
Abstract
Objective:
Celastrol is a compound extracted from a medicinal plant Tripterygium wilfordii which has a broad-spectrum anti-inflammatory effect in traditional medicine. However, the effect of celastrol on acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) is still unknown.
Methods:
We reported that celastrol alleviated LPS-induced acute lung injury by H&E staining, MPO activity and the expression of cytokines in broncho-alveolar lavage fluid. The effect of celastrol on bone marrow-derived macrophages (BMDMs) after LPS treatment was measured by ELISA and Western blotting.
Results:
In vivo, celastrol reduced the LPS-induced lung edema and MPO activity of lung tissue. Furthermore, the production of inflammatory cytokines IL-6, TNF-α, and KC in bronchoalveolar lavage was reduced. In vitro, upon treatment of LPS, celastrol dose-dependently inhibited the expression of iNOS in BMDMs. Meanwhile, the expression of IL-6, TNF-α, and KC in BMDMs were also inhibited by celastrol treatment. Furthermore, we found that celastrol attenuated the phosphorylation of p38 MAPK and MK2, and inhibited the interaction between p38 MAPK and MK2.
Conclusion:
Our data indicate that celastrol has an anti-inflammatory effect on LPS-induced inflammatory response in vivo and in vitro, suggesting celastrol is a promising compound for the treatment of ALI and ARDS.
Introduction
Acute respiratory distress syndrome (ARDS) is a clinical syndrome caused by non-cardiogenic pulmonary edema and characterized with acute lung injury, with high mortality rate. 1 The pathogenesis of ARDS is complex and hasn’t been completely unveiled. Bacterial pneumonia, acute lung injury (ALI) and sepsis are common disorders associated with the development of ARDS.2–4 Sepsis can induce severe inflammatory response and cause multi-organ dysfunction, especially lead to acute lung injury. 5 Lipopolysaccharide (LPS), a glycolipid of Gram-negative bacterial outer membranes, is known as a common endotoxin that leads to ALI and sepsis. The inflammatory response including nitroxide (NO) production and cytokine generation is the main cause of LPS-induced ALI.6,7 Due to the causes of ALI are complicated and the pathogenesis of ALI is still unknown, the clinical treatment is primarily supportive with lung protective ventilation and a conservative fluid management.8,9 There is still no effective medicine for ALI in ARDS. Therefore, it is important to find an effective compound for treating ALI and ARDS.
Dysregulated inflammation is a major pathological change of ARDS in clinical, following with diffuse alveolar injury, lung edema and neutrophil infiltration.10–12 The lung inflammation in ARDS includes the activation of inflammatory cells and the release of inflammatory mediators. 13 Activated neutrophils migrate into alveolar space and produce pro-inflammatory cytokines to exacerbate pulmonary inflammation. 14 In addition to infiltrated neutrophils, macrophages also play a crucial role in the pathogenesis of ARDS. In lung tissue, alveolar macrophages, and tissue-resident macrophages are important immune cells, which protect against pathogen infection. However, activated macrophages also induce the production of cytokines, reactive oxygen, and nitrogen species, which induce endothelial cell and alveolar epithelial cell injury. 15 The high levels of nitrites/nitrates and NO end-products, as well as elevated iNOS, were detected in the plasma and bronchoalveolar lavage fluid (BALF) of ARDS patients.16,17 In addition, inflammatory cytokines have been involved in the development of ARDS. 18 Interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6 are increased in the plasma and bronchoalveolar lavage fluid (BALF) in ALI, which lead to alveolocapillary permeability enhancement and neutrophil infiltration in alveolar and epithelial space. 19 The recruitment and activation of neutrophils further aggravate the inflammation and induce alveolar damage. Therefore, inflammatory cells are potential target for the treatment of ARDS and ALI.
Celastrol is a compound derived from the root bark of Tripterygium wilfordii, which has anti-inflammatory and anti-oxidant effects. 20 It has been known that celastrol is beneficial to several immune-related diseases, such as colitis and cancer.21,22 Celastrol attenuates the DSS-induced colon injury and modulates inflammatory cytokine production and intestinal homeostasis. 21 Additionally, celastrol also has neuroprotective effect on acute spinal cord injury by inhibiting microglial activation and pyroptosis. 23 Although celastrol has protective effects on several diseases, it remains unknown whether celastrol has effects on ALI and ARDS.
In the present study, we found that celastrol alleviated the lung tissue damage and inflammatory response induced by LPS. In addition, celastrol inhibited the expression of inflammatory cytokines and iNOS in LPS-induced BMDMs. Furthermore, celastrol also inhibited the phosphorylation of p38 MAPK and MK2. In conclusion, our study indicates that celastrol has a protective effect on LPS-induced acute lung injury, which is a promising compound for treating ARDS.
Methods
Reagents and animals
Celastrol (C0869), dimethylsulfoxide (DMSO) (D2650), LPS (L2630), and protease inhibitor cocktail (P8340) were obtained from Sigma-Aldrich (St Louis, MO, USA). The ELISA kits for TNF-α (MTA00B), IL-6 (M6000B), and KC (MKC00B) were purchased from R&D Systems (Minneapolis, MN, USA). Antibodies against iNOS (13120), P-p38 MAPK (4511), p38 MAPK (8690), P-MK2 (3042), MK2 (3041), GAPDH (5174), Myc-Tag (9B11), and Flag-Tag (D6W5B) were purchased from Cell Signaling Technology (Danvers, MA, USA). The Hematoxylin and Eosin staining Kit, cell counting kit-8, Bradford protein assay kit and RIPA lysis buffer (P0013K) were purchased from Beyotime (Shanghai, China). Electrochemiluminescence (32209) and Protein G beads (10003D) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The pcDNA 3.1 plasmids harboring Myc-p38 MAPK or Flag-MK2 were purchased from Genomeditech (Shanghai, China).
C57BL/6 mice (6–10 weeks old, specific pathogen-free) were purchased from SLAC Laboratory Animal Corporation (Shanghai, China). Animals were bred and housed at the Shanghai Jiao Tong University Laboratory Center. The procedures involving mice were approved by the Institutional Animal Care and Use Committee at Shanghai Jiao Tong University.
LPS-induced acute lung injury model
Mice were randomly divided into three groups: Vehicle groups, LPS + Vehicle groups and LPS + celastrol groups. Celastrol was dissolved in vehicle (10% DMSO, 60% cremophor, 20% ethanol, and 10% PBS). ALI was induced in 8-week-old male mice with intratracheal injection of LPS (40 µg in 50 µL PBS, 2 mg/kg) for 6 h. For LPS + Vehicle groups and LPS + celastrol groups, mice were administered with an intraperitoneal injection of vehicle or celastrol (20 mg/kg). Mice were anesthetized with pentobarbital sodium (50 mg/kg) by intraperitoneal injection to collect the lung tissue and BALF for analysis. To saving the consumption of mice, we used five mice per group to perform the experiment and got the significant difference between groups.
Histological analysis
The left lobe of lung from each group was fixed by 4% paraformaldehyde, embedded in paraffin, cut into 5 µm sections. The sections fixed in slides were stained with hematoxylin-eosin (H&E) according to the manufacture’s instruction.
Myeloperoxidase (MPO) estimation
The largest right lobes of lungs were homogenized in 50 mmol/L potassium phosphate buffer containing 0.5% hexadecyl trimethyl ammonium bromide (HTAB) and centrifuged to collect the pellet. Then the pellet was resuspended by 0.5% HTAB. The HTAB supernatant was repeatedly frozen and thawed at −80°C three times, frozen for 20 min. After the last thaw, the supernatant was homogenized again, and centrifuged to collect the supernatant. Supernatants were diluted in reaction solution containing 3, 3′, 5, 5′-tetramethylbenzidine (TMB) and H2O2 and incubated at 25°C for 5 min, then measured the change of absorbance at 655 nm over 5 min. MPO activity was defined as the change in the absorbance value per minute at 25°C by 1.0 (ΔA min−1), and MPO activity was expressed as unit mass of MPO enzyme activity (ΔA g−1 min−1).
Analysis of protein concentration in BALF
The BALF was centrifuged at 4°C after collection. The supernatant was collected for analysis of protein concentration using the Bradford protein assay kit according to instruction.
Cytokines concentrations by ELISA
The expression of cytokines TNF-α, IL-6, and KC in BALF and BMDMs medium were measured by using ELISA assay kits according to the manufacture’s instruction.
Bone Marrow-derived Macrophage isolation
BMDMs were obtained from male C57BL/6 mice. Mice were sacrificed by cervical dislocation and execution. The femurs and tibias were separated from legs. The bones were cut off at both ends, and the cells were pipette into a sterile conical tube after filtered through a 40-µm filter. After collection and red blood cell lysis, the cells were cultured in DMEM medium (100 units/mL of penicillin and 100 μg/mL streptomycin), 10% FBS. BMDMs were derived by MCSF (10 ng/mL). At day 3, the cells were cultured in new medium for 4 days. At day 7, BMDMs were planted into 6-wells plates or 12-wells plates for detection.
Cell viability assay
The effect of celastrol on cell viability of BMDMs was detected by cell counting kit-8 (CCK-8) assay. BMDMs were planted in 96-well culture plates with the density of 5000 cells/well and incubated for overnight. Cells were treated with different concentrations of celastrol at 0, 2, 10, and 50 μM for 24 h. Then10 μL of CCK-8 solution was added into each well and incubated for 2 h at 37°C. The absorbance at 450 nm was measured by a microplate reader (ThermoFisher Scientific, USA).
Western blotting
BMDMs were planted in 6-well plates and incubated overnight. Cells were treated with celastrol (0, 2, 10, and 50 µM) for 15 min before LPS (100 ng/mL) stimulation for 18 h or 30 min. Cultured cells were collected in ice-cold RIPA buffer containing a cocktail of protease inhibitors. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred to a nitrocellulose (NC) membrane. The membranes were incubated in 5% skim milk at room temperature for 1.5 h and incubated by specific antibodies at 4°C overnight (1:1000 dilution), followed by incubation with secondary antibodies for 2 h at room temperature. The blots were visualized using an enhanced electrochemiluminescence detection system (solution A: solution B = 1:1). Quantification of Western blots was analyzed with ImageJ software.
Immunoprecipitation
HEK293T cells were transfected with the pcDNA 3.1 plasmids of Myc-p38 MAPK and Flag-MK2 for 42 h. The cells were then treated with celastrol (50 μM) for 6 h, washed with cool PBS and lysed on ice with RIPA lysis buffer with protease inhibitor cocktail. Cell lysate was obtained and part of it was used as input for loading control. The remaining lysate was incubated with anti-Myc antibody for 4 h. Protein-G beads were added and shaken overnight at 4°C. The beads were then eluted with PBS and protein was released by adding loading buffer. Immunoprecipitated sample and total cell lysates fractions (input) were analyzed by immunoblotting.
Statistical analysis
Data are expressed as means ± SEM. The data were analyzed by one-way ANOVA and Tukey post hoc test was performed to determine the significance of differences between two groups. P < 0.05 was considered statistically significant. Analysis and graphing were performed using Prism software (ver. 5.0; GraphPad, San Diego, CA, USA).
Results
Celastrol alleviates LPS-induced ALI
Celastrol is an extract compound derived from medicinal herbs Celastraceae family, and has various bioactivities. However, the effect of celastrol on acute lung injury is still unknown. Therefore, we established the mouse model of LPS-induced acute lung injury. Mice were pretreated with celastrol or vehicle by intraperitoneal (i.p.) injection for 30 min, and then intratracheally (i.t.) challenged with LPS at 2 mg/kg for 6 h to induce lung injury. Hematoxylin-eosin staining (H&E staining) was performed to observe the histopathological damage in the lung. As shown in Figure 1(a), the lung of vehicle-treated group had normal architecture and had no histopathological damage. LPS challenge induced severe lung injury. Whereas, pre-treatment with celastrol at 20 mg/kg significantly alleviated LPS-induced lung injury, with less infiltration of inflammatory cells (Figure 1(a)).
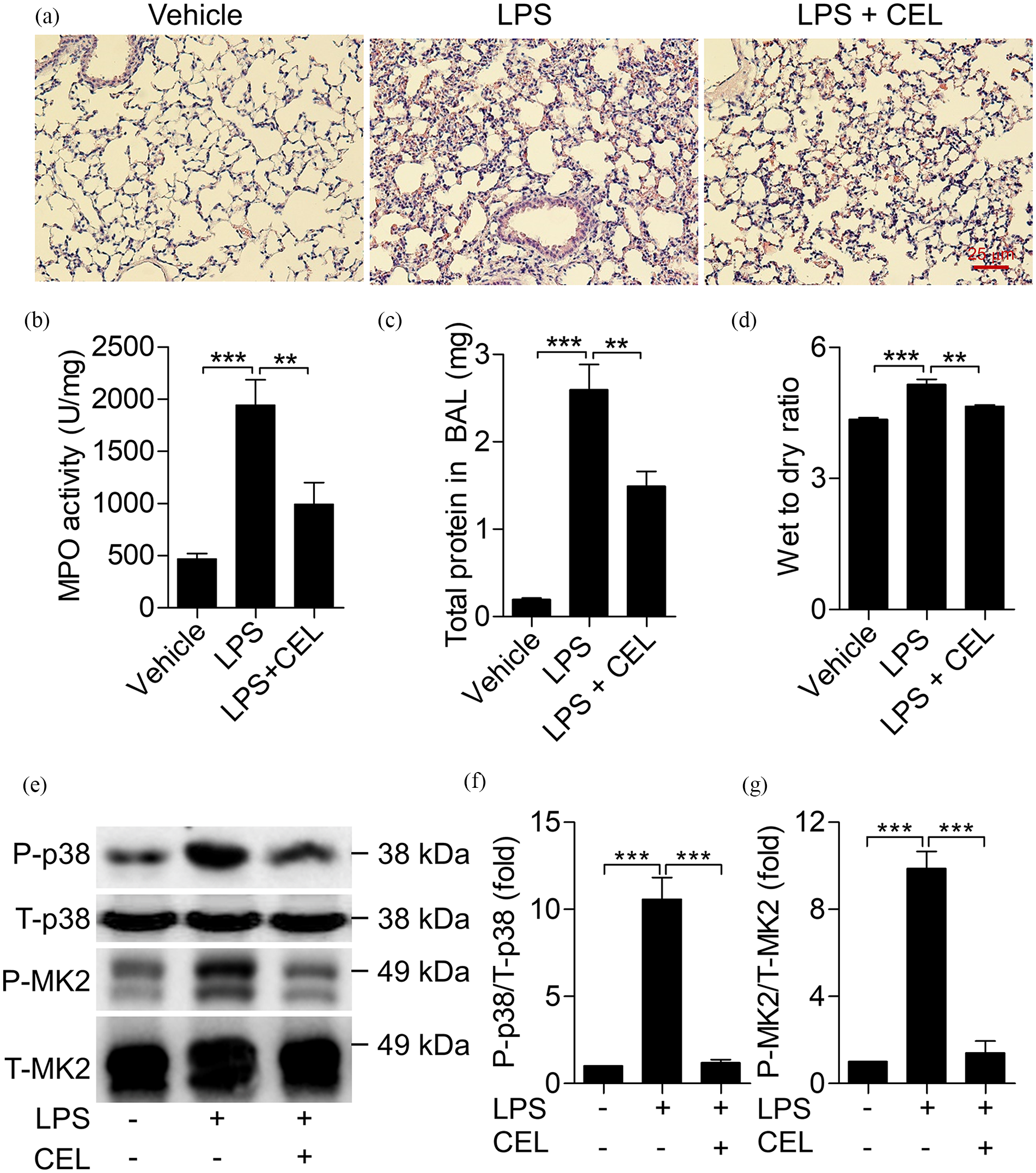
Celastrol alleviates LPS-induced acute lung injury. Mice were intratracheally challenged with LPS (40 µg in 50 µL PBS) for 6 h to induce lung injury. Mice was intraperitoneally injected with celastrol (20 mg/kg) for 30 min before LPS treatment. Lung tissue and BALF were collected for analysis. (a) Hematoxylin Eosin (H&E) staining of the lung tissues (×200). (b) MPO activities of the lung tissues were measured. (c) Total protein concentration of BALF was measured. (d) Wet to dry ratio of the lung tissues. (e) The protein levels of P-p38 MAPK, p38 MAPK, P-MK2, and MK2 in lung were detected by western blot. (f and g) The quantification of the blots in (e) was shown. Data shown are means ± SEM from one representative of three independent experiments (n = 5 mice per group per experiment). Shown are **p < 0.01, ***p < 0.001.
MPO activity of lung tissue is an important marker of pulmonary neutrophil infiltration, which indicates the inflammatory injury of lung. As shown in Figure 1(b), the increased MPO activity after LPS induction was remarkably reduced by administration of celastrol. The total protein concentration in BALF was increased 3-fold after LPS induction. Whereas, with celastrol treatment, the protein concentration in BALF was reduced by around 50% (Figure 1(c)). Celastrol treatment also reduced the wet to dry ratio of lung tissue which was induced by LPS stimulation, indicating that celastrol alleviates the LPS-induced lung edema (Figure 1(d)). Furthermore, we detected the phosphorylation of p38 MAPK and MK2 in lung tissue. The expression of P-p38 MAPK and P-MK2 were induced in LPS-induced ALI lung (Figure 1(e)–(g)). Consistent with above results, the phosphorylation of p38 MAPK and MK2 in ALI lung were decreased by celastrol treatment (Figure 1(e)–(g)). These results suggest that celastrol prevents against LPS-induced acute lung injury.
Celastrol alleviates the inflammatory response
To further determine the protective effect of celastrol on LPS-induced ALI, we detected the production of pro-inflammatory cytokines in BALF. The expression of TNF-α in BALF was increased approximately 8-fold after LPS treatment for 6 h (Figure 2(a)). And the expression of IL-6 and KC was also dramatically increased after LPS induction (Figure 2(b) and (c)). In response to celastrol treatment at 20 mg/kg, the expression of pro-inflammatory cytokines TNF-α, IL-6, and KC was reduced about 50%. These in vivo data suggest that celastrol alleviates LPS-induced pulmonary inflammation.

Celastrol reduces the inflammatory response in LPS-induced acute lung injury. (a–c) The protein levels of TNF-α, IL-6, and KC in BALF were measured by ELISA. Data shown are means ± SEM from one representative of three independent experiments (n = 5 mice per group per experiment). Shown are *p < 0.05, **p < 0.01, ***p < 0.001.
Celastrol inhibits the LPS-induced cytokine production in BMDMs
To explore the protective effect of celastrol on LPS-induced ALI, we assessed whether celastrol can inhibit macrophage activation. With stimulation of LPS (100 ng/mL) for 3 h, activated BMDMs produced inflammatory cytokines, including TNF-α, IL-6, and KC. Pretreatment with celastrol at 2, 10, and 50 µM for 15 min reduced the production of TNF-α, IL-6, and KC in a dose-dependent manner (Figure 3(a)–(c)). Furthermore, we detected the effect of celastrol on BMDMs cell viability in different concentration. After the treatment of celastrol for 24 h, the cell viability didn’t decrease significantly, even in 50 μM (Figure 3(d)). These data indicate that celastrol inhibits the production of inflammatory cytokines after LPS stimulation.

Celastrol inhibits the LPS-induced cytokine production in BMDMs. BMDMs were pre-treated with celastrol for 15 min. And then treated with LPS (100 ng/mL) for 3 h. The cell medium was collected. (a–c) The protein levels of TNF-α, IL-6, and KC in BMDMs medium were measured by ELISA. (d) Cell viability of BMDMs were detected by CCK-8 assay after treated with celastrol (0, 2, 10, 50 μM) for 24 h. All quantitative data shown are mean ± SEM based on triplicate measurement. *p < 0.05, **p < 0.01, ***p < 0.001.
Celastrol inhibits the LPS-induced expression of iNOS in BMDMs
As TLR4 ligand, LPS can induce BMDMs to generate inflammatory mediators, such as NO, reactive oxygen species (ROS), and inflammatory cytokines. 24 When macrophages are activated, the generation of NO is related to the expression of iNOS. As shown in Figure 4, the expression of iNOS was 15-fold after LPS stimulation at 100 ng/mL for 18 h. Pretreatment of celastrol at 2, 10, and 50 µM for 15 min reduced the expression of iNOS in LPS-induced BMDMs in a dose-dependent manner. When BMDMs were treated with celastrol at 50 µM, upon treatment with LPS, the expression of iNOS was reduced to 5-fold. These results indicate that celastrol inhibits the LPS-induced expression of iNOS in BMDMs.
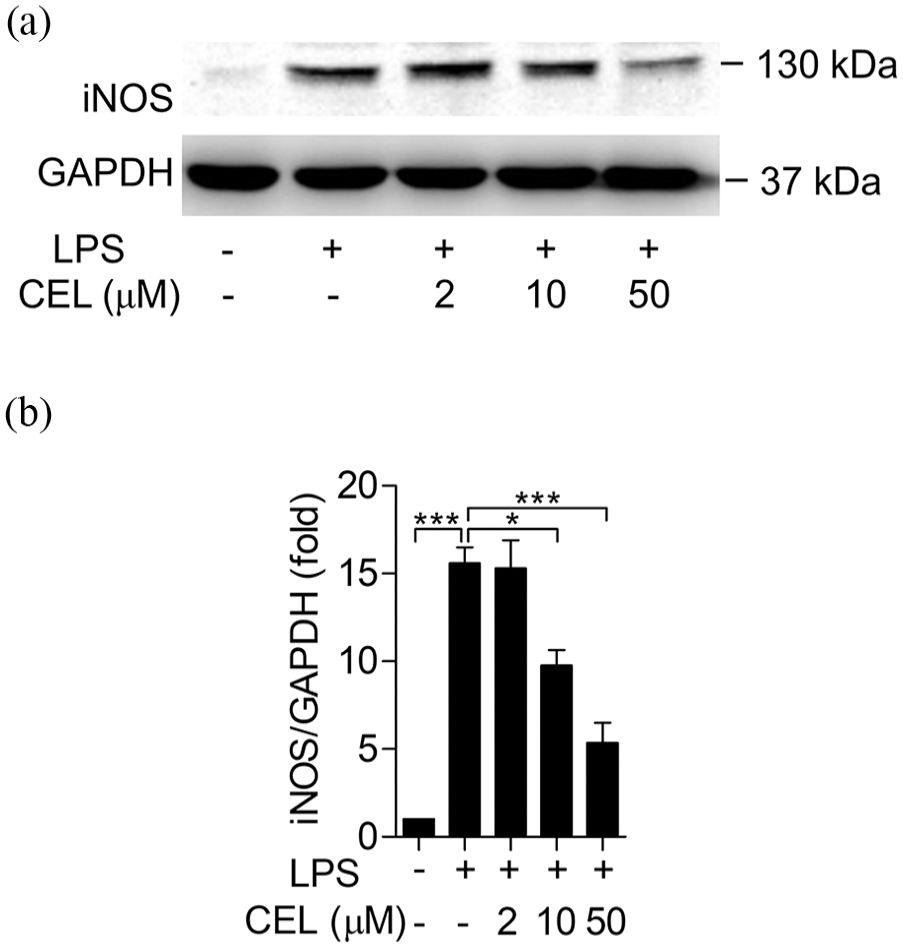
Celastrol inhibits the LPS-induced expression of iNOS in BMDMs. BMDMs were pre-treated with celastrol for 15 min. And then treated with LPS (100 ng/mL) for 18 h. Cells were collected and western blotting was performed. (a) The protein level of iNOS in BMDMs was detected. GAPDH was used as loading control. (b) The quantification of the blots in (a) was shown. All quantitative data shown are mean ± SEM based on triplicate measurement. *p < 0.05, ***p < 0.001.
Celastrol inhibits the phosphorylation of p38 MAPK and MK2 and the interaction between p38 MAPK and MK2
To determine the anti-inflammatory effect of celastrol on macrophage activation, we detected the activation of p38 MAPK and MK2. BMDMs were treated with celastrol at 0, 2, 10, and 50 µM for 15 min, followed by LPS (100 ng/mL) challenge for 30 min. As shown in Figure 5, the phosphorylation of p38 MAPK was increased approximately 6-fold and the phosphorylation of MK2 was induced 50-fold when BMDMs were treated with 100 ng/mL of LPS. With the pretreatment of celastrol, the phosphorylation of p38 MAPK and MK2 was significantly decreased. To further determined how celastrol inhibited the inflammation induced by LPS, we transfected HEK293T cells with expression vectors of Myc-p38 MAPK and Flag-MK2, and then we immunoprecipitated of lysates with anti-Myc. We found that celastrol inhibited the interaction between p38 MAPK and MK2 (Figure 5(d)). In conclusion, celastrol attenuates LPS-induced activation of p38 MAPK and MK2 in BMDMs, also inhibits the interaction between p38 MAPK and MK2.

Celastrol attenuates the LPS-induced phosphorylation of p38 MAPK and MK2. BMDMs were pre-treated with celastrol for 15 min. And then treated with LPS (100 ng/mL) for 30 min. (a) The protein levels of P-p38 MAPK, p38 MAPK, P-MK2, and MK2 in BMDMs were detected by western blot. (b and c) The quantification of the blots in (a) was shown. (d) Immunoassay of HEK293T cells transfected with expression vector pf Myc-p38 MAPK or Flag-MK2, followed by immunoprecipitation of lysates with anti-Myc. All quantitative data shown are mean ± SEM based on triplicate measurement. *p < 0.05, ***p < 0.001.
Discussion
In our study, we found that celastrol alleviated LPS-induced acute lung injury. In vitro, upon treatment with LPS, celastrol inhibited the expression of iNOS and inflammatory cytokines in BMDMs. Furthermore, the phosphorylation of p38 MAPK and MK2 was reduced in response to celastrol pretreatment. These results suggest that celastrol, a compound extracted from medical herbs, alleviates LPS-induced ALI and inflammatory response.
Celastrol is extracted from the traditional medicinal plant Tripterygium wilfordii and has multiple anti-inflammatory activities. 25 Celastrol has multiple molecular targets in different diseases. During the development of hepatic fibrosis, celastrol inhibits the inflammation by activating AMPK-SIRT3 signaling, which attenuates liver fibrosis. 26 Inflammasome plays a critical role in IL-1β production and inflammatory response. Celastrol abolishes the NLRP3 inflammasome activation and reduces inflammasome-mediated inflammatory response. 27 Celastrol reduces lung injury by suppressing the Ednrg/Kng1 signaling pathway and alleviates chronic obstructive pulmonary disease (COPD). 28 According to the published paper and our study, celastrol don’t lead to the damage of lung and also display lower toxic effect on cells or mouse models.29–31 In our research, we found that celastrol alleviated LPS-induced lung injury in mice. The elevated production of iNOS and cytokines induced by LPS was decreased after celastrol treatment. This result is consistent with previous study about the anti-inflammation effect. These experimental results indicated that celastrol has an anti-inflammatory effect on inflammatory response and inflammatory disease. Therefore, celastrol is a potential agent for ARDS treatment.
Inflammatory cells play a key role in ARDS and sepsis-induced lung injury. 14 In the initial stage, the dysregulation inflammation induces acute lung injury. 32 The products of microbial or danger associated molecular patterns (DAMP) bind to Toll-like receptors (TLRs) on the lung epithelium and pulmonary macrophages and active these cells. Because tissue resident macrophages are the first line to defense against the pathogenic microorganisms, 33 activated macrophages release various inflammatory cytokines such as IL-6 and TNF-α,34,35 which lead to neutrophil and macrophage infiltration in the lung of sepsis and ARDS. LPS-induced mouse acute lung injury is a classical model to mimic the critical pathological processes in human ARDS, including neutrophil infiltration and protein-rich BALF.24,36 In clinical research, it has been reported that the block of IL-6 and TNF-α contribute to inhibit inflammatory response in many inflammatory diseases, such as colitis and arthritis. Celastrol can reduce the production of iNOS and decrease the expression of inflammatory cytokines IL-6, TNF-α, and KC, suggesting celastrol not only alleviates ALI, but also relieves other inflammatory diseases.
Mitogen-activated protein kinase (MAPK) is a crucial signaling pathway in inflammation, which leads to inflammatory response and the production of cytokines. 37 The p38 MAPK and its downstream MK2 are important serine/threonine-protein kinases in MAPK, which are associated with inflammatory response, cell migration, and transcriptional regulation. 38 LPS, the agonist of TLR4, can mediate inflammation, which is an interacting process for pathogen and cell. 24 After the activation of TLR4 by LPS, a series of cascades are initiated following the activation of myeloid differentiating factor 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF). Then, the signaling leads to the activation of NF-κB and MAPK.39,40 MK2, as a substrate of p38 MAPK, is activated by p38 MAPK, and modulates the stability and translation of cytokine mRNA to induce the synthesis and expression of pro-inflammatory cytokines such as IL-6 and TNF-α.41,42 Depletion of MK2 protects against sepsis-induced ALI, and decreases the expression of IL-6 and TNF-α. 43 Therefore, p38 MAPK or MK2 inhibitors can inhibit the expression of IL-6 and TNF-α in monocytes. 44 NF-κB and MAPK are crucial targets of anti-inflammatory agents. Many native compounds have anti-inflammatory effects by inhibiting the activation of p38 MAPK and MK2, such as berberine 45 and Hesperdin. 46 In our study, celastrol inhibited the activation of p38 MAPK and MK2, which was associated with the decreased expression of IL-6, TNF-α, and KC in response to LPS treatment. Our results provide the direct evidence that celastrol have a protective effect on the LPS-induced lung inflammation by inhibiting the activation of p38 MAPK and MK2, which confirms the key role of MK2 and p38 MAPK on inflammatory cytokine production in ARDS and MK2 and p38 MAPK are crucial targets for the treatment of ARDS.
Conclusions
Our results unveil that celastrol protects against LPS-induced acute lung injury. Celastrol inhibited the production of IL-6, TNF-α, and KC and reduced the production of iNOS in response to LPS treatment. Meanwhile, the activation of p38 MAPK-MK2 signaling pathway was inhibited by celastrol treatment. Therefore, celastrol is a potent and effective compound for the treatment of ARDS and other inflammatory diseases.
Footnotes
Author contributions
F.Q. conceived the study. Z.C., X.Y., L.Z., and L.S. designed, performed and interpreted experimental data. X.Y. and F.Q. wrote the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (81773741, 81770633, 81973329, 82073858 and 81573438).
Ethics approval
The in vivo experiments were carried out according to the National Institutes of Health Guide for the Care and Use of Laboratory Animal approved by the Biological Research Ethics Committee of Anhui Medical University (LLSC20200649). The date of Ethical Approval is March 1, 2020.
Animal welfare
The present study followed international, national, and/or institutional guidelines for humane animal treatment and complied with relevant legislation.