
Abstract
Background
Phosphodiesterase-5 (PDE-5) inhibition increases intracellular cyclic guanosine monophosphate (cGMP), a second messenger molecule implicated in migraine pathogenesis. Given the clinical overlap between migraine and post-traumatic headache (PTH), we investigated whether pharmacologic elevation of cGMP induces migraine-like headache in individuals with persistent PTH.
Methods
Adults with persistent PTH and no pre-trauma history of migraine were enrolled in a randomized, double-blind, placebo-controlled, 2-way crossover study. Each participant received a single 100-mg oral dose of sildenafil or placebo on two experimental days, separated by a 1-week washout interval. Headache outcomes were assessed using structured diaries over 12 h post-ingestion. The primary outcome was the incidence of migraine-like headache during this observation window. The secondary outcome was the baseline-corrected area under the curve (AUC) for headache intensity scores over the same period.
Results
A total of 21 participants (mean age 42.3 years; 57% female) with persistent PTH completed both experimental days. Migraine-like headache occurred in 15 participants (71%) after sildenafil administration, compared with 4 (19%) following placebo (P = 0.003). Headache intensity scores, as quantified by the AUC, were significantly higher after sildenafil than after placebo (P < 0.001).
Conclusions
Pharmacologic elevation of cGMP via PDE-5 inhibition elicits migraine-like headache in individuals with persistent PTH, despite no pre-trauma history of migraine. These findings provide the first experimental evidence linking intracellular cGMP-dependent signaling to headache provocation in this patient population. The observed response implicates cGMP-dependent mechanisms in the pathogenesis of PTH and identifies this pathway as a potential target for future therapeutic development.
This is a visual representation of the abstract.
Introduction
Persistent post-traumatic headache (PTH) is a prevalent and disabling sequela of mild traumatic brain injury (mTBI) (1,2), often presenting with clinical characteristics that overlap with migraine, including moderate-to-severe headache, photophobia, phonophobia, and nausea (3,4). Despite this phenotypic resemblance, the underlying neurobiological mechanisms remain poorly defined (1,2), and no targeted therapies have been approved to date.
Human provocation models have begun to delineate molecular triggers in individuals with persistent PTH (5–9), suggesting the presence of pathophysiological features that partially converge with those observed in migraine (10). This pathophysiological overlap is supported by provocation models involving migraine-associated neuropeptides such as calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase-activating polypeptide (PACAP) (5,7). Both of these peptides are known to reliably induce migraine attacks in people with migraine (11,12). When administered to individuals with persistent PTH, these neuropeptides elicit migraine-like headache (5,7), while only mild, transient headache is reported in healthy adults (11,12). These effects are mediated via intracellular cyclic adenosine monophosphate (cAMP)-dependent signaling (10,13), implicating second-messenger cascades in the activation and sensitization of post-traumatic trigeminovascular circuits (5,7). Consistent with this mechanism, cilostazol—a selective phosphodiesterase-3 (PDE-3) inhibitor that increases intracellular cAMP levels (14–16)—triggers migraine attacks in people with migraine and induces migraine-like headache in those with persistent PTH (9).
Together, these findings implicate intracellular cAMP-dependent signaling as an important pathogenic driver across both headache disorders. Given the overlapping downstream effectors of cAMP- and cyclic guanosine monophosphate (cGMP)-dependent signaling, (10,13), attention has turned to the potential contribution of cGMP-mediated mechanisms. The nitric oxide (NO)-soluble guanylyl cyclase (sGC)-cGMP axis, regulated by phosphodiesterase-5 (PDE-5), modulates vascular tone and nociceptive messaging via shared targets, including certain vascular potassium channels (17,18). In people with migraine, sildenafil—a PDE-5 inhibitor that increases intracellular cGMP (17)—consistently induces migraine attacks indistinguishable from their spontaneous attacks (17). However, whether this mechanism elicits a comparable response in individuals with persistent PTH remains unknown.
The absence of experimental data in persistent PTH presents an opportunity to test whether PDE-5 inhibition can reveal cGMP-dependent signaling in this patient population. To address this, we conducted a randomized, double-blind, placebo-controlled, 2-way crossover study in adults with persistent PTH who had no pre-trauma history of migraine. Participants received a single oral dose of sildenafil or placebo, and headache outcomes were monitored over 12 h. This study aimed to determine whether cGMP elevation via PDE-5 inhibition is capable of inducing migraine-like headache in persistent PTH and thereby clarify the extent to which cGMP-dependent mechanisms contribute to the pathogenesis of PTH.
Methods
Design
This study applied a randomized, double-blind, placebo-controlled, 2-way crossover design involving adult participants with persistent PTH. The study was conducted at the research facilities of a tertiary academic medical center in Denmark, between January and June 2023. The study protocol was approved by the relevant ethics committee (Identifier: H-21067665), and all study-related assessments and procedures adhered to the principles outlined in the Declaration of Helsinki (19). The protocol was prospectively registered with ClinicalTrials.gov (Identifier: NCT05669885). All authors vouch for the completeness and accuracy of the data and for adherence to the study protocol.
Participants
Eligible participants were adults aged 18 to 65 years with a diagnosis of persistent PTH attributed to mTBI, in accordance with the International Classification of Headache Disorders, 3rd edition (ICHD-3). Furthermore, it was required that the mTBI had occurred ≥12 months before enrollment, and eligible participants had to report ≥4 monthly headache days during the preceding 3 months. Exclusion criteria included a history of moderate-to-severe TBI, multiple mTBIs, whiplash injury, or other headache disorders (except for infrequent episodic tension-type headache). Participants were also excluded if they had initiated, changed, or discontinued any preventive headache medication within 2 months prior to enrollment.
Eligibility was established based on self-report, corroborated by a review of electronic medical records. All participants provided written informed consent prior to undergoing any study-related assessments or procedures.
Randomization and masking
Participants were randomly assigned to receive sildenafil or placebo in a two-way crossover design, with each participant serving as their own control. Block randomization was implemented in blocks of four participants to ensure balanced treatment allocation. The random sequence was generated using a computer-based random number generator. The final block contained only one participant due to the odd sample size (n = 21).
Randomization codes were stored in sealed envelopes and only broken in the case of medical emergency. Allocation concealment, drug preparation, and blinding procedures were executed by pharmacy personnel uninvolved in outcome assessments or data analysis. Sildenafil (Vizarsin®) and the calcium-based placebo were each encapsulated in identical gelatin capsules to ensure indistinguishable appearance and taste. Investigators, participants, and outcome assessors were all blinded to drug allocation.
Procedures
Each participant completed two experimental sessions, spaced one week apart to allow a sufficient washout period and minimize pharmacologic carryover. Upon arrival, participants underwent a semi-structured interview, neurological examination, and baseline vital sign measurements. To stabilize physiological baselines, participants rested in a supine position for 15 min prior to any recordings.
A single oral dose of 100 mg sildenafil or matched placebo was administered in each session. During the 60-min post-dosing period, participants remained in a temperature- and light-controlled clinical environment under medical supervision. Headache intensity, associated symptoms, heart rate, and mean arterial blood pressure were assessed every 10 min during this in-hospital window. Participants were discharged afterward and instructed to complete hourly entries in an electronic headache diary over the next 11 h.
Participants were rescheduled for a new test session if they had taken any acute headache medication within 48 h before drug or placebo administration, reported migraine-like headache at baseline, or reported a baseline headache intensity greater than 3 on an 11-point numeric rating scale. Rescue medication was permitted if the headache became intolerable, in which case time and type of medication were recorded. Experimental criteria for migraine-like headache are shown in Supplemental material Table 1.
Demographics and baseline characteristics of the study population.

n, number of subjects; SD, standard deviation; IQR, interquartile range; TBI, traumatic brain injury.
Statistical analysis
The target sample size was determined a priori to detect an absolute difference of 40% in the incidence of migraine-like headache between sildenafil and placebo. The calculation assumed that 50% of participants would develop migraine-like headache exclusively after sildenafil administration, compared with 10% exclusively after placebo. These assumptions were informed by findings from prior human provocation studies in migraine populations showing substantial active–placebo differences (20–22). A minimum of 21 participants was required to achieve 80% statistical power at a one-sided α = 0.05, considering the crossover design and within-subject comparisons.
The primary outcome was the difference in the incidence of migraine-like headache following sildenafil versus placebo during the 12-h observation period. The secondary outcome assessed differences in area under the curve (AUC) of headache intensity scores between sildenafil and placebo over the same period. The outcome analyses were conducted per protocol and included only participants who completed both experimental sessions. All adverse events were prospectively monitored and recorded in both arms.
Baseline demographic and clinical characteristics were summarized using descriptive statistics. Continuous variables were reported as means ± standard deviations for normally distributed data or as medians with interquartile ranges for non-normally distributed data. Normality was assessed by visual inspection of histograms and confirmed using the Shapiro-Wilk test. Categorical variables were presented as absolute counts and percentages.
The primary outcome—the incidence of migraine-like headache within 12 h after administration—was compared between arms using McNemar's test for paired data. For the secondary outcome, the AUC for headache intensity scores over the 12-h period was calculated using the trapezium rule and adjusted for baseline values. Baseline-corrected AUCs were compared using the Wilcoxon signed-rank test. Paired t-tests were employed to assess within-subject percentage changes in mean arterial blood pressure and heart rate between sildenafil and placebo sessions. Potential carryover effects were evaluated using Fisher's exact test. All statistical analyses were performed using R version 4.5.1.
Results
Participants
From January to June 2023, 145 individuals were screened, of whom 21 met all eligibility criteria and completed both experimental days (Figure 1). Participants had a mean age of 42.3 ± 11.9 years, and 57% (n = 12) were female. The median time since mild traumatic brain injury was 5.4 years (IQR, 3.2–7.9), and participants experienced an average of 23.5 ± 9.3 monthly headache days. Most participants (20 of 21; 95%) had a migraine-like phenotype, and one-third (7 of 21; 33%) reported current use of preventive headache medication(s). A detailed summary of demographic and clinical characteristics is presented in Table 1.

Study flow diagram. n; number.
Headache response
Sildenafil induced migraine-like headache in 15 (71%) of 21 participants, compared with 4 (19%) following placebo (P = 0.003; Table 2). Among these, 11 participants developed migraine-like headache only after sildenafil administration. All
Clinical characteristics of study participants after sildenafil and placebo.

No, number; TTH, tension-type headache; Bilat, bilateral; Unilat, unilateral; Throb, throbbing; Pres, pressing; Comb, combined throbbing and pressing; NA, not applicable.
Headache characteristics are reported as: localization (unilateral or bilateral) / pain intensity (11-point numeric rating scale, where 0 = no headache and 10 = worst imaginable headache) / headache quality (throbbing, pressing, or combined) / aggravation by routine physical activity (denoted as “+” for presence and “–” for absence).
Participant-reported assessment of whether the headache following sildenafil or placebo administration resembled their usual migraine-like headache, if applicable.
Migraine-like headache was classified based on the criteria defined in Table 1.
Associated symptoms include nausea, photophobia, and phonophobia. Worsening of these symptoms is defined as an increase in severity at headache onset compared to baseline (time of infusion start), rated on a 4-point Likert scale (0 = none, 1 = mild, 2 = moderate, 3 = severe).
Baseline-corrected AUC for headache intensity scores was significantly higher following sildenafil compared to placebo (P < 0.001; Figure 2A). Rescue medication was required in 8 participants post-sildenafil and in 3 participants after placebo.
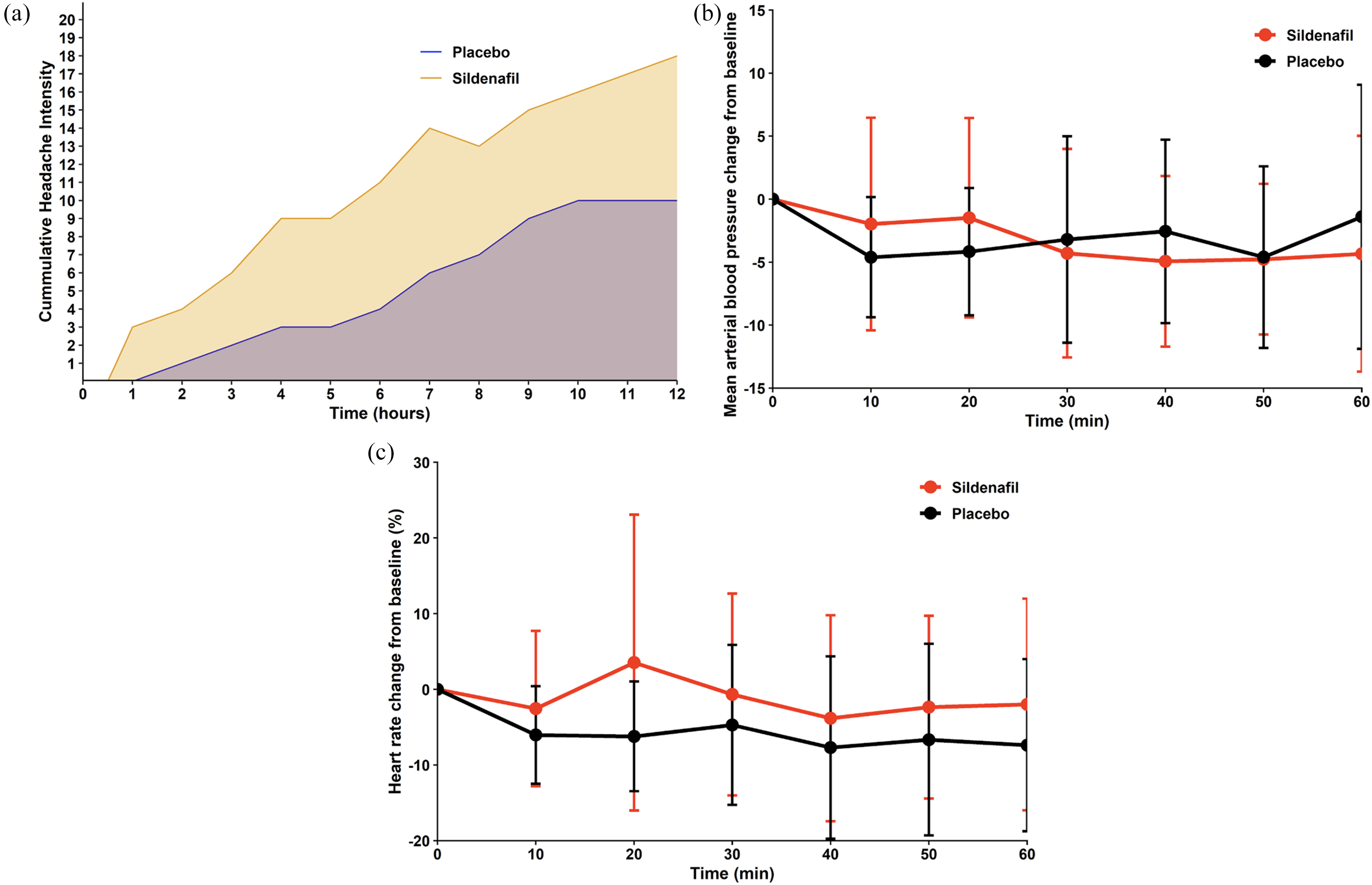
Median peak headache intensity following sildenafil was rated as 5 (IQR, 4–6) on an 11-point scale, and the median time to peak intensity 240 min (IQR, 120–360). Headaches were most often of bilateral location (76%; n = 16) and with pressing quality (67%; n = 14). Photophobia (62%; n = 13), phonophobia (24%; n = 5), and nausea or vomiting (29%; n = 6) were commonly reported, and routine physical activity exacerbated the headache in more than half of participants (57%; n = 12).
Adverse events and rescue medication use
The most common adverse events were palpitations, warm sensations, and facial flushing. Palpitations and warm sensations were each reported by 6 (29%) participants after sildenafil and 4 (19%) after placebo. Facial flushing was reported by 5 (24%) participants after sildenafil administration, of whom 3 (14%) developed a migraine-like headache, and by 4 (19%) participants after placebo, of whom 2 (10%) developed a migraine-like headache. Rescue medication was required for headache relief in eight participants after sildenafil administration and in three participants after placebo.
Hemodynamic variables
During the 60-min in-hospital observation period, the baseline-corrected AUC for mean arterial pressure was lower following sildenafil administration, compared with placebo (P = 0.002; Figure 2B). In parallel, the baseline-corrected AUC for mean heart rate was higher after sildenafil than after placebo (P = 0.008; Figure 2C).
Discussion
This randomized double-blind, placebo-controlled, 2-way crossover study offers the first experimental evidence that pharmacologic elevation of cGMP via PDE-5 inhibition provokes migraine-like headache in individuals with persistent PTH. Among participants with no pre-trauma history of migraine, a single oral dose of sildenafil induced migraine-like headache in 15 (71%) of 21 cases, with symptom profiles closely resembling their spontaneous exacerbations. These findings position cGMP-dependent signaling as a novel mechanistic pathway in PTH and suggest a potential therapeutic target in this patient population.
Existing evidence and added value
The role of second messengers—particularly cAMP and cGMP—in the pathogenesis of headache disorders is increasingly supported by experimental provocation studies (10,13). Agents that elevate intracellular cAMP, such as CGRP, PACAP, and cilostazol (a PDE-3 inhibitor), have consistently triggered migraine attacks in people with migraine (11,12,15) as well as migraine-like headache in those with persistent PTH (7,9,23). For example, intravenous CGRP induced migraine-like headache in 21 (70%) of 30 participants with persistent PTH (5), while PACAP triggered similar responses in 20 (95%) of 21 participants (7). These findings are paralleled in people with migraine, reinforcing the relevance of cAMP signaling across both migraine and persistent PTH (9).
Extending this framework to cGMP, the present study shows that sildenafil—a PDE-5 inhibitor that increases intracellular cGMP—provoked migraine-like headache in 15 (71%) of 21 participants with persistent PTH. This rate mirrors earlier findings in people with migraine, where 10 (83%) of 12 participants developed attacks after sildenafil administration (17). The similarity in response profiles suggests that cGMP-dependent signaling contributes to headache pathogenesis in a manner analogous to cAMP.
Together, these observations support a model in which converging second messenger pathways—mediated by cAMP and cGMP—underlie the provocation of migraine-like headache in both migraine and persistent PTH. This expanded mechanistic understanding highlights the potential for therapeutic strategies that target both signaling cascades in order to more effectively treat headache disorders across etiologies.
Hypothesized mechanisms and sites of action
Migraine-like headache elicited by pharmacologic enhancement of cGMP signaling is hypothesized to arise through three distinct but potentially overlapping mechanisms, each engaging the NO–sGC–cGMP pathway at different anatomical sites (Figure 3A–C).

Hypothesized mechanisms and sites of action of sildenafil-induced migraine-like headache. The figure illustrates a proposed mechanistic pathway through which sildenafil may induce migraine-like headache in individuals with persistent post-traumatic headache. (A) Sildenafil penetrates the vascular smooth muscle cells of meningeal arteries, where it selectively inhibits phosphodiesterase 5 (PDE-5), reducing the breakdown of cyclic guanosine monophosphate (cGMP). The resulting elevation in intracellular cGMP activates protein kinase G (PKG), which phosphorylates several downstream targets, including ATP-sensitive potassium KATP channels and large-conductance calcium-activated potassium BKCa channels. Phosphorylation promotes channel opening, leading to potassium efflux and subsequent meningeal vasodilation. Elevated extracellular potassium increases the resting membrane potential of adjacent nociceptive neurons, causing depolarization. Additionally, the vasodilation may mechanically stimulate and sensitize nearby perivascular nociceptors. (B) Sildenafil directly activates meningeal nociceptors of the trigeminal nerve. By crossing the neuronal membrane and inhibiting PDE-5, it modulates intracellular cGMP levels, ultimately contributing to the depolarization of these nociceptive neurons. (C) Sildenafil directly activates nociceptive neurons in the trigeminocervical complex (TCC). Sildenafil may likewise promote depolarization via cGMP-dependent pathways.
Before considering potential mechanisms, it is important to address whether early systemic hemodynamic changes could account for the observed migraine-like headache phenotype. Mean arterial pressure and heart rate were recorded only during the first 60 min after drug administration, whereas headache outcomes were assessed over a 12-h post-ingestion period. Although sildenafil induced modest, statistically significant hemodynamic changes during this early window, migraine-like headache onset and peak intensity most often occurred several hours later, indicating a temporal dissociation. Moreover, electrophysiological data demonstrate that local dural activation of the NO–cGMP pathway, independent of systemic administration, is sufficient to sensitize meningeal nociceptors, arguing against early hemodynamic changes as a primary driver of migraine-like headache (24).
The first proposed mechanism involves vessel-to-neuron signaling within the meninges, in which vascular changes promote activation of adjacent trigeminal afferents. NO activates sGC in vascular smooth muscle cells, leading to elevated cGMP levels that are further amplified by PDE-5 inhibition (17,25–27). The accumulation of cGMP activates protein kinase G, which in turn opens ATP-sensitive potassium (KATP) channels and large-conductance calcium-activated potassium (BKCa) channels, resulting in hyperpolarization and relaxation of vascular smooth muscle (6,8,10,13,21,22). The resulting dilation of meningeal arteries is hypothesized to mechanically deform perivascular trigeminal fibers, thereby activating mechanosensitive nociceptors (10,13). Concurrent K+ efflux through these channels might chemically sensitize perivascular sensory neurons, augmenting the overall nociceptive drive (10,13).
Supporting this mechanism, levcromakalim—a vascular KATP channel opener—induces migraine attacks in people with migraine (21) and migraine-like headache in those with persistent PTH (6). Genetic evidence further implicates this pathway, as mice with loss-of-function mutations in the Kir6.1 subunit of vascular KATP channels exhibit reduced sensitivity to NO donor–induced pain-like behavior (28). In addition, pharmacologic inhibition of vascular KATP channels with glibenclamide attenuates pain responses to both systemic and dural NO donors, underscoring the role of vascular KATP channels in cGMP-mediated nociceptive signaling (29).
The second hypothesized mechanism posits direct peripheral sensitization of trigeminal afferents that innervate the meninges. PDE-5 is expressed in trigeminal ganglion neurons (30), and sildenafil has been shown to increase neuronal cGMP levels (30), extending the duration of NO-mediated signaling. Furthermore, electrophysiological studies demonstrate that systemic or local administration of NO donors induces delayed mechanical hypersensitivity in meningeal nociceptors (31). However, it remains uncertain whether these effects in preclinical models reflect direct neuronal sensitization or are secondarily mediated via vessel-to-neuron signaling.
The third hypothesized mechanism involves direct modulation of nociceptive messaging via cGMP signaling within the trigeminocervical complex. Because sildenafil crosses the blood–brain barrier (32), its central effects are biologically plausible. Systemic NO donors increase c-Fos expression in the trigeminocervical complex (33), and electrophysiological recordings from this region show increased spontaneous firing and enhanced responses to dural and cutaneous stimuli following NO donor administration (34). These responses are dose-dependent and can be attenuated by triptans, consistent with migraine pharmacodynamics (34). Taken together, these findings suggest that cGMP not only amplifies afferent input from the periphery but also modulates second-order nociceptive circuits within the CNS (34). However, whether cGMP-mediated activation of CNS pathways constitutes a primary initiating mechanism or represents downstream amplification of peripheral input remains unresolved and is a key focus for future research.
Therapeutic implications and future directions
Targeting PDE-5 signaling represents a promising, mechanistically driven strategy for treating persistent PTH. Experimental evidence showing that sildenafil provokes migraine-like headache in individuals with persistent PTH underscores the relevance of cGMP elevation in headache pathogenesis. While sildenafil also affects PDE-1 and PDE-6 (35,36), their roles in nociceptive messaging remain poorly defined.
However, whether this response reflects a sildenafil-specific pharmacological effect or a broader cGMP-dependent mechanism remains uncertain. Replication of these findings using other modulators of the cGMP pathway—including additional PDE-5 inhibitors, sGC activators, NO-donor systems, or agents with distinct downstream profiles—will be essential to determine whether the observed effects reflect a sildenafil-specific pharmacology or a broader cGMP-dependent mechanism. Moreover, because vascular diameter was not directly assessed in this study, the precise contribution of meningeal vasodilation relative to downstream neuronal cGMP signaling remains uncertain. Future studies capable of disentangling vascular and neuronal components of cGMP pathway activation will be crucial for establishing the dominant mechanism through which sildenafil induces migraine-like headache in persistent PTH.
Therapeutic approaches that modulate cGMP—either alone or in combination with cAMP-targeting strategies—could yield additive or synergistic benefit. However, systemic activation of PDE-5 carries the risk of off-target effects due to its expression in vascular smooth muscle and other tissues (37). Excessive cGMP reduction, for example, could lead to vasoconstriction or disrupt homeostatic signaling in non-neuronal systems.
Developing isoform-specific or tissue-targeted PDE-5 modulators offers a pathway toward greater therapeutic precision. Selective inhibition in vascular or trigeminal pathways could maximize efficacy while minimizing systemic side effects. Advancing this approach will require rigorous preclinical and clinical testing to establish safety, optimize delivery, and assess long-term outcomes in individuals with persistent PTH.
Strengths and limitations
This study has several strengths, including a randomized, double-blind, placebo-controlled, 2-way crossover design that allowed each participant to serve as their own control. This approach minimizes interindividual variability and enhances statistical power. Furthermore, our eligibility criteria ensured a homogenous population of participants with persistent PTH and no pre-trauma migraine history, thereby isolating the effect of PDE-5 inhibition in a well-defined clinical phenotype. In addition, structured diaries and standardized symptom assessments enabled precise temporal mapping of headache characteristics.
Several limitations should be acknowledged. First, the in-hospital observation period was limited to 60 min due to logistical constraints, which is shorter than the expected time to peak plasma concentration for 100-mg oral sildenafil (typically 60 to 90 min). Second, the use of rescue medication—though necessary for ethical reasons—could have influenced headache duration or intensity, potentially confounding post-discharge symptom trajectories. Lastly, unmeasured environmental factors could have impacted interparticipant variability in headache expression.
Conclusions
Selective PDE-5 inhibition, by elevating intracellular cGMP, induces migraine-like headache in individuals with persistent PTH and no pre-trauma migraine history. This controlled provocation offers the first direct experimental evidence implicating cGMP-dependent signaling in PTH pathogenesis. These findings establish cGMP as a mechanistically distinct and therapeutically actionable target in persistent PTH.
Key findings
In a randomized, placebo-controlled crossover trial, sildenafil, a PDE-5 inhibitor, reliably induced migraine-like headache in individuals with persistent post-traumatic headache (PTH).
The provoked headache response provides the first direct experimental evidence that intracellular cGMP signaling contributes to headache generation in PTH, extending pathophysiological insights beyond clinical observation.
These findings implicate cGMP-dependent pathways in PTH pathogenesis and suggest that modulation of this signaling cascade could represent a novel therapeutic avenue for future investigation.
Supplemental Material
sj-docx-1-cep-10.1177_03331024261419409 - Supplemental material for Hypersensitivity to phosphodiesterase-5 inhibition in post-traumatic headache: Evidence of cGMP-dependent signaling
Supplemental material, sj-docx-1-cep-10.1177_03331024261419409 for Hypersensitivity to phosphodiesterase-5 inhibition in post-traumatic headache: Evidence of cGMP-dependent signaling by Haidar M Al-Khazali, Rune H Christensen, Anna G Melchior, Messoud Ashina and Håkan Ashina in Cephalalgia
Footnotes
Abbreviations
Acknowledgments
Not applicable.
Author contributions
Study concept and design: H.A.; Data acquisition and analysis: H.MA, R.H.C., A.G.M., H.A.; Drafting of the manuscript: H.MA, R.H.C., H.A.; Critical revision of the manuscript: H.MA, R.H.C., A.G.M., MA, H.A.; Funding acquisition: H.A.
Consent to participate
All participants provided written informed consent prior to undergoing any study-related assessments or procedures.
Consent for publishing
Authors agree to publish the article in Cephalalgia.
Data availability statement
Upon reasonable request, the corresponding author will provide the necessary data and materials to interested researchers for the purpose of academic scrutiny, reproducibility, and further scientific investigation.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: H.M.A. reports personal fees from Abbvie, Lundbeck, and Pfizer, outside of the submitted work. RH.C. reports personal fees from Abbvie, Lundbeck, Teva, and Pfizer, outside of the submitted work. MA is also a Primary Investigator for ongoing clinical trials sponsored by AbbVie, Lundbeck, and Pfizer. MA also serves as an Associate Editor of Brain and The Journal of Headache and Pain. HA has received personal fees from AbbVie, Lundbeck, Pfizer, and Teva, outside of the submitted work. A.G.M. declares no potential conflicts of interest.
Ethical considerations
The study protocol was approved by the relevant ethics committee (Identifier: H-21067665), and all study-related assessments and procedures adhered to the principles outlined in the Declaration of Helsinki.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding was provided by the Danish National Research Foundation (DNRF188 to MA) and Lundbeck Foundation (R310-2018-3711 to MA; R403-2022-1352 and R481-2024-1392 to HA). The funding sources were not involved in the design or conduct of the study; in the collection, analysis, or interpretation of the data; or in the preparation, review, or approval of the manuscript.
Open practices
Not applicable.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.