
Abstract
Objective:
This study aims to comprehensively investigate the expression profiles of interleukins in prostate adenocarcinoma (PRAD) and their relationship with immune cell infiltration, tumor progression, and patient prognosis. By establishing an interleukin-related risk score, we seek to enhance the understanding of the tumor immune microenvironment and facilitate the development of tailored immunotherapeutic strategies for PRAD patients.
Introduction:
Interleukins can nurture a tumor promoting environment and simultaneously regulate immune cell infiltration. However, the potential roles of interleukins in the prostate adenocarcinoma immune landscape remain abstruse.
Methods:
We comprehensively investigated the interleukin expression patterns and tumor immune landscape of prostate adenocarcinoma patients. And explored the interleukin expression patterns with immune infiltration landscape. The interleukin score was established using LASSO cox regression analysis. Multivariate Cox regression analysis was employed to assess the prognostic value of the interleukin score.
Results:
We identified two distinct interleukin clusters, characterized by different immune cell infiltration, tumor promoting signaling pathways activation and prognosis. The interleukin score was established to estimate the prognosis of individual prostate adenocarcinoma (PRAD) patient. Further analysis demonstrated that the interleukin score was an independent prognostic factor of PRAD. Finally, we investigated the predictive value of interleukin score in the programmed cell death protein (PD-1) blockade therapy of patients with prostate adenocarcinoma. At the same time, the differences in related genes among different prostate cell lines were also identified.
Conclusions:
This study demonstrated the correlation between interleukin and tumor immune landscape in prostate adenocarcinoma. The comprehensive evaluation of interleukin expression patterns in individual prostate patients contribute to our understanding of the immune landscape and helps clinicians selecting proper immunotherapy strategies for prostate patients.
Keywords
Introduction
Prostate adenocarcinoma is the most common malignancy in men worldwide and metastasis is the leading cause of cancer death. 1 Although patients with localized tumors have encouraging prognosis, the 5-year survival rate for metastatic patients descends to 30%. 2 Standard treatment advanced prostate adenocarcinoma relies on androgen-deprivation therapies, which could restrict tumor growth in the early stage but eventually lead to the recurrence and generation of castration-resistant prostate adenocarcinoma. 3 Moreover, treatment failure is usually connected with formation of even more invasive subtypes, like neuroendocrine prostate adenocarcinoma.4,5 Hence, figuring out the driver genes that promote the conversion from localized to metastatic prostate adenocarcinoma would contribute to develop novel agents for prostate adenocarcinoma patients.
The interleukin and interleukin receptors play essential roles in tumor progression and cancer dissemination. 6 After interacting with their corresponding receptors, interleukins activate various oncogenic signaling pathways such as STAT3/RORγ, PI3K, and MAPK signaling.7–9 Many interleukins and interleukin receptors like IL-6, IL-23, and IL-17 have been proved to play important roles in prostate tumor growth and androgen-deprivation therapy resistance.8,10,11 Interleukins promote tumor progression by either directly acting on tumor cells or employing regulatory effects on immune cells or stromal cells in tumor microenvironment.12,13 Immunotherapies like PD-1 or CTLA-4 inhibition have shown promising effects in many solid tumors’ treatment, however, their efficacy in prostate adenocarcinoma seems to be limited.14,15 Considering the critical roles of interleukin and interleukin receptors in mediating immune cell trafficking and activation, they could be novel targets for immunotherapy agent development.
In the present study, we systematically explored mutation and transcriptional profiles and clinical significance of interleukin and interleukin receptors in prostate adenocarcinoma patients. Tumor subtypes with different prognosis and immune cell infiltration status was identified based on interleukin and interleukin receptors expression. Subsequently, an interleukin related risk model was constructed to predict tumor recurrence. We further investigated the relationship between this risk model and tumor mutation burden, immune cell infiltration and target therapy response. Our study provided a better understanding of interleukin and interleukin receptors in PRAD, which would help to select proper immunotherapies.
Material and methods
Patient and samples
mRNA expression data (raw counts) of 492 and 388 PRAD patients with matched clinical annotations including tumor stage, gender, and recurrence free time were downloaded from the Cancer Genomes Atlas (TCGA) and Gene expression Omnibus (GEO) database respectively. Gene somatic mutation data (MAF files) of PRAD was obtained from TCGA. The IDs of the GEO database is GSE21034 and GSE1168918.
Identification of PRAD subtypes
Interleukins and interleukin receptors were summarized from previous study and a total of 67 genes was obtained. 6 Unsupervised clustering algorithm was employed for cluster analysis of these genes. And then R package Consensus-Clusterplus package was used for the analysis and the repetition was set to 100 to ensure the credibility of the classification.
Calculation of immune cell and stromal cell infiltration
ESTIMATE algorithm, which could portrait the general infiltration level of stromal and immune cell, was used to calculate immune score and stromal score. Single sample GSEA was used to assess the infiltration of 28 immune cells. Moreover, microenvironment cell population counter (MCP-counter) was also used to evaluate the absolute abundance of immune and stromal cells in the tumor microenvironment.
Pathways and enrichment analysis
To explore biological processes and potential pathways correlated with interleukin signature, Kyoto Encyclopedia of Genes and Genomes (KEGG) and gene ontology (GO) enrichment analyses were conducted using R package “clusterProfiler.”
Tumor mutational burden (TMB) analysis
Tumor mutational burden was defined as mutations per million bases. We obtained tumor mutation data from TCGA database. Subsequent analysis was conducted by R package “maftools.”
Drug and immunotherapy sensitivity prediction
Drug sensitivity prediction was conducted as previously described. 16 Gene expression profile and somatic data of human cancer cell lines (CCLs) were downloaded from the Broad Institute Cancer Cell Line Encyclopedia (CCLE) project (https://portals.broadinstitute.org/ccle/). 17 Drug sensitivity data of CLLs were obtained from the Cancer Therapeutics Response Portal (CTRP v.2.0, released October 2015, https://portals.broadinstitute.org/ctrp) and PRISM Repurposing dataset (19Q4, released December 2019, https://depmap.org/portal/prism/). The CTRP contains the drug sensitivity data of 481 compounds in 835 CCLs and the PRISM contains the sensitivity data of 1448 compounds in 482 CLLs. Both datasets use the area under the dose-response curve (AUC) values as a calibration of drug sensitivity and lower AUC values suggest increased sensitivity to treatment. K-nearest neighbor (k-NN) imputation was used to impute the missing AUC values. The sensitivity to anti-PD-1 and anti-CTLA-4 therapy was predicted by comparing the gene expression of tumor subtypes with 47 melanoma patients treated with immunotherapy using subclass mapping (https://www.genepattern.org/).
Construction of interleukin related risk score
Univariate cox analysis had been used to identify genes correlated with recurrence (p < 0.05). Thereafter, recurrence related genes were analyzed with the LASSO multivariate Cox regression algorithm using R package “glmnet.” The default maximum iterations (1000) were used for the LASSO fitting process. Then, the key genes and coefficients in the risk score signature were identified according to the most suitable penalty parameter λ. The optimal λ value was determined based on the minimum mean cross-validated error and the most regularized model within one standard error of the minimum. The risk score formula was Risk score =
Single-cell data analysis
Download the GSE176031 single-cell dataset in .h5 format along with annotation results obtained from TISCH. Utilize R software, particularly MAESTRO and Seurat, to process and analyze the data. Re-cluster the cells by applying the t-SNE method.
Cell culture
Human prostate epithelial cells RWPE-1 and human prostate carcinoma cells PC3 were obtained from the Wuhan Procell Life Science and Technology Company Limited. All cell lines were authenticated before purchase by standard short tandem repeat DNA-typing methodology. All cell lines were maintained at 37°C, 5% CO2 and cultured in standard medium as recommended by the company. The RWPE-1 and PC3 cells were cultured in DMEM medium (Gibco, C11995500BT) supplemented with 10% fetal bovine serum (FBS, Gibco, 10270-106).
Reverse transcription and real time PCR (RT-qPCR)
RNA was isolated using TRIzol reagent (Invitrogen, 15596018), and its concentration was quantified using a Nanodrop Spectrophotometer (Thermo Scientific). The isolated RNAs were then reverse transcribed into cDNAs with the Evo M-MLV RT Kit (Accurate Biotechnology, AG11705). These cDNA samples underwent RT-qPCR analysis using SYBR Green qPCR mix (ABclonal, RK21203). The results were normalized to the ACTIN and analyzed using the 2-DDCt method. The primers of the six genes were listed in Table 1.
Primer name and sequence.

Statistical analysis
All statistical analyses were conducted using R software (https://www.r-project.org/). The unpaired Student’s t-test was used to compare two cohorts with normally distributed variables. One way analysis and Kruskal-Wallis test of variance were used as parametric and non-parametric methods for cohorts comparing. The surv-cutpoint function of survminer package was used to determine the optimal cutoff point in each dataset. The chi-square test is used to prove the correlation between parameters. Survival curves were plot with Kaplan-Meier method and p value was calculated with log rank test. And p value less than 0.05 was considered as statistically significant.
Results
Somatic alternation landscape of interleukins
To study the genomic features of interleukins in prostate adenocarcinoma, we portrayed the mutation and CAN frequency of 492 PRAD patients in the TCGA cohort (Figure 1(a) and (b)). The DNA mutation of interleukins occurred in about 46% of patients and mutation frequency ranged from 0% to 9%. The interleukins with the highest mutation frequency are IL17C (9%), IL7(7%), IL-34(6%), IL-21(4%), and IL-2(3%) and their most common mutations are deep deletion, amplification, missense mutation, and truncating mutation. Importantly, log-rank test indicates that patients with interleukins altered had worse overall survival (p = 0.046; Figure 1(g)). At the same time, the chi-square test also shows that patients with interleukins altered have relatively higher Gleason scores, indicating that their prognosis is relatively poor (Supplemental Table 1). Moreover, patients in the altered cohort had significant higher biochemical recurrence (15.04% vs 8.65%, p = 0.0279) and primary lymph node positive rates (88.05% vs 80.83%, p = 0.0358; Figure 1(h) and (i)). Besides, patients in the mutated cohort had higher tumor mutation burden (TMB), mutation count and fraction genome altered. These indicates that mutation and copy number variation of interleukins might co-occurrence with other genes (Figure 1(c)–(f)). In summary, these results suggest that mutation and copy number variation of interleukins means worse overall survival and higher biochemical recurrence rate.

The alternations of interleukins in PRAD. (a) Landscape of genomic alternations of the interleukins in PRAD. Each row represents a gene, and each column represents a patient. The frequency of alternations in top 20 interleukins. (b) Gene alternation frequency of PRAD patients in TCGA. (c) Histogram of the proportion of different mutation modes in PRAD. (d–f) The difference of TMB, fraction genome altered frequency and mutation count between altered and unaltered cohort. (g) The overall survival of PRAD patient between altered and unaltered cohort. (h) Histogram of the frequency of biochemical recurrence in altered and unaltered cohort. (i) Histogram of the frequency of primary lymph node presentation in altered and unaltered cohort.
Identification of interleukin subtypes in PRAD
In the previous steps, we demonstrated that patients with altered in interleukin cohort have a worse prognosis. Next, we will further explore which interleukins play a key role in this. Many interleukins had similar effects in promote tumor progression and anti-tumor immunity. To investigate whether interleukin co-expressed with each other, we calculated the correlation of these genes. Results showed that the expression of most interleukins significantly correlated with each other (Supplemental Figure 1). This indicates that interleukins might cooperate with each other to regulate the progression of prostate adenocarcinoma. Subsequently, based on the expression levels of interleukins from TCGA, two distinct expression patterns were identified using the unsupervised clustering method, including 148 cases in the interleukin cluster1 and 347 cases in interleukin cluster2 (Figure 2(a)). T-SNE analysis showed that patients could roughly divided into two cohorts, which further corroborated two distinguished subtypes (Figure 2(b)). Moreover, we found patients in cluster 1 had worse recurrence outcomes in the cohort (Figure 2(c)). In order to prevent potential causal logic reversal that may arise from a single queue, an external queue was introduced for further validation. Similar results were found in external independent cohort (Figure 2(d) and (e)). The relationship between clinical factors in TCGA cohort and interleukin subtypes were also explored (Figure 2(g)). Patients of cluster 1 had a higher proportion of high Gleason score (GS ⩾8), advanced tumor stage and biochemical recurrence. Chip-square test revealed significant difference of these clinical features (Figure 2(f)). The transcriptomic profile of differently expressed interleukins in two subtypes was portrayed in the heatmap. These further demonstrated that dysregulation of interleukins could contribute to the progression of prostate adenocarcinoma.

Consensus clustering of interleukins in PRAD. (a) Consensus matrices of patients in the TCGA cohort for k = 1 using 100 iterations of unsupervised consensus clustering method (K-means) to ensure the clustering stability. (b) t-SNE analysis of interleukin related subtypes in TCGA cohort. (c) Comparison of prognosis of patients in different PRAD subtypes in TCGA cohort. (d) t-SNE analysis of interleukin related subtypes in GSE21034. (e) Comparison of prognosis of patients in different PRAD subtypes in GSE21034. (f) Heatmap of interleukin in TCGA cohorts. Interleukin related cluster, recurrence status, overall survival time, N stage, T stage, M stage, Gleason score, and age were used as patient annotations. (g) Sankey plot summarized the relationship among the cluster, Gleason score, T stage, and recurrence status.
The immune landscape of interleukin subtypes
To determine the difference in the activation of biological process between two clusters, GSVA analysis was carried out. Cluster 1 was remarkably enriched in pathways related to immune activation including “primary immunodeficiency,” and pathways related to tumor progression including “basal cell carcinoma” (Figure 3(a)). Considering the close relationship between interleukin subtypes and immune activity, we calculated the stromal and immune score in tumor samples using ESTIMATE algorithm based on gene expression profiles. Stromal and immune score reflects the tumor associated stromal and immune cell infiltration level. Cluster 1 exhibited significantly higher stromal score (Wilcoxon test, p < 0.05) and immune score (Wilcoxon test, p < 0.05; Figure 3(b)–(d)). Furthermore, to better characterize the infiltration of immune cells in two clusters, MCP-counter and ssGSEA were conducted. MCP-counter results show that cluster 1 had significantly higher immune cell infiltration, including T cells, CD8+T cells, cytotoxic lymphocyte, B lineage, NK cells, Monocytic lineage, Myeloid dendritic cells, neutrophils, endothelial, and fibroblasts (Figure 3(e)). Similar results were found in cluster 1 using ssGSEA analysis (Figure 3(f)). Moreover, we conducted an investigation into the disparities in human leukocyte antigen (HLA) and major histocompatibility complex (MHC) expression between the two study cohorts. Remarkably, we observed a consistently elevated expression of HLA and MHC molecules in cluster 1 (Figure 3(g) and (h)). Because the two clusters differed significantly regarding immune infiltration, we evaluated the correlation with the five common immune checkpoints. Cluster 1 had higher expression of PD-L1, CTLA4, and HAVCR2 (Supplemental Figure 2(a)–(c)). Cluster 1 was associated with a higher TIDE score (Supplemental Figure 2(d)). Overall, Cluster 1 is characterized by more robust immune activation, higher immune cell infiltration, and greater immune checkpoint expression, suggesting a more immunologically active tumor microenvironment compared to Cluster 2.
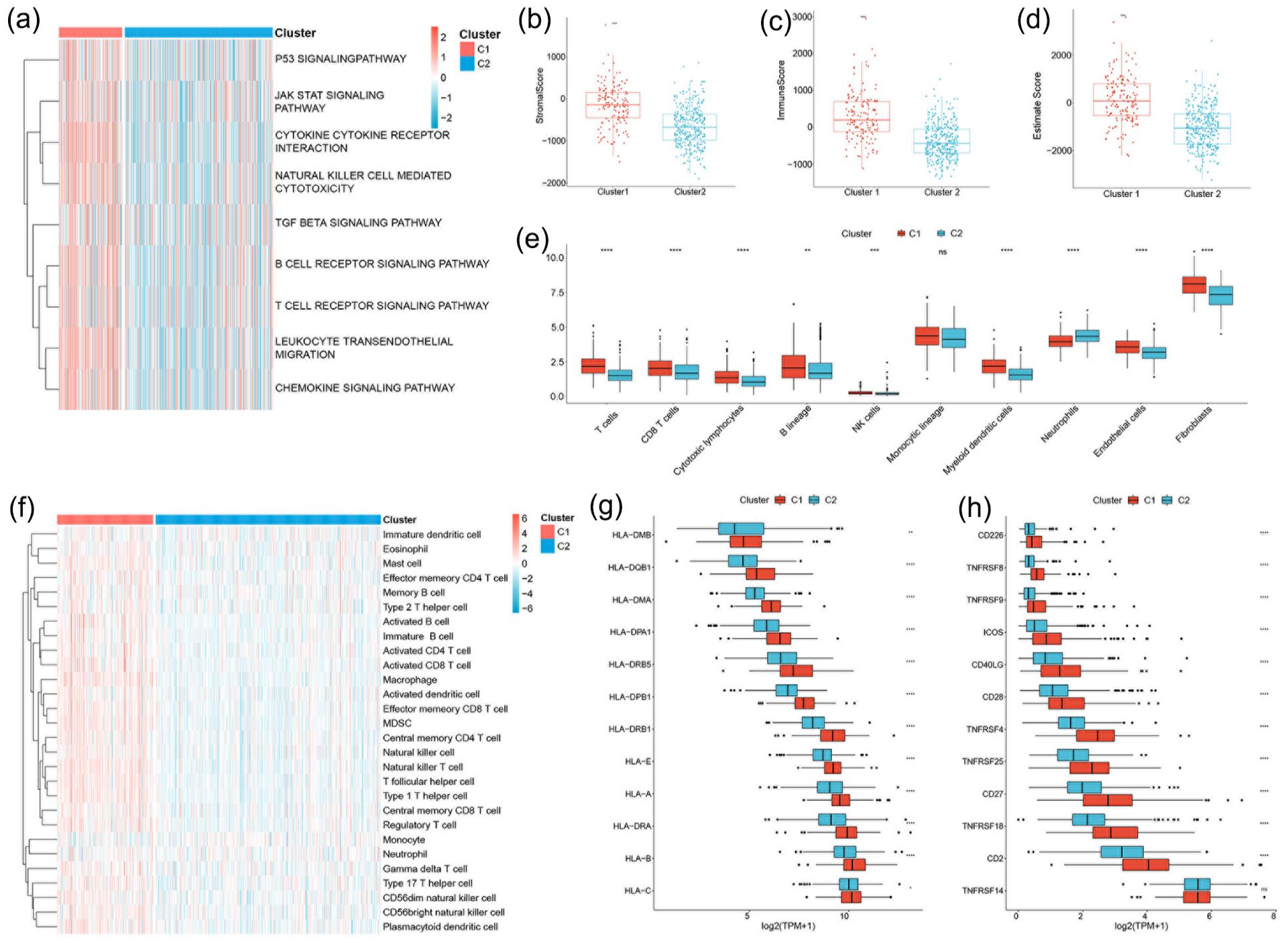
Biological characteristics and immune landscape of interleukin subtypes in the TCGA cohort. (a) Heatmap of the activation status of biological process in different subtypes evaluated by GSVA. Red and blue represents the activation and inhibition of biological process respectively. (b–d) The difference of stromal score, immune score and estimate score in two interleukin subtypes. (e) The difference of immune cell infiltration between two interleukin subtypes evaluated by MCP-counter. (f) The immune cell infiltration landscape between two interleukin subtypes calculated using ssGASEA. (g–h) Gene expression of HLA and MHC gene sets between two interleukin subtypes. Statistical significance at the level of ns ⩾0.05, *<0.05, **<0.01, ***<0.001, and ****<0.0001.
Construction of the interleukin gene signature
We developed an interleukin related risk score to evaluate the prognosis of individual patient. The LASSO cox regression analysis was applied to identify optimal prognostic signatures from interleukin and their corresponding receptors. After taking variables into the LASSO cox regression model with minimized lambda, five interleukins or receptors including IL4R, CSF1R, IL2RA, IL11, and IL1F10 was used to build the interleukin related score (Figure 4(a)). The expression of these genes is significantly correlated with poor prognosis. The interleukin related signature was calculated using the following formula: interleukin related risk score (IRRS) = (0.0014*IL4R) + (0.0046*CSF1R) + (0.068*IL2RA) + (0.1353*IL11) + (1.1291*IL1F10) (Supplemental Figure 3). The median danger score is 1.468369, which is used to differentiate between high-risk and low-risk cohorts. Notably, patients with high-risk scores were predominantly found in interleukin cluster 1, indicating that this group may have poorer prognoses. These findings suggest a significant association between the risk scores and interleukin clusters, indicating that the combination of these two approaches may enhance our understanding of the biological features of prostate adenocarcinoma and patient prognosis. The correlation of IRRS, interleukin subtypes and recurrence status were exhibited by the Sanky plot (Figure 4(f)). The high IRRS cohort accounts for higher percentage of the patients with recurrence and cluster1 accounts for higher proportion of patients with high IRRS (Figure 4(b)). Moreover, the validity of IRRS to predict patient prognosis was validated in the external cohort (Figure 4(c) and (d)). These patients were divided into high and low risk cohorts based on median risk score. Relatively sufficient clinical data were selected for further analysis. The distribution of the risk score and recurrence status in the datasets was displayed (Figure 4(e)). In addition, the AUC values of prognosis prediction reached 0.636 in the training cohort and 0.716 in the validation cohort. These results indicates that the interleukin risk score could be used to predict the prognosis of prostate adenocarcinoma patient.

Construction of interleukin related score. (a) The distribution of risk score, recurrence status, and hub gene expression level. (b–d) Kaplan-Meier curves for patients with high or low interleukin score in the TCGA, GSE21034, and GSE1168918. (e) Multivariate cox regression analysis of interleukin core and other clinical characteristics. (f) Sankey plot summarized the relationship among the risk core, cluster, Gleason score, T stage, and recurrence status.
Immune landscape was significantly associated with the expression of interleukins
As the expression of interleukins correlate with immune filtration. We further compared the infiltration of immune and stromal cells using three different algorithms (Figure 5(a)–(c)). These algorithms showed similar results. Samples in the high risk cohort had significantly higher degree of immune cell and stromal cell infiltration. Moreover, for better understanding the correlation between risk score, Pearson correlation were calculated between risk score and immune infiltration (Figure 5(d) and (e)). Gene mutations had been demonstrated to shape the immune environment of tumor cells. We explored the mutation patterns in the high and low risk cohort (Figure 5(f)). We observed that the expression levels of immune modulators were predominantly higher in the high-risk cohort compared to the low-risk cohort, with a majority of them exhibiting stimulatory effects. Furthermore, for the purpose of conducting a more comprehensive examination of the high-risk cohort, we conducted enrichment analysis employing three distinct algorithms. Subsequently, we successfully identified a profound association with immunization (Figure 6(a)–(c)). The majority of the high-risk cohort is enriched in pathways similar to adaptive immune response and immune receptor activity. They are all highly associated with immunity. Finally, we utilized single-cell data from public databases to conduct further research on the genes in the IRRS. The results revealed a strong correlation between these genes and immunity. Specifically, IL4R is highly expressed in mast cells, progenitor cells, and monocyte-macrophage (Supplemental Figure 4), while CSF1R shows prominent expression in monocyte-macrophage (Supplemental Figure 5). IL2RA is notably expressed in regulatory T cells, CD8+ T cells, and plasma cells (Supplemental Figure 6). These findings confirm the close association between these genes and immune function, indirectly supporting the relationship between the IRRS and immunity.

Relationship between risk score and immune cell infiltration. (a) The correlation between risk core and immune score in TCGA cohort. (b) The correlation between risk score and stromal score in TCGA cohort. (c) The correlation between risk score and estimate score in TCGA cohort. (d) The difference of immune cell infiltration level in high and low risk cohort identified by MCP-counter analysis. (e) The immune cell landscape in high and low risk cohort evaluated by ssGSEA. (f) Regulation of immunomodulators in high and low risk cohort. From left to right: mRNA expression (median normalized expression levels); expression versus methylation (gene expression correlation with DNA-methylation beta value); amplification frequency (the difference between the fraction of samples in which an immunomodulator is amplified in a particular subtype and the amplification fraction in all samples); and the deletion frequency (as amplifications) 75 IM by interleukin score.

Enrichment analysis of highly expressed genes in the high interleukin score cohort. (a) Cellular Component. (b) Biological process. (c) Molecular function.
Association between interleukin score and mutations
To investigate the differences in genomic mutations between the high- and low-risk cohort, we downloaded simple nucleotide variation data (Figure 7(a) and (b)). TTN (17%), TP53 (16%), and SPOP (9%) were the top three genes with the highest mutation frequencies in the high-risk cohort, while SPOP (13%), TTN (10%), and TP53 (9%) were the top three genes in the low-risk cohort. By comparing two cohort s, we found that the high-risk cohort had higher tumor mutation burden (Figure 7(c)). Subsequent investigations revealed that the high-risk cohort exhibited a noticeably higher proportion of fraction genome altered, as well as both focal and broad copy number alternations copy number variations, in comparison to the low-risk cohort (Figure 7(d) and (e)). The aforementioned studies have demonstrated a higher prevalence of genetic mutations in tumors within the high-risk cohort.

Association between interleukin score and mutations. (a–b) Oncoprint of mutation status of top 20 genes in high (a) and low (b) risk cohort. (c–d) The tumor mutation burden (c) and fraction genome altered (d) in high and low risk cohort. (e) Focal and broad copy number alternations among the high and low risk cohort. Statistical significance at the level of ns ⩾0.05, *<0.05, **<0.01, ***<0.001, and ****<0.0001.
Interleukin gene signature in the role of anti-PD1 therapy
Considering the correlation between interleukin and immune cell infiltration, we analyzed the sensitivity of these individuals to immunotherapy (Figure 8(a)). We found that PD-1 therapy was effective in the high-risk cohort. This can serve as the foundation for subsequent experiments. Two different approaches were adopted to identify candidate agents with higher drug sensitivity in high PPS score patients. The analyses were performed using CTRP and PRISM-derived drug response data, respectively. First, differential drug response analysis between PPS score-high (top decile) and PPS score-low (bottom decile) cohorts was conducted to identify compounds with lower estimated AUC values in PPS score-high cohort (log2FC > 0.10). Next, Spearman correlation analysis between AUC value and PPS score was used to select compounds with negative correlation coefficient (Spearman’s r < −0.30 for CTRP or −0.35 for PRISM). These analyses yielded five CTRP-derived compounds (including birinapant, PF−184, etoposide, clofarabine, and luvastatin) and three PRISM-derived compounds (including LY2606368, alvocidib, and MK-1775). All these compounds had lower estimated AUC values in PPS score-high cohort and a negative correlation with PPS (Figure 8(b) and (c)).

Immunotherapy response prediction and identification of candidate agents with higher drug sensitivity in high interleukin score patients. (a) The response of high and low interleukin score patients to PD-1 and CTLA-4 immunotherapy. (b) The results of Spearman correlation analysis and differential drug response analysis of five CTRP derived compounds. (c) The results of Spearman correlation analysis and differential drug response analysis of three PRISM derived compounds.
The expression of CSF1R, IL2RA, IL11, IL1F10, and IL4R in different prostate cell lines
To verify the results of our data analysis, we extracted total RNA from prostate adenocarcinoma cell line PC3 and the normal epithelial prostate cell line RWPE-1 and measured the mRNA expression levels of IL4R, CSF1R, IL2RA, IL11, and IL1F10. RT-qPCR assays showed that the mRNA expression levels of CSF1R, IL2RA, IL11, and IL1F10 were significantly higher in PC3 cells than in RWPE-1 cells (Figure 9(a)–(d)). The mRNA levels of IL4R in RWPE-1 cell were significantly increased (Figure 9(e)). All five genes show significant differences between normal epithelial cells and tumor cells. This proves the feasibility of the model.

The mRNA expression levels of CSF1R (a), IL2RA (b), IL11 (c), IL1F10 (d) and IL4R (e) in different cell lines (RWPE-1 and PC3) were measured by RT-qPCR. Results were normalized to reference gene ACTIN. Data are shown as the mean ± SEM, two-tailed unpaired t test was used for statistical calculation for each marker, n = 4 independent experiments.
Discussion
The resistance to immune elimination and the formation of pro-tumoral inflammation circumstance are two of the critical hallmarks of cancer. 18 Interleukins mediate key communications between tumor and immune cells in the tumor microenvironment (TME).6,19 Chronic inflammation has been acknowledged as one of the drivers for tumor formation in prostate adenocarcinoma.20,21 After tumorigenesis, the role of interleukin spans tumor growth, metastasis and therapeutic failure. 6 Delineating the landscape of interleukin in tumor immune regulation and progression facilitated the development novel, individualized and highly effective drugs. In the present study, we portrayed the mutation and expression pattern of interleukin and explored their role in tumor immune infiltration and therapeutic resistance, which, in turn, would promote our understanding of the prostate adenocarcinoma microenvironment and provide more effective immunotherapeutic strategies for patients with prostate adenocarcinoma.
Previous studies have demonstrated that genetic alternations of interleukin contribute to the progression of tumor. For example, IL8 correlates with prostate adenocarcinoma progression and castration resistance and IL-8 rs4073 polymorphism indicates a higher risk of prostate adenocarcinoma.22,23 The genetic alternation analysis suggested a high frequency of copy number alternations of interleukin in PRAD cohort. Moreover, patients in the mutation cohort had a higher tumor mutation burden, mutation count and fraction genome altered. These indicate mutation of interleukins might co-occur with other genes. Importantly, patients with IL gene alternations had a poorer prognosis and a higher frequency of biochemical recurrence, suggesting a critical role for interleukin in the development and progression of prostate adenocarcinoma.
In the present study, based on the expression of interleukins, we identified two distinct subtypes with significant different prognosis and immune landscape. Cluster 1 has worse prognosis than cluster 2. Further analysis of clinical traits showed that cluster 1 correlated with advanced tumor stage and higher Gleason score, which in part explained the poor prognosis of this cluster. Differentially expressed genes of these two clusters were subjected to gene enrichment analysis to explore the potential reason for these differences. The results suggested that cluster 1 was prominently enriched in signaling pathways correlate with immune activation like B cell receptor signaling pathway. Therefore, we explored the associations between two interleukin related subtypes tumor immune infiltration. Cluster 1 showed significant higher immune cell infiltration based on three different evaluation algorithm. In fact, immune infiltration analysis shows that nearly all kinds of immune cells are highly infiltrated in cluster 1. Indeed, the relationship between immune cells and tumor cells is heterogeneous and different cells might have opposite roles. Tumor infiltrating cells are pivotal mediators of anti-tumor immune response, however, unlike other solid tumors, prostate adenocarcinoma is one of the two kinds of tumors in which higher CD8+ T cells correlates with worse prognosis. Consistent with previous studies, cluster 1 had significant higher infiltration of CD8+ T cells. B cell is another important mediator of prostate cancer progression. Compared with benign prostatic tissues, prostate tumor had significant higher abundance of B cell infiltration. Moreover, the recruitment of B cells into tumor microenvironment has been linked with the inception of castration resistant and distant metastasis. Macrophages is also one of the crucial immune cell populations with two distinct phenotypes, M1 and M2. Transition from phenotype M1 to M2could be induced by IL-4, IL-6, or IL-13, and correlated with early biochemical relapse. Myeloid derived suppressor cell is another protagonist in tumor promoting immune environment. Importantly, the intra-tumor infiltration of myeloid derived cells could induce resistance of prostate adenocarcinoma cells to androgen deprivation therapy by secreting IL-23. Similar with macrophages and T helper cells, neutrophils are characterized by phenotype plasticity and could be polarized into anti-tumor “N1” or tumor promoting N2 subtypes by diverse cytokines like IL-1B. Despite not an immune cell, cancer associated fibroblasts are critical members in the tumor immunosuppressive circuity. As noted, interleukin is closely associated with tumor immunity. In the present study, pro-tumourigenic immune cells including macrophages, Tregs, neutrophils, and fibroblasts showed higher abundance in cluster 1 than cluster 2. Accordingly, the major histocompatibility complexes and T cell stimulators are increased in cluster 1.
Considering the influence of interleukin and corresponding subtypes on the clinical outcomes, an interleukin related risk score based on five prognostic signature was constructed using LASSO Cox regression analysis. The interleukin related score could be calculated for individual patient according to the aforementioned formula. And the high and low IRS subtypes were stratified based on optimal cutoff point. In the TCGA PRAD cohort, high risk significantly correlated with worse prognosis. Moreover, we performed log rank test in external validation cohort, GSE21034 and GSE116918, confirmed the practicability of IRS in evaluating patient prognosis. Univariate and multivariate Cox regression analysis proved that IRS was an independent prognostic factor. Additionally, further analysis indicates that IRS significantly correlated with the infiltration of immune cells. The key gene plays an important role in this process. IL1F10 has the potential to upregulate regulatory T cells (Tregs) and influence various components of both the host immune system and the cancer microenvironment. Studies have shown that IL-10 can improve the prognosis of colorectal cancer patients. Furthermore, IL-10 may contribute to the prognosis of colorectal cancer by regulating CD8+ tumor-infiltrating T cells and modulating the expression of PD-L1. 24 IL11 inhibits the activity of monocytes and macrophages. 25 IL2RA and IL4 are members of the γ chain (γc) family, primarily functioning as growth and proliferation factors for both progenitor and mature cells. Additionally, they play roles in lineage-specific cell differentiation. 26 The proliferation, differentiation, and survival of mononu dependent on signals received through the and survival of mononuclear phagocytes are dependent on signals received through the macrophage colony-stimulating factor receptor (CSF1R). 27 This is consistent with our findings when analyzing the immune profile of the high- and low-risk cohorts.
A heightened mutational load may enhance the immunogenic potential of tumor cells and promote the infiltration of immune cells into the tumors. 28 This is due, in part, to the accumulation of genetic and epigenetic alternations contribute to the immunogenicity of the tumor cells. Tumor mutation burden, termed as the total number of nonsynonymous mutations per sequenced coding area of a tumor genome, has been reported as a biomarker for immunotherapy. 29 Patients with advanced stage of tumor treated with immune check point inhibitors suggested that higher somatic TMB was correlated with better overall survival. 30
Immune checkpoint inhibitors, for example, monoclonal antibodies targeting CTLA4, protein PD-1, and its ligand PD-L1, have changed the therapeutic paradigm for a lot of cancers. 31 But the clear role of these therapies for prostate adenocarcinoma has yet to be clarified.32,33 In the present study, we found that high risk cohort correlates with higher immune cell infiltration and higher mutation frequency. In according with previous studies, we found that patients with high risk score are more sensitive to PD-L1 immunotherapy. This could partly be explained by higher TMB and immune cell infiltration level in the high risk cohort. However, PD-1/PD-L1 blockade combined with other therapeutic methods is a novel and promising treatment strategy contributes to improving the treatment efficacy of prostate adenocarcinoma. 15 And interleukin targeted therapy is one of the most promising. The research on targeted therapy utilizing interleukins is currently undergoing a thriving transformation.34–36
Subsequent study has yielded a promising list of eight drugs that can exhibit potential efficacy against prostate adenocarcinoma. Among these identified drugs, Etoposide and Clofarabine have demonstrated promising efficacy in preclinical trials for treating prostate adenocarcinoma.37,38 These drugs have exhibited significant effectiveness in inhibiting tumor growth and proliferation in prostate adenocarcinoma. Furthermore, Lovastatin has demonstrated potential as a chemosensitizer for paclitaxel-resistant prostate adenocarcinoma cells. 39 By inhibiting the enzyme CYP2C8, Lovastatin can enhance the sensitivity of drug-resistant prostate adenocarcinoma cells to paclitaxel. This suggests that Lovastatin can be used as an adjunct therapy to improve the effectiveness of paclitaxel treatment in patients with drug-resistant prostate cancer. Birinapant and Alvocidib are used for other cancers.40,41 This provides a potential basis for our future drug choices.
Conclusion
This study explored the link between interleukins, immune cell infiltration, and immunotherapy response, revealing a strong correlation. Our study established the IRRS model as a viable prognostic indicator for prostate adenocarcinoma (PRAD), revealing its correlation with the immune microenvironment and predictive value for immunotherapy response. This finding was further corroborated in an external, independent PRAD cohort, underscoring its potential as a tool for prognosis assessment and guiding therapeutic decisions. By meticulously examining interleukin expression profiles within PRAD, our research deepens insights into the intricate relationship between cytokines and immune cell activation, thereby facilitating the tailored selection of immunotherapeutic approaches for PRAD patients.
However, it faced limitations: immune cell quantification relied on algorithms due to technical restrictions; the biological mechanisms behind interleukin effects on PRAD prognosis remain elusive, necessitating further functional studies. Additionally, the absence of clinical cohorts limits validation of interleukin scores’ prognostic value and their correlation with tumor immune infiltration in PRAD. Large-scale prospective trials are needed for confirmation.
Supplemental Material
sj-docx-1-iji-10.1177_03946320251328476 – Supplemental material for Interleukin expression patterns and immune cell infiltration in prostate adenocarcinoma: Implications for recurrence risk
Supplemental material, sj-docx-1-iji-10.1177_03946320251328476 for Interleukin expression patterns and immune cell infiltration in prostate adenocarcinoma: Implications for recurrence risk by Jialong Zhang, Cong Huang, Xu Wang, Jun He, Hongzhi Wang and Chaozhao Liang in International Journal of Immunopathology and Pharmacology
Supplemental Material
sj-docx-2-iji-10.1177_03946320251328476 – Supplemental material for Interleukin expression patterns and immune cell infiltration in prostate adenocarcinoma: Implications for recurrence risk
Supplemental material, sj-docx-2-iji-10.1177_03946320251328476 for Interleukin expression patterns and immune cell infiltration in prostate adenocarcinoma: Implications for recurrence risk by Jialong Zhang, Cong Huang, Xu Wang, Jun He, Hongzhi Wang and Chaozhao Liang in International Journal of Immunopathology and Pharmacology
Footnotes
Acknowledgements
All authors would like to thank the TCGA and GEO databases for the availability of the data.
Author contributions
Chaozhao Liang and Hongzhi Wang conceived and designed the experiments. Jialong Zhang, Cong Huang, and Xu Wang contributed to the data interpretation, data analysis, experimentation, and writing of the draft. Jun He was responsible for the feeding of cells. Chaozhao Liang and Hongzhi Wang checked the manuscripts. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is funded by The National Natural Science Foundation (82370776 82170787), Anhui Translational Medicine Foundation (202204295107020007), and Anhui Educational Foundation (2022AH051172).
Ethics approval
Ethical approval was not sought for the present study because our study only used cell experiments and public database analysis.
Informed consent
Not applicable.
Trial registration
Not applicable.
Open access
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source.
Code availability
The underlying code for this study is not publicly available but may be made available from the corresponding author upon reasonable request.
Data availability
All data supporting the findings of this study are available within the paper and its supplementary materials.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.