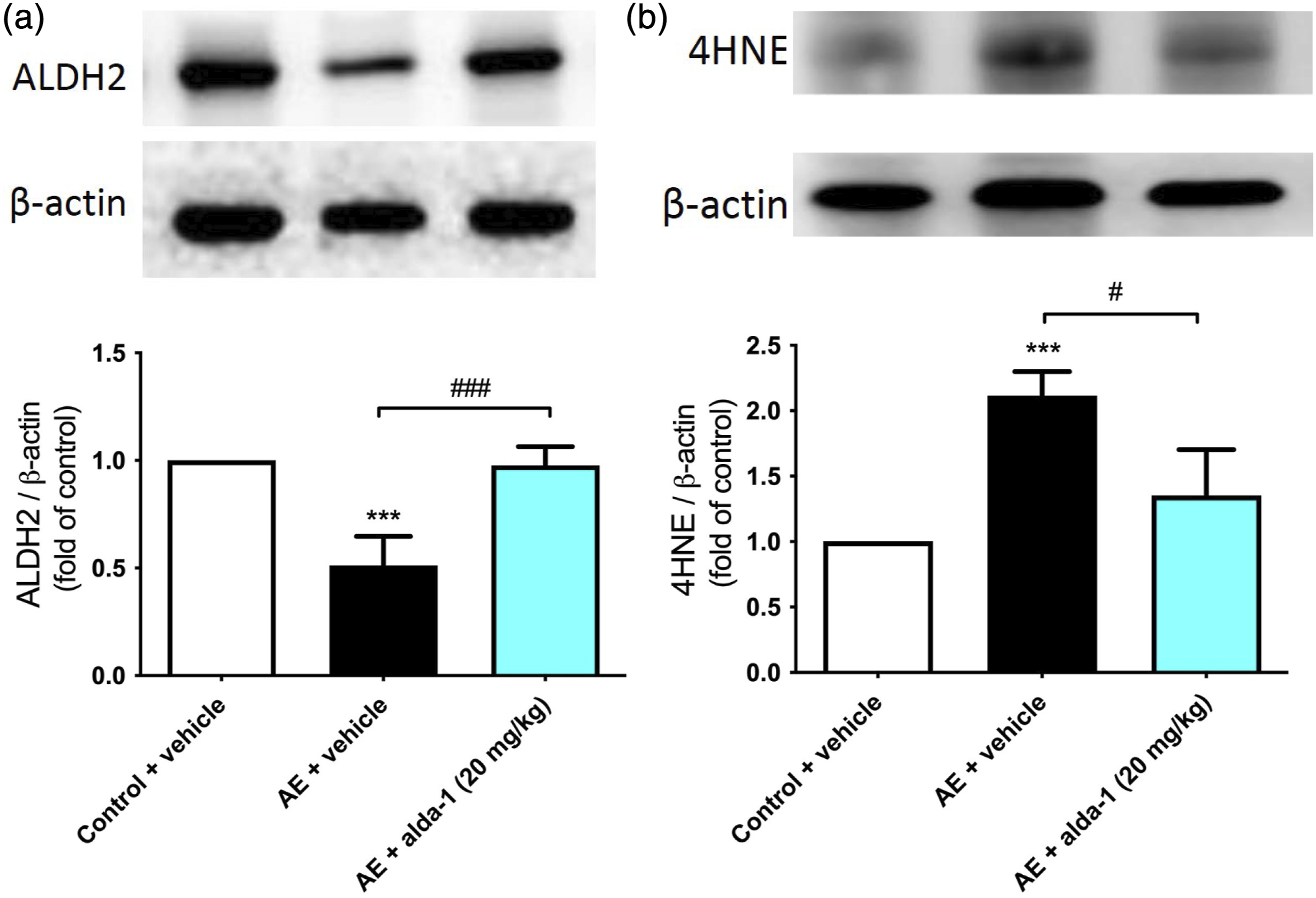
Alda-1 administration ameliorates AE-induced MPO and MDA expression in lung tissue and the production of proinflammatory cytokines in BALF
AE pathogenesis is characterized by the presence of oxidative stress. To evaluate oxidative damage in lung tissue, MPO expression and MDA levels were determined as indicators of peroxynitrite formation. Examination of lung tissue samples revealed a marked rise in the count of MPO-stained cells and MDA levels in the AE group compared to the control + vehicle group. Nevertheless, the administration of Alda-1 at a dosage of 20 mg/kg significantly mitigated this increase, signifying the potential of Alda-1 to alleviate oxidative damage. In addition, BALF samples were obtained from rats subjected to Alda-1 treatment and control rats following AE-induced injury, and the concentrations of TNF-α and CINC-1 were assessed using ELISAs. Consistent with the increased infiltration of neutrophils, the concentrations of TNF-α and CINC-1 in BALF were significantly increased following AE injury. However, the administration of 20 mg/kg Alda-1 effectively lowered the TNF-α and CINC-1 levels that were elevated as a result of AE induction. These findings support the observation that Alda-1 administration mitigates the overall oxidative stress and inflammatory response in AE injury (Figure 4). Alda-1 administration resulted in a reduction in AE-induced (a) MPO activity and (b) MDA levels in lung tissue and (c) TNF-α and (d) CINC-1 levels in BALF. Immunohistochemical staining of consecutive paraffin-embedded sections from AE-induced rats displayed a heightened count of MPO-positive cells, as indicated by the black arrow at 200× magnification. Furthermore, there was an increase in the levels of MDA within lung tissue, as well as elevated TNF-α and CINC-1 levels in BALF after AE injury. Notably, the administration of Alda-1 at a dose of 20 mg/kg significantly mitigated these effects. The data, presented as the mean ± SD (n = 6 in each group), showed statistical significance (*p < .05, ***p < .001) compared to the control + vehicle group and (##p < .01, ###p < .001) compared to the AE group.
Alda-1 administration attenuates AE-induced apoptotic marker expression, the PI3K/Akt pathway, and NF-κB activation
This experiment investigated Bcl-2, known for its antiapoptotic properties, and examined the activity of caspase-3, which serves as an indicator of the level of apoptosis during AE injury. We conducted Western blot analysis on lung tissues to assess the levels of cleaved caspase-3 and Bcl-2 proteins. Notably, the rise in cleaved caspase-3 expression and the decrease in Bcl-2 protein levels observed after AE-induced injury were substantially mitigated by the administration of Alda-1 at a dosage of 20 mg/kg. Given that AE stimulation has been documented to trigger the activation of the NF-κB pathway, which is subject to modulation by the PI3K/Akt signaling cascade,5,22 we conducted Western blot analysis on lung tissues from both groups to evaluate the status of the PI3K/Akt pathway and NF-κB expression. As illustrated in Figure 5(c)–(f), the levels of PI3K, p-Akt, and NF-κB expression within the nucleus exhibited a significant increase, whereas the expression levels of IκB-α in the cytosol markedly decreased in the AE group when compared to the control + vehicle group. However, the heightened levels of PI3K/Akt and NF-κB signaling observed following AE-induced injury were effectively attenuated by the administration of Alda-1 at a dosage of 20 mg/kg. The administration of Alda-1 resulted in a reduction of AE-induced lung apoptosis, as demonstrated by Western blot analysis of (a) Bcl-2 expression and (b) cleaved caspase-3 protein expression in lung tissue. The expression levels of Bcl-2 and cleaved caspase-3 were normalized to β-actin, and representative blots are provided. AE significantly reduced Bcl-2 expression and induced cleaved caspase-3 protein expression in lung tissue. However, the treatment with Alda-1 at a dosage of 20 mg/kg substantially reversed these apoptotic effects induced by AE. Moreover, Alda-1 exhibited inhibitory effects on AE-induced activation of the PI3K/Akt and NF-κB signaling pathways. Protein levels of (c) PI3K, (d) Akt and p-Akt, (e) IκB-α, and (f) NF-κB were analyzed, with Lamin B1 and β-actin serving as loading controls for nuclear and cytoplasmic proteins, respectively. After AE exposure, there was a significant increase in PI3K, p-Akt, and NF-κB protein levels, while IκB-α protein levels significantly decreased in lung tissue. Notably, Alda-1 treatment effectively attenuated the expression of p-Akt and PI3K proteins, nuclear translocation of NF-κB, and the degradation of IκB in the context of AE-induced injury. The data, presented as the mean ± SD (n = 6 in each group), exhibited statistical significance (*p < .05, **p < .01***, p < .001) compared to the control + vehicle group and (#p < .05, ##p < .01, ###p < .001) compared to the AE group.
Discussion
Our study demonstrated that Alda-1, an ALDH2 agonist, provides protection against AE-induced ALI. The administration of Alda-1 led to a reduction in lung edema, alleviated alveolar damage, and resulted in decreased apoptosis and oxidative stress in the context of AE-induced ALI. The mechanisms responsible for the protective effects of Alda-1 encompassed the upregulation of ALDH-2 expression and the removal of reactive aldehydes such as 4-HNE and MDA. Furthermore, Alda-1 inhibited the activation of the PI3K/Akt and NF-κB signaling pathways, resulting in reduced production of proinflammatory cytokines and subsequent ALI.
In the context of AE injury, the generation of excessive ROS is a significantly harmful occurrence that contributes to the destruction of alveoli. 5 The presence of ROS triggers the production of reactive aldehydes, such as 4-HNE and MDA, which further intensify lung damage. ALDH2 serves as the primary enzyme responsible for detoxifying these reactive aldehydes. Mice deficient in ALDH2 exhibited elevated levels of 4-HNE, exacerbating oxidative damage related to stroke. 8 Alda-1 has been associated with the alleviation of various oxidative stress-related conditions, including intestinal, liver, kidney, and brain IR injury, aortic aneurysm formation, aging-induced retinal damage, and experimental autoimmune encephalomyelitis, among others.23-30 In the context of the lungs, Alda-1 can decrease MDA and ROS levels in lung tissue, thereby reducing sepsis-induced lung injury. 31 The administration of Alda-1 has the potential to mitigate the severity of lung IR injury by eliminating 4-HNE in human pulmonary alveolar epithelial cells. 11 Moreover, Alda-1 exhibits the potential to ameliorate immune cell infiltration, protein leakage, and alveolar permeability associated with oxidative stress in hyperoxia-induced ALI. 19 Furthermore, the development of lung injury and dysfunction of the lung microvascular endothelial barrier caused by inhaling acrolein, a highly reactive aldehyde, was prevented and reversed by Alda-1. 18 In line with prior research, our study demonstrated a reduction in ALDH2 expression during AE injury, hampering its effectiveness in eliminating 4-HNE and MDA. As a result, the accumulation of these harmful reactive aldehydes contributes to subsequent lung damage. Nevertheless, the administration of Alda-1 significantly enhanced ALDH2 activity, preventing the buildup of 4-HNE and MDA linked to AE, consequently leading to reduced lung injury. This investigation presents the first evidence indicating that Alda-1 has the potential to mitigate AE-induced ALI in rats by activating ALDH2.
The apoptotic pathway is characterized by the initiation of the caspase cascade, and the critical regulation of apoptosis hinges on the cleavage of caspase-3. In addition, Bcl-2, a regulator of programmed cell death, exhibits strong antiapoptotic properties that promote cell survival. An increasing body of evidence suggests that the excessive production of ROS can lead to apoptosis in alveolar cells. This phenomenon is associated with elevated Bcl-2 expression levels and a concurrent reduction in caspase-3 expression.9,20 An earlier investigation revealed that 4-HNE triggers apoptosis in human pulmonary alveolar cells and augments the permeability of the alveolar capillary barrier. 11 Tsai et al. 25 found that inhibiting ALDH2 leads to heightened accumulation of 4-HNE and increased expression of cleaved caspase-3 in human aortic smooth muscle cells. In contrast, overexpressing ALDH2 can lead to elevated Bcl-2 expression levels and reduce myocardial apoptosis in mice. 32 In a previous study, it was identified that the administration of Alda-1 could elevate Bcl-2 expression, thus ameliorating hepatocyte apoptosis and mitigating liver injury induced by IR. 23 Additionally, Alda-1 treatment resulted in decreased expression of cleaved caspase-3 in the context of liver and heart IR injury.24,33 In a murine model of spinal cord injury, treatment with Alda-1 diminished caspase-3 activity, leading to a decrease in neuronal cell apoptosis. 34 Alda-1 also attenuated apoptosis induced by hyperoxia in lung vascular endothelial cells. 35 Furthermore, the administration of Alda-1 led to a significant increase in Bcl-2 expression and a decrease in caspase-3 expression within lung tissue following intestinal IR injury. 30 Our current findings are consistent with previous research, indicating that Alda-1 administration can enhance Bcl-2 expression levels and reduce cleaved caspase-3 expression during AE-induced injury. These results suggest that Alda-1 can mitigate AE-induced apoptosis in lung tissues.
NF-κB, a well-known transcription factor, controls proinflammatory responses, cell proliferation, and cellular differentiation. The induction of oxidative stress prompts significant activation of the NF-κB pathway, which has been acknowledged as a pivotal inflammatory signaling mechanism in AE-induced ALI.5,9,20 Furthermore, the accumulation of 4-HNE has been documented to trigger the activation of the NF-κB pathway and exacerbate oxidative stress-related pulmonary vascular remodeling. 36 In a prior study, Alda-1 administration diminished the expression of NF-κB induced by angiotensin II in vascular smooth muscle cells. 25 In a rat model of pulmonary arterial hypertension, Alda-1 administration was observed to downregulate the NF-κB signaling pathway as well. 36 In addition, the PI3K/Akt signaling pathway is of vital importance in transmitting intracellular signals and governing numerous cellular processes. 37 A previous study found that the PI3K/Akt signaling pathway participates in the regulation of IκB-α, leading to the activation of the NF-κB pathway in lipopolysaccharide-stimulated macrophages. 38 Lee et al. 39 revealed that the generation of ROS in mitochondria by 4-HNE leads to the activation of the PI3K/Akt pathway, which subsequently triggers the activation of NF-κB in vascular smooth muscle cells. Furthermore, according to the findings by Patil et al., 19 the administration of Alda-1 suppressed p-Akt signaling in lung tissue under hyperoxia conditions. In alignment with prior investigations, we observed a substantial rise in the expression of PI3K/Akt and NF-κB signaling markers in the lung tissue of rats experiencing AE-induced injury. Remarkably, this effect was subsequently counteracted by the administration of Alda-1. This finding indicates that Alda-1 could safeguard against AE-induced ALI by suppressing the expression of the downstream inflammatory signaling pathways of PI3K/Akt and NF-κB.
Limitations
One limitation of our study is that the precise mechanisms through which Alda-1 upregulates ALDH2 expression and subsequently suppresses inflammatory signaling pathways in the context of AE-induced ALI remains unclear. Further investigations are needed to comprehensively understand the underlying molecular processes involved. Additionally, the translational potential of Alda-1 in clinical settings needs to be further explored through well-designed clinical trials to assess its safety and efficacy in treating AE-induced ALI in humans.
Conclusion
In conclusion, Alda-1, an ALDH2 agonist, has the potential to protect against AE-induced ALI by enhancing ALDH2 expression, reducing 4-HNE accumulation, and suppressing inflammatory signaling pathways. Further research is necessary to validate its efficacy in human subjects.
Footnotes
Authors’ contribution
Shu-Yu Wu: experimental conduction; data curation; formal analysis; and writing original draft. Shi-Jye Chu: methodology; research design; writing review and editing. Shih-En Tang: investigation; resources; and project administration. Hsin-Ping Pao: experimental conduction; formal analysis. Wen-I Liao: conceptualization; methodology; resources; research design; supervision; and writing review and editing.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This research received partial support from Tri-service General Hospital, National Defense Medical Center, Taipei, Taiwan (grants: TSGH-E-111209 and TSGH-E-112214) and the Ministry of National Defense-Medical Affairs Bureau, Taiwan (grants: MAB-106-066, MND-MAB-C-11116-111058 and MND-MAB-C03-112011).