
Abstract
Introduction
Systemic sclerosis (SSc), also known as scleroderma, is an immune-mediated systemic rheumatic disease that poses considerable clinical challenges due to its high morbidity and mortality.1,2 It is characterized by vasculopathy, immune system abnormalities, and excessive deposition of collagen (fibrosis) in many tissues throughout the human body, causing hardening and thickening. 3 SSc is heterogeneous and multisystemic, and hence it is not common but also shows uncertain development and outcomes of lethality or reduced quality of life.1,4,5 Therefore, exploring the molecular characteristics and mechanism of SSc is important to improve understanding for better management of this disease. 6
A novel type programmed cell death called ferroptosis, which has distinct physical and biochemical characteristics, is driven on by lipid peroxidation via an iron-dependent route. 7 This unique modality of cell death is regulated by multiple cellular metabolic events, so ferroptosis is involved in various organ injuries and degenerative pathologies.8,9 Recently, several studies reported an association between ferroptosis and the immune system. As a biomarker, the ferroptosis-related gene (FRG) signatures can be used to diagnose, forecast, and treat a variety of diseases. 10 A proinflammatory response to ferroptosis has been linked to the emergence of necroinflammatory illnesses in the past. 11 The function of FRGs in SSc is yet unclear. Thus, there is a pressing need for a deeper comprehension of SSc as well as for the development of new biomarkers and treatment targets.
In this study, differentially expressed genes (DEGs) were identified by comparing SSc and normal samples in the GSE125362 and GSE76807 data sets, which were then intersected with the ferroptosis database with the purpose of obtaining the key FRGs. Moreover, investigations of the protein-protein interaction (PPI) network and functional enrichment were performed. The Search Tool for the Retrieval of Interacting Genes (STRING) and Cytoscape software were used to analyze modules. We analyzed SSc microarray data and selected differential gene expression profiles to identify critical pathways and hub genes. We constructed a multifactor regulatory network depending on key hub genes and evaluated immune infiltration. The results might contribute to a better understanding of the molecular pathogenesis of SSc.
Our primary findings involved two aspects: description of FRGs in SSc patients and creation of an SSc regulatory network based on the seven hub genes reported in this study. This study was novel in reporting a potential relationship between ferroptosis and SSc via bioinformatics analysis.
Materials and methods
Microarray data
NCBI-GEO (http://www.ncbi.nlm.nih.gov/geo) 12 is a public functional genomics data repository. Two gene expression data sets, GSE125362 and GSE76807, were downloaded. GSE125362 data set contained eight SSc skin samples and four normal skin samples. GSE76807 contained 10 SSc skin samples and 5 normal skin samples. Besides, we downloaded the FRGs from the FerrDb database (http://www.zhounan.org/ferrdb/).
Basic information of the two microarray data sets from GEO.

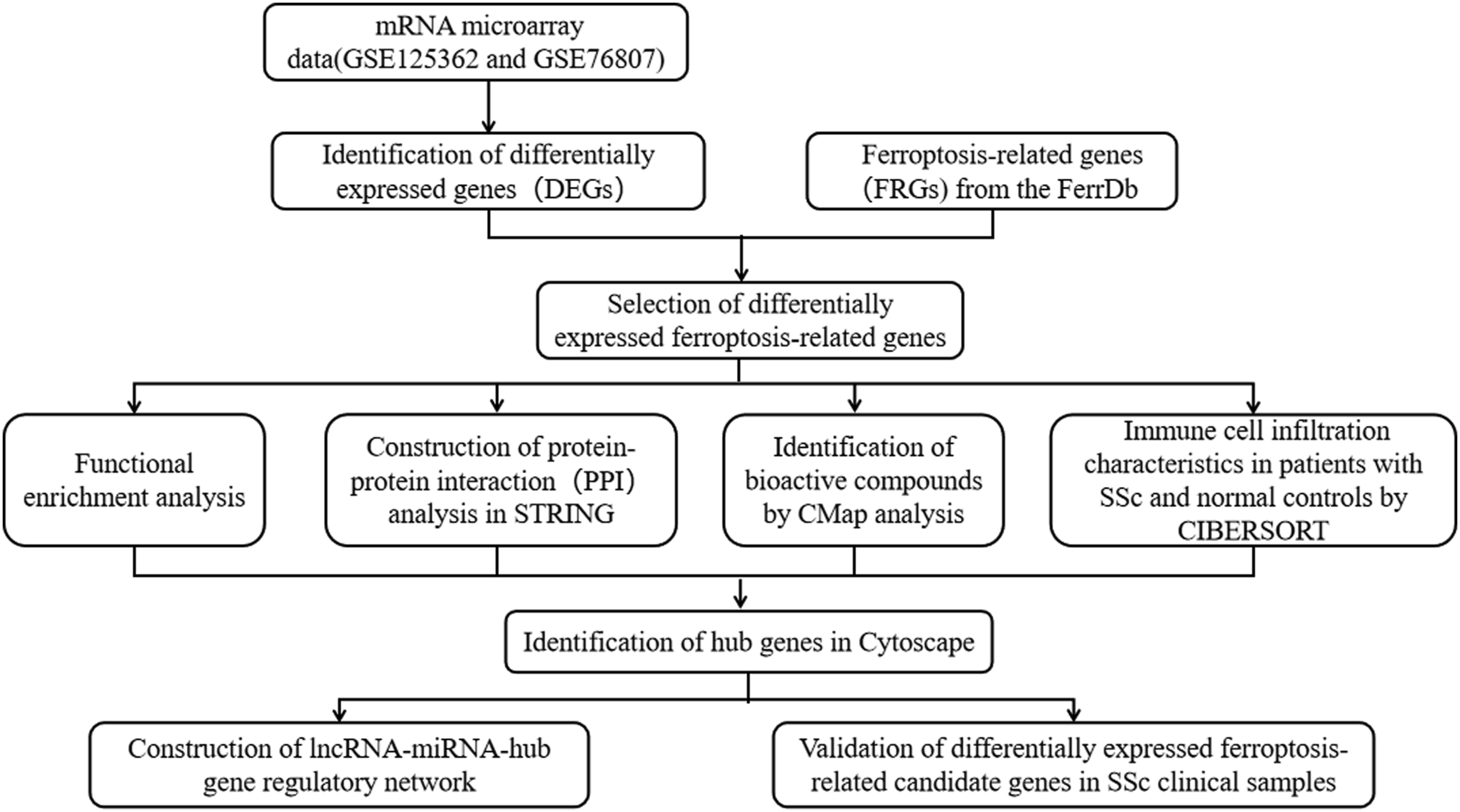
Flow diagram of strategy for data preparation, preprocessing, and analysis.
Identification of FRGs
The initial data was examined using the R language to find DEGs. The data normalization process, which included background correction and conversion to raw data, was conducted out using the normexp method. 13 The DEGs analysis of the data was then performed utilizing Limma package. Besides, the DEGs were screened with the thresholds of |log2 (fold-change)| > 1 and p-value <.05. The heatmap and volcano plot were drawn using the “Heatmap” and “ggplot2” packages in the R software (version 4.0.0). 14 The DEGs and FRGs were intersected to obtain the differentially expressed FRGs.
Functional enrichment and PPI analysis
Online biological information can be found in the DAVID program (http://david.ncifcrf.gov/, version 6.8). 15 The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a database tool for analyzing large-scale molecular data sets produced by high-throughput experimental techniques to better understand high-level biological processes and systems. 16 A key bioinformatics tool for annotating genes and examining their biological processes is the Gene Ontology (GO) (http://geneontology.org/page/download-ontology). The Cytoscape software (version 3.8.1) and STRING (http://string-db.org) were used to establish the PPI analysis of the differentially expressed FRGs. First, the STRING database was used to construct the PPI network. The PPI network was visualized using Cytoscape (version 3.8.1). Subsequently, genes with the top 5% of interaction scores were screened as hub genes using the MCC algorithm of the Cytohubba plugin in Cytoscape software.
miRNet, NetworkAnalyst, and encyclopedia of RNA interactomes
A helpful online resource focused on miRNAs and the chemicals that interact with them is entitled miRNet. 17 For the investigation of gene expression and the creation of interacting networks, NetworkAnalyst is a comprehensive and potent database. 18 The Encyclopedia of RNA Interactomes (ENCORI) focuses on predicting RNA interactions, 19 which was used in this study to anticipate the compounds that lncRNAs targeting selected miRNAs. The top three lncRNAs were chosen based on the strong stringency screening criterion (the number of Ago CLIP-seq experiments was no less than five).
Profiling infiltrating immune cells using CIBERSORT in the skin
To analyze the immune infiltration differences between SSc and normal groups and to evaluate the function of immune microenvironment, we used the online CIBERSORT algorithm (https://cibersort.stanford.edu/). The GSE125362 series matrix files were retrieved from the GEO database. Analysis was done on the variances in 22 immune cells between SSc and normal skin.
CMap analysis
The possible agents for SSc were identified using the Connectivity Map (CMap). The identified DEGs were queried using the Connectivity Map online tool (L1000 platform; https://clue.io/l1000-query). The connectivity scores were determined after proffering a list of 14 upregulated and 10 downregulated hub genes. The proximity between the expression profiles was measured using a score from −1 to 1: a positive score indicated a promoted impact, while a negative score signified an inhibited impact.
Patients
We collected clinical samples from five patients with SSc and five healthy individuals at Fujian Provincial Hospital. Eligible patients had ages between 18 and 80 years, and satisfied the ACR/EULAR 2013 classification criteria for systemic sclerosis. 20 These patients formed the case and control groups, respectively (Table S1). All protocols were approved by the Ethics Committee of the Fujian Provincial Hospital.
RNA extraction and quantitative real-time PCR
Primer sequences used for qRT-PCR.

qRT-PCR: quantitative real-time polymerase chain reaction.
Enzyme-linked immunosorbent assay
The plasma of the participants was collected, and the protein levels of IL-6 (limits of detection: 1.5–1000.0 pg/mL) was detected by the ELISA kit (Roche) according to the manufacture’s protocol.
Statistical analysis
The R software (version 4.0.0) was used for the statistical analysis of the bioinformatics data. The Student’st test was used to evaluate gene expression levels in clinical samples. Statistical significance was set at p < .05.
Results
DEG identification
The differential expression of mRNA between patients with SSc and controls was determined using microarray analysis. After union, 1084 genes were obtained involving 808 DEGs from the GSE125362 data set and 382 DEGs from the GSE76807 data set; 106 DEGs were common between the two data sets. The Venn figure displays that 24 FRGs, including 14 upregulated and 10 downregulated genes, were all substantially differentially expressed between the SSc and control groups when absolute values of logFC >1 and p values 0.05 were met (Figure 2(a); Table S2). The 24 FRGs are plotted in Figure 2(b) and (c). Identification of differentially expressed ferroptosis-related genes (FRGs). (a) Venn diagram of FRGs based on the two GEO datasets. (b) Volcano plot of the 24 FRGs in GSE76807. (c) Volcano plot of the 24 FRGs in GSE125362. Red, upregulation; green, downregulation. (d) Heatmap of 24 FRGs.
GO annotation and KEGG pathway enrichment analyses
To explore the underlying mechanism of ferroptosis in SSc, we used the online tool DAVID to analyze the GO and KEGG of 24 FRGs, shown in Figure 3. GO biological process analysis revealed that the 24 FRGs were significantly associated with negative regulation of cell proliferation, inflammatory response, and positive transcription from the RNA polymerase II promoter. The cell component terms of GO analysis included nucleus, extracellular exosome, and extracellular region. The changes in molecular function were primarily attributed to protein binding, identical protein binding, and iron-ion binding. KEGG pathway analysis revealed that the FRGs were primarily associated with necroptosis, the NOD-like receptor signaling pathway, lipids, and atherosclerosis (Table S3). GO annotation and KEGG pathway analyses. The FRG enrichment of BP, MF, CC and KEGG pathways (p < .05).
PPI and modular analysis
The PPI network complex, with 30 nodes and 50 edges, received a total of 24 FRGs (Figure 4(a)). Seven hub genes were investigated after we applied Cytohubba plugin for additional investigation (Table S4). In Figure 4(b), the particle genes are displayed. PPI network and the significant modules of FRGs. (a) The PPI network of FRGs. (b) Hub genes screened by the cytoHubba plugin.
Construction of the networks between hub genes using miRNAs, transcription factors, and signal molecules
The interactive network analysis was conducted to predict upstream or downstream molecules of seven hub genes. To study how transcription factors (TFs) and miRNAs interact in human skin tissues, the seven hub genes were uploaded to the miRNet. In addition, 246 miRNAs were predicted, and 3 miRNAs, hsa-miR-7-5p, hsa-miR-148b-3p, and hsa-miR-107, were chosen for further study (Figure 5(a)). There were 106 connections between 73 TFs and 7 hub genes in the TF-hub gene regulatory network. The five TFs with the strongest connection to hub genes were IRF8, NFKB1, RELA, EP300, and STAT1. Upstream lncRNAs of the three miRNAs were searched for using the ENCORI database. Two lncRNAs were anticipated: XIST (targeting hsa-miR-7-5p, hsa-miR-148b-3p, and hsa-miR-107) and NEAT1 (targeting hsa-miR-148b-3p and hsa-miR-107) (Figure 5(b)). The interaction network of hub genes in miRNet and Network Analyst. (a)The network of hub genes with transcription factors. The yellow nodes represent hub genes, and the green nodes represent transcription factors. The five transcription factors that connect with at least four hub genes are labeled. (b) The predicted lncRNA-miRNA-hub gene regulatory network. The red diamonds represent lncRNAs, blue diamonds represent miRNAs.
Immune cell infiltration characteristics in patients with SSc and normal controls
Different immune cells’ status of infiltration into epidermal tissues varied noticeably (Figure 6(a)). The majority of invading cells, particularly in SSc skin tissues, were resting mast cells. Activated natural killer (NK) cells, resting dendritic cells, and resting NK cells all demonstrated varying levels of infiltration in SSc patients as compared to healthy controls. Resting dendritic and NK cells were downregulated in SSc skin tissues, whereas activated NK cells were upregulated (Figure 6(b)). Resting dendritic cells positively correlated with resting NK cells (r = 0.06). Activated NK cells had inverse correlations with resting NK and dendritic cells (r = −0.39 and −0.28, respectively). Resting NK cells were strongly negatively associated with CXCL2, IL6, and CYBB (r = −0.68, −0.63, and −0.61, respectively). Resting dendritic cells were also negatively associated with IL6, CXCL2, and CYBB (r = −0.63, −0.61, and −0.52, respectively). However, activated NK cells were strongly positively associated with TLR4 and CYBB (r = 0.82 and 0.75, respectively). The remaining several types of immune cells also had weakly to moderately negative correlations with the expression of most of the hub genes as displayed in the heatmap. The associations between hub gene expression and differently infiltrating immune cells are shown in Figure 6(c). Immune infiltration of SSc skin tissues compared with normal tissues. (a) Stack bar chart of the proportions of immune cell infiltration. (b) Box plot of proportions of immune cell infiltration. The significance markers are shown as follows: ns, p > .05; *, p < .05. (c) Heatmap of correlations of FRGs with differentially infiltrated immune cells.
Identification of bioactive compounds by CMap analysis
Top 10 compounds predicted via a connectivity map to have activity against SSc.

Validation of differentially expressed ferroptosis-related candidate genes in SSc clinical samples
According to the interaction scores by using the Cytohubba plugin in Cytoscape software, we examined the top three differentially expressed candidate hub genes and detected their expression in clinical samples by qRT-PCR and ELISA (Figure 7). Our results showed that IL-6 and CYBB in the clinical samples of SSc demonstrated statistically significant changes in expression. IL-6 was higher expressed in the SSc group than in the control group. CYBB was lower expressed in the SSc group than in the control group and the expression of NOX4 showed no significant differences. Verification of the expression of the ferroptosis-related candidate genes in SSc clinical samples (a) Raising protein levels of IL-6. (b) Increased expression of CYBB. (c) Unchanged levels of NOX4. The blue and red boxes represent SSc and healthy samples, respectively. The expressions were assessed by ELISA in A and by RT-qPCR in B-C. p-values were calculated using the Student’s t test. IL-6: interleukin-6; NOX4: NADPH oxidase 4; CYBB: cytochrome b-245 beta chain; NS: non-significant.
Discussion
SSc, a refractory autoimmune disease that affects patients, is highly teratogenic and disabling, posing a formidable challenge to clinicians and researchers. Uncovering the mechanisms driving SSc pathogenesis is critical for developing novel therapeutics for this patient population. Although numerous etiological studies on SSc have been performed, the immunological pathogenesis of SSc and relevant therapeutic targets remain undetermined. Previous studies have shown that ferroptosis is closely related to inflammation and the immune system. However, the role of ferroptosis in the development and progression of SSc has not been previously documented.
In order to investigate the potential roles of 24 differentially expressed FRGs, enrichment analyses were performed in this work. The enrichment analysis showed that these genes were mainly related to cell proliferation, inflammatory response, phospholipid peroxidation, and apoptotic processes. These elements had a close connection to the development of SSc. The FRGs were predominantly linked to the necroptosis pathway, according to KEGG pathway analysis. When apoptosis signaling was blocked, the necroptotic pathways were activated and the dying cells had the potential to initiate innate immune responses via the production of damage-associated molecules, resulting in an inflammatory response. 21 Besides, some studies demonstrated that the necroptosis pathways played important roles in autoimmune diseases.22,23
Next, we identified seven ferroptosis-related hub genes, including CYBB, IL-6, TLR4, CXCL2, JUN, NOX4, and LY96. In clinical samples, we found that the expression levels of IL-6 and CYBB were consistent with the biological information of the mRNA chip. IL-6 is involved in inflammatory processes in numerous diseases. 24 It is highly expressed in SSc tissues, induces leukocyte aggregation, and promotes SSc inflammation by regulating the expression of chemokines. 25 However, there are no distinct reports concerning the involvement of IL-6 in ferroptosis in SSc. Our study might serve as a basis for further research on the regulation of ferroptosis in SSc by IL-6; nevertheless, further follow-up experiments are warranted to confirm our hypothesis. Yang et al. demonstrated that CYBB can transport electrons through the plasma membrane to generate ROS and activate ferroptosis. 26 Martin et al. revealed that CYBB was induced in Bleomycin mice and repressed by EHP-101 (an oral lipid formulation of dual PPARγ/CB2 receptors activator) treatment, which is mainly regulated by TGF-β. 27 But the functions of CYBB in SSc have not yet been investigated. Our results might provide directions for additional research on these topics.
A practical method for predicting cross-talk networks and vetting possible biomarkers in SSc is provided by bioinformatics. Several miRNAs and lncRNAs have been implicated in the initiation and development of SSc. 28 The majority of earlier research on the connection between noncoding RNAs and ferroptosis, however, concentrated on different malignancies. Using the miRNet database, this study built hub gene networks using miRNAs and transcription factors and discovered three important miRNAs, including hsa-miR-107, hsa-miR-148b-3p, and hsa-miR-7-5p. Mollazadeh reported that the lentivirus overexpressing miR-148b-3p increased the osteogenic differentiation capability of human bone marrow–derived mesenchymal stem cells, and this miR could be applied as a therapeutic modulator to optimize bone function. 29 Furthermore, a lack of miR-7-5p expression led to increased levels of transferrin receptor, promoting the uptake of iron and the production of lipid reactive oxygen species and demonstrating that DOX-induced ferroptosis occurred in cardiomyocytes. 30 miR-7-5p was involved in mitochondrial homeostasis impairment and endothelial dysfunction. 31 Using the ENCORI database, we also predicted the upstream lncRNAs of three miRNAs, and we identified the two lncRNAs with the strongest experimental support. In fibroblast-like synoviocytes and osteoclasts, the proinflammatory and proliferative pathways are upregulated due to the sequestration effect exerted by lnc-XIST overexpression on miRNAs. 32 NEAT1 promoted the activation of inflammasomes in macrophages. 33 Hence, we hypothesized that the XIST/hsa-miR-7-5p or hsa-miR-107 or hsa-miR-148b-3p/hub gene axes and NEAT1/hsa-miR-107 or hsa-miR-148b-3p/hub gene axes might modulate ferroptosis in SSc pathophysiology. The foundation for investigating the underlying pathways of ferroptosis in SSc pathogenesis would be laid by finding five significant transcription factors: TRF8, NFKB1, RELA, EP300, and STAT1.
Less studies have examined immune infiltration in SSc, despite the fact that immune infiltration in malignancies continues to catch the attention of researchers. However, other research found that SSc skin tissues had an increase in activated NK cells and a decrease in resting dendritic and NK cells. When TLRs are stimulated, NK cells release pro-inflammatory cytokines such TNF-α, IL-6, and macrophage inflammatory protein-1 and interact with other immune cell types, including DCs. 34 DCs provide antigens and stimulate immature T and B lymphocytes. Research revealed that cellular and humoral immunity were involved in the etiology of SSc. 35 Various immune cells could infiltrate skin tissues. Circulating autoantibodies are common in scleroderma. An elevated T cell ratio was found in peripheral blood samples of patients with scleroderma. Activated T cell clones might release chemotactic lymphokines for fibroblasts and stimulate fibroblast proliferation and collagen synthesis, causing endothelial cell damage and fibrosis. 36 As a result, the infiltration of immune cells played a sophisticated role in the pathogenesis of SSc, and more research is urgently required to clarify the results of this study.
Ten drugs (nafcillin, minoxidil, ketorolac, daunorubicin, apafant, toremifene, ranolazine, warfarin, cortisone-acetate, and dinaciclib) were identified as potential treatment options based on the CMap analysis. Dinaciclib is a novel cyclin-dependent kinase inhibitor (CDKI) with significant activity against various cancers in vitro and in vivo. Recently, its use to treat other immune-mediated diseases, including systemic lupus erythematosus and rheumatoid arthritis, is under exploration.37,38 Pharmacological inhibitors of CDK likewise prevented and reversed TGF-β responses in fibroblast monolayers and in ex vivo human skin organ cultures, ameliorated collagen overproduction in SSc fibroblasts. 39 Direct immunomodulatory action of CDKI on T and B cells may be one of the mechanisms underlying the beneficial effects. Cyclin-dependent kinase inhibitors represent a novel and promising therapeutic strategy for immune-mediated diseases.
There were glaring gaps in this investigation. In the beginning, the investigation’s clinical sample size was modest. The expression of hub genes and their relationship to immune cells were not further validated by basic tests. Further research into the phenotypes associated with ferroptosis in SSc may have a theoretical foundation thanks to this discovery.
Conclusions
Seven ferroptosis-related hub genes were discovered (CYBB, IL-6, TLR4, CXCL2, JUN, NOX4, and LY96). The lncRNA-miRNA-hub gene regulatory network was created after it was projected that three miRNAs, two lncRNAs, and five transcription factors would target these hub genes. Hub genes relevant to ferroptosis were strongly linked to immune infiltration in the skin tissues of SSc patients. A potential treatment for scleroderma is dinaciclib.
Supplemental Material
Supplemental Material - Comprehensive analysis of ferroptosis-related hub gene signatures as a potential pathogenesis and therapeutic target for systemic sclerosis: A bioinformatics analysis
Supplemental Material for Comprehensive analysis of ferroptosis-related hub gene signatures as a potential pathogenesis and therapeutic target for systemic sclerosis: A bioinformatics analysis by Chenmin Wu, Jianwen Liu, Zhihan Chen, Yanfang Wu, and Fei Gao in International Journal of Immunopathology and Pharmacology.
Footnotes
Acknowledgment
We thank the participants of the study.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by Natural Science Foundation of Fujian, grant no. 2023J011199.
Ethical approval
The studies involving human participants were reviewed and approved by the Ethics Committee of Fujian Provincial Hospital (Approval number: K2022-03-005).
Informed consent
The participants provided their written informed consent to participate in this study.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.