
Abstract
Objective
Numerous studies indicate that immune-related genes (IRGs) are closely related to tumorigenesis and tumor progression. We aimed to establish a robust IRGs-based signature to predict risk of recurrence for patients with laryngeal squamous cell carcinoma (LSCC).
Methods
Gene expression profiles were acquired to select differentially expressed IRGs (DEIRGs) between tumor and adjacent normal tissues. Functional enrichment analysis was performed to explore the biological roles of DEIRGs in LSCC. Univariate Cox analyses and LASSO regression model were used to construct a IRGs-based signature with the ability to predict recurrence for LSCC patients.
Results
A total of 272 DEIRGs were identified, of which 20 DEIRGs were significantly associated with recurrence-free survival (RFS). Subsequently, we constructed an eleven-IRGs signature that could classify patients into high-risk or low-risk groups in TCGA-LSCC training cohort. Patients in high-risk groups suffered from shorter RFS (log-rank p = 9.69E−06). Besides, the recurrence rate of high-risk group was significantly higher than that of low-risk group (41.1% vs. 13.7%; Fisher’s exact test p < 0.001). The predictive performance was validated in an independent cohort (GSE27020, log-rank p = 1.43E-03). Person correlation analysis showed that the risk scores calculated by eleven-IRGs signature were significantly associated with filtrating immune cells. Furthermore, three immune checkpoint molecules were significantly over-expressed in the high-risk group.
Conclusion
Our findings for the first time constructed a robust IRGs-based signature to precisely predict risk of recurrence and further provided a deeper understanding of IRGs regulatory mechanism in LSCC pathogenesis.
Introduction
Laryngeal carcinoma (LC) is a prevalent and severe malignancy, which is representative of >20% of all head and neck cancer.1,2 Laryngeal squamous cell carcinoma (LSCC) is currently the most common pathological classification of LC. 3 Currently, a variety of treatments, such as surgery, radiation therapy, and chemotherapy, are available to improve clinical outcome for LSCC patients.4,5 Despite advances in therapies, nearly half of LSCC patients experience disease recurrence caused by chemotherapy resistance and high invasiveness. 6 For clinicians, it is a difficult task to predict recurrence and survival risk as tumor heterogeneity and diverse clinical behaviors. Therefore, a valid and convenient predictive model is urgently needed to improve recurrence risk prediction.
Tumor microenvironment (TME) plays a critical regulatory role in cancer progression and treatment responses. As the essential components of the tumor microenvironment, tumor-infiltrating immune cells have been validated to be involved in tumor growth, invasiveness, and metastasis. Many researches have reported that pronounced immunosuppressive tumor activity is closely related to tumorigenesis. In addition, immunodeficiency also affects recurrence and prognosis.7,8 Recently, cancer immunotherapy has emerged as an effective way to improve clinical outcomes.9-11 For example, targeting programmed death protein (PD-1) and programmed death ligand 1 (PD-L1) with antibodies against PD-1 rescues effector T cell function, maintaining tumor cell-killing capacity of T cell. 12 For LSCC patients with high expression of PD-L1, antibodies against PD-1 are efficacious in reducing the risk of recurrence and improving clinical outcome.13-15 Therefore, exploring the roles of immune-related genes (IRGs) in LSCC progression is beneficial to discovering more effective immunotherapy targets.
With the development of chip sequencing technology, many studies have explored the function of IRGs using gene expression data. Gao et al. found four IRGs with a higher prognostic performance in renal clear cell carcinoma. Prognostic scores calculated by four IRGs effectively reflect the level of immune cell infiltration. 16 Xu et al. comprehensively analyzed the expression data and clinical data and then developed an individualized immune signature to improve prognostic evaluation for bladder cancer patients. 17 Many studies have confirmed that IRGs as key regulators were involved in the occurrence, metastasis, diagnosis, and treatment of LSCC.18,19 Mo et al. analyzed the role of IRGs to develop an immune-related prognostic signature, which can make risk classification for LSCC patients. 20 IRGs signature has gradually become an effective method to explore the relationship between immune cells and tumor progression. However, few signatures based on IRGs are developed to predict the risk of recurrence of patients with LSCC. Therefore, developing a IRGs-based signature to estimate risk of recurrence is beneficial to understanding the potential predictive value of tumor-infiltrating immune cells and exploring new immunotherapy targets.
In this study, using gene expression profiles and clinical information of LSCC extracted from TCGA database, we comprehensively analyzed the expression of IRGs in LSCC and its potential predictive value of recurrence. We further constructed a IRGs-based signature to predict the risk of recurrence in LSCC patients. Besides, we analyzed the association between risk scores calculated by IRGs-based signature and LSCC immune cell infiltration, and further evaluated the correlation with three immune checkpoint molecules. Our study explored the potential clinical utility of IRGs on predicting risk of recurrence and laid a foundation for potential LSCC immunotherapy targets.
Materials and methods
Data acquisition and processing
Clinical characteristics of patients with laryngeal squamous cell carcinoma.

Differentially expressed IRGs (DEIRGs) analysis
We derived a list of immune-related genes (IRGs) from the Immunology Database and Analysis Portal (ImmPort) Database. Student t-test was applied to select the DEIRGs between the tumor and normal tissue samples. The threshold was set to false discovery rate (FDR) < 0.05 and |log2fold change (FC)|>1. Volcano plots and heatmaps were generated using the “ggplot2” and “pheatmap” packages in R software, respectively.
Functional enrichment analysis and protein–protein interaction network
In order to investigate the biological processes that the DEIRGs may be involved in, functional enrichment analysis were performed using Database for Annotation, Visualization and Integrated Discovery (DAVID, available at: http://www.david.abcc.ncifcrf.gov/). Besides, to illustrate the key cellular activities in carcinogenesis, a protein–protein interaction (PPI) network was built by The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, available at: https://string-db.org/). A strict criterion (combined score >0.9) was set to identify reliable interactions among the DEIRGs. The PPI network was visualized using Cytoscape (available at: www.cytoscape.org). Then, hub genes with high degrees of connectivity were selected by cytoHubba application.
Establishment of the IRGs-based signature for LSCC
DEIRGs, which were significantly associated with relapse-free survival (RFS) using univariate Cox regression analysis, were selected to establish the prognostic signature by the least absolute shrinkage and selection operator (LASSO) method in TCGA-LSCC training cohort. The Lasso-penalized Cox model is a popular method to achieve the feature selection and obtain an optimal model subsequently. Regression coefficients of each DEIRG in the model were determined by Lasso-penalized Cox model. The risk scoring model is:
Risk score = ∑ n i=0GiβiGi is the normalized expression value of gene i in risk scoring model. βi is the regression coefficient of gene i, which represents the contribution of gene i to risk score. Based on the risk scoring model, patients whose risk scores were larger than the median risk score were classified into high-risk group, otherwise classified into low-risk group.
Evaluation of relationship between IRGs-based signature and immune cell infiltration, and immune checkpoint molecules
Tumor Immune Estimation Resource (TIMER, available at: https://cistrome.shinyapps.io/timer/) is an online tool to analyze and visualize the abundances of tumor-infiltrating immune cells. Based on gene expression data, the algorithm estimated the abundance of six subtypes of tumor-infiltrating immune cells (including B cells, CD4 T cells, CD8 T cells, macrophages, neutrophils, and dendritic cells) for each sample from TCGA public database. Estimation results of immune cell infiltration abundance for LSCC patients were retrieved from TIMER to analyze the correlation between immune cell infiltration and risk scores. Meanwhile, Estimation of Stromal and Immune cells in Malignant Tumor tissues using Expression data (ESTIMATE) was a method to evaluate the infiltration level of stromal and immune cells for samples from TCGA. 21 We extracted the immune scores and stromal scores of LSCC samples to calculate the difference between high-risk group and low-risk group, respectively. To investigate the correlation between risk scores and three important immune checkpoint molecules (including PD-1, PD-L1, and CTLA4), we performed T-test to observe the expression difference of three immune checkpoint molecules between high-risk and low-risk groups, respectively.
Statistical analysis
Statistical analysis for all data in the current study was implemented by R software. T-test was used to observe the gene expression difference between different groups. Multivariable Cox regression analysis was used to assess whether the risk score was independent of other clinical features, such as stage, age, and gender. Hazard ratios (HRs) and 95% confidence intervals (CIs) were computed based on the Cox regression analysis. The Kaplan–Meier method was used to assess the differences in survival time of low- and high-risk LSCC patients, and the log-rank test was used to determine the statistical significance of observed differences between groups. The receiver operating characteristic (ROC) curve was generated using the “pROC” package. The difference in mortality rate between different risk groups was tested by Fisher’s exact test. p < 0.05 was regarded as statistically significant.
Results
Identification of differentially expressed IRGs
Using a threshold of |log2FC| >1 and FDR <0.05, 3267 differentially expressed genes were identified between LSCC and adjacent normal tissues in GSE127165 (Figure 1(a)). Then, a list of IRGs containing 1811 genes were downloaded from ImmPort database, of which 272 IRGs were significantly differentially expressed (including 143 up-regulated IRGs and 129 down-regulated IRGs) and filtrated for further analysis (Figure 1(b)). Volcano plots of differentially expressed genes (A) and heat maps of differentially expressed immune-related genes (B).
Functional enrichment of differentially expressed IRGs
Using the DAVID database, we performed functional enrichment analysis (including GO annotation and KEGG pathway enrichment) to explore the underlying biological processes related to 272 DEIRGs. Functional enrichment results are shown in Figure 2. GO functional annotation revealed that DEIRGs were significantly involved in many tumor-related terms (Figure 2(a)). As expected, inflammatory pathways were most frequently enriched. “immune response,” “extracellular region,” and “cytokine activity” were the most frequent biological terms among biological processes, cellular components, and molecular functions, respectively. KEGG pathway enrichment indicated that 272 DEIRGs were mainly enriched in “Cytokine-cytokine receptor interaction,” “Chemokine signaling pathway,” and “Jak-STAT signaling pathway” (Figure 2(b)). Enrichment of Gene Ontology (GO) terms (A) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (B) of differentially expressed immune-related genes.
Protein–protein interaction network
With a strict criterion (combined score >0.9), a PPI network consisting of 94 nodes and 166 edges was constructed using STRING online tool to explore the interactions between 272 DEIRGs. The PPI network was visualized using Cytoscape software (Figure 3(a)). The top 10 IRGs were generated by cytoHubba, including IL1B, PIK3R1, CSF2, STAT1, IL12A,IL1A, CCL22, CXCL10, OASL, and RSAD2 (Figure 3(b)). We found that most genes were closely involved in tumorigenesis and progression. The protein-protein interaction network constructed by differentially expressed immune-related genes (A) and top 10 immune-related genes generated by cytoHubba (B).
Development and validation of the IRGs-based signature
Univariate cox analysis of DEIRGs.

Note: DEIRGs, differentially expressed immune-related genes; HR, hazard ratio; CI, confidence interval; DEdir, differentially expressed direction.
Gene information entering into the signature.

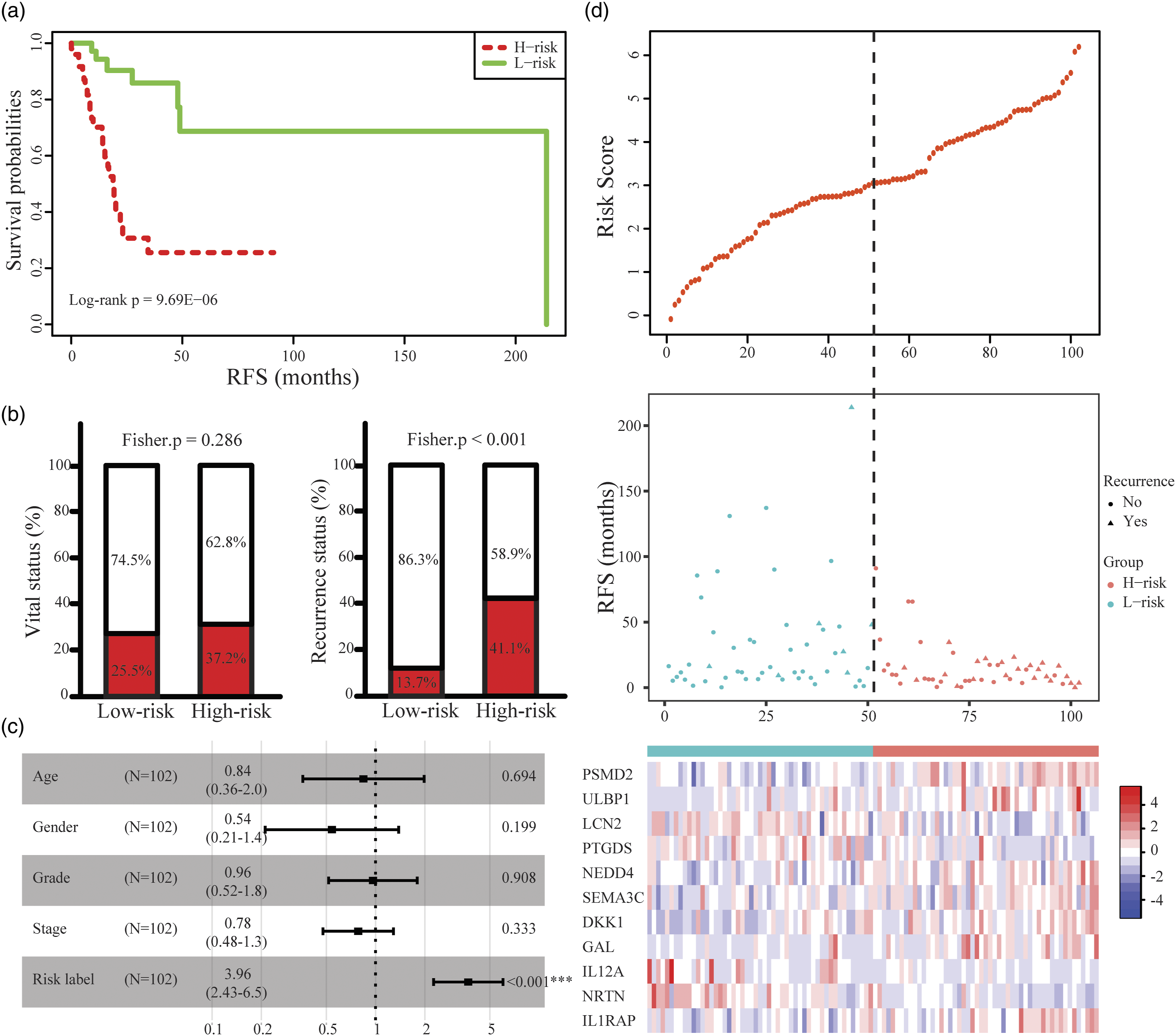
Construction of the eleven-IRGs signature in TCGA-LSCC training cohort. (A) Kaplan–Meier curve of the recurrence-free survival between low-risk and high-risk groups. (B) The ratio of death and recurrence in high-risk and low-risk groups, respectively. (C) Forest plot of the multivariate Cox regression analysis. (D) Risk score distribution, survival status and expression heat map of eleven genes corresponding to each sample above.

Predictive performance of the eleven-IRGs signature in validation cohort (GSE27020). (A) Kaplan-Meier curve of the recurrence-free survival between low-risk and high-risk groups in GSE27020. (B) The ratio of recurrence in high-risk and low-risk groups. (C) Forest plot of the multivariate Cox regression analysis. (D) Risk score distribution, survival status and expression heat map of eleven genes corresponding to each sample above.
Relationship of the IRGs-based signature with immune cell infiltration and immune checkpoint molecules
We utilized the TIMER database to reveal the association between eleven-IRGs signature and filtrating immune cells. As shown in Figure 6, eleven-IRGs risk scores were significantly negatively correlated with levels of infiltrating B cell (r = −0.41, p < 0.01), CD4 + T cell (r = −0.26, p = 0.04) and CD8 + T cell (r = −0.33, p = 0.01). Meanwhile, Using ESTIMATE algorithm to evaluate the infiltration level of stromal and immune cells, we observed that patients with higher risk scores tended to be lower immune scores but higher stromal scores (Figures 7(a)–(b)). Besides, the expression values of three immune checkpoint molecules (including PD-1, PD-L1 and CTLA4) were compared between high-risk group and low-risk group. We found that patients within high-risk group presented with a lower expression level for all three immune checkpoint molecules (Figures 7(c)-(e)). The association between risk scores based on eleven-IRGs signature and immune cell infiltration in TCGA-LSCC patients. Comparison of immune score, stromal score, and expression of three immune checkpoint molecules between high-risk and low-risk groups. (A) Immune score. (B) Stromal score. (C) PD-1. (D) PD-L1. (E) CTLA4.

Discussion
Growing evidences have confirmed the contributions of tumor microenvironment to human diseases.22-24 Recently, many researches have revealed that IRGs could act as a key regulator in LSCC progression.25-27 Many efforts have been made to elaborate the clinical utility of IRGs on prognostic stratification for LSCC patients,20,28 but few researches explored the predictive ability of IRGs for the risk of recurrence. In this study, we identified 272 DEIRGs between LSCC and adjacent normal tissues. Functional enrichment analysis results showed that DEIRGs were most frequently involved in inflammatory biological processes. Besides, other cancer-associated processes, such as “positive regulation of cell proliferation,” “G protein-coupled receptor signaling pathway,” and “signal transduction” were enriched. For example, Thomas et al. found that two G protein-coupled receptors ligands, including prostaglandin E2 and bradykinin, could activate the epidermal growth factor receptor pathway by increasing selective autocrine release of transforming growth factor-α, which plays key roles in LSCC carcinogenesis. 29 KEGG enrichment analysis also showed that DEIRGs were significantly enriched in “Cytokine-cytokine receptor interaction,” “Chemokine signaling pathway,” and “Jak-STAT signaling pathway.” Furthermore, ten hub IRGs were selected from PPI network. All genes have been reported to be closely related to the progression and prognosis of various tumors.30-33 For example, PIK3R1, known as the regulatory subunit of class 1A PI3Ks, has been identified to be involved in cancer progression. Wang et al. 34 observed that PIK3R1 was down-regulated in LSCC tissues and cell lines. By directly binding to 3′-untranslated region (UTR), MiR-17-5p could positively regulate PIK3R1 expression to modulate the activation of PI3K/AKT signaling pathway. These researches suggested that IRGs could play critical roles in LSCC development, including proliferation, apoptosis, migration, and invasion.
Several risk-prediction models have been constructed in recent years.35,36 For example, Cui et al. 37 constructed a risk-prediction nomogram for predicting recurrence probability based on eight clinicopathologic variables. Nevertheless, the risk-prediction nomogram on the basis of clinical and pathologic variables exists limitations due to tumour heterogeneity of LSCC. With the development of molecular biology and high-throughput sequencing, molecular factors may provide in-depth understanding of the mechanism of LSCC progression and improve the predict recurrence probability of LSCC. In our study, we focused on IRGs closely related to patient RFS and then developed a risk-prediction model consisting of eleven IRGs to precisely predict recurrence probability. Among the eleven IRGs, Dickkopf-1 (DKK1) is a secreted protein and Wnt signaling pathway inhibitor. Researches have revealed that DDK1 is over-expressed in LSCC tissues and can be served as an unfavorable survival predictor, which may be a promising therapeutic target for LSCC patients.38,39 Another IRG, namely, interleukin 1 receptor accessory protein (IL1RAP), acts as an IL1R co-receptor to mediate the activation of IL-1 responsive genes. 40 In recent years, many researchers have found that IL1RAP as a key tumor-associated antigen might be used as an immunotherapy target. Pasvenskaite et al. 41 found significant IL1RAP polymorphisms in LSCC carcinogenesis, which could be act as predictors for the clinical course of LSCC. Although other IRGs, such as PSMD2, LCN2, NEDD4, and IL12A, have not clearly reported related to the progression of LSCC, a large number of research studies have found that these IRGs play critical roles in cancer progression.42-44 Treatment of larynx cancer has changed dramatically over the past several years. New targeted therapies have appeared to increasing local control rates and overall survival. In the TREMPLIN trial, researchers found that target therapies such as cetuximab showed evidence of a benefit in resectable LSCC. 45 However, adequate evidence for patients with unresectable LSCC is not available currently. Our study could lay a foundation of new specific therapeutic targets within signature to limit the development of LSCC. Targeted therapies play a role in chemoradiotherapy, but further comparative trials must be performed. Great advances have been made in larynx cancer treatment, 46 but we must continue improving our knowledge and quality standards to benefit as many patients as possible.
Although our eleven-IRGs signature showed impressive performance in LSCC recurrence prediction, there were several limitations in our study. First, the selection of predictors was limited to IRGs. Clinicopathologic factors, such as age, grade, smoking, alcohol, and TNM stage, were also important indicators in progression of LSCC. A combination of clinicopathologic factors and molecular factors may be a more powerful pattern for disease risk classification. Second, although this study was validated using an independent GEO cohort, prospective studies and multicenter prospective cohorts with large sample sizes are still needed to make a further clinical validation for the eleven-IRGs signature. Third, there are differences based on anatomical subsites in laryngeal carcinoma with distinct presentations and prognosis. However, as the details of sublocations in laryngeal carcinoma were not publicly available, we could not really explore the association of IRG signature and sublocations in depth. Finally, lack of mechanism studies hindered better application of the eleven-IRGs signature. Experimental research of each IRG within the eleven-IRGs signature could be performed to provide helpful information for understanding of their functional roles.
Conclusion
To summarize, we comprehensively utilized expression profiles and clinical data derived from TCGA database to identify DEIRGs, and for the first time constructed a novel eleven-IRGs signature for predicting recurrence in LSCC patients, which presents better predictive capacity and may aid the clinical decision-making process. These IRGs may act as molecular biomarkers to provide helpful information for selection of therapeutic strategies.
Footnotes
Acknowledgements
We thank the Gene Expression Omnibus and The Cancer Genome Atlas Database for sharing a large amount of data. We also thank all the R programming package developers.
Author contributions
Xiu Hong, Bing Liu, Yang Li and Yuqiang Hu contributed to data analysis, interpretation, and drafting. Qian Lv, Ying Liu, and Jun An contributed to data collection. Xiuli Wang and Rui Li involved in study design, study supervision, and final approval of the article. All authors read and approved the final article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Health Commission of Jiangsu Province [grant number: Z2021016], Xuzhou Health Commission [grant number: XWKYHT20210590] and Xuzhou Science and Technology Bureau [grant number: KC20135].
Data availability
The source data of this study were derived from the public repositories, as indicated in Materials and Methods. All data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethical approval
All information from TCGA and GEO databases is freely available to the public, so the approval of the medical ethics committee board was not applicable.