
Abstract
Aim
To assess whether the timing of atogepant administration influences its tolerability and effectiveness over 12 weeks in patients with episodic and chronic migraine in a real-world setting.
Methods
This is a post-hoc analysis of the STAR study, a prospective, Italian, multicenter study evaluating atogepant 60 mg for migraine prevention. Data were collected at baseline (T0) and after the first 12 weeks (T3) of treatment. Patients were grouped by administration timing (morning vs. evening) and by administration with or without food. Changes in monthly headache days (MHDs), monthly migraine days (MMDs), and Migraine Disability Assessment (MIDAS) were measured. Tolerability was evaluated via adverse events (AEs). Linear mixed-effects models (LMMs) were used.
Results
Eighty-one patients (86% females, mean age 50.8 ± 13.7 years) were included. At T3, MMDs decreased from 16.6 to 9.7 (p < 0.001) and MHDs from 19.8 to 11.9 (p < 0.001); 60% of patients achieved ≥50% reduction in MMDs. AEs occurred in 34 (42%) participants. Atogepant was taken in the morning by 57% and in the evening by 43% of patients. Fifty-seven out of 81 participants (70.4%) took atogepant with food. No significant differences in MMDs, MHDs, or AEs emerged between morning and evening users. Evening users had higher baseline MIDAS scores (estimated marginal means [EMMs]: 69.9 vs. 39.9, p = 0.034) that showed a greater reduction compared to morning users (F(1,63) = 6.29, p = 0.015), reaching similar final scores after 12 weeks (EMMs: 25.1 vs. 23.8). No difference in atogepant effectiveness and tolerability according to intake with or without food, except for a reduction in MHDs for patients who took atogepant without food (EMMs from 21.3 to 9.9 vs with food: EMMs from 18.4 to 12.7; F(1,79) = 8.553, p = 0.005).
Conclusions
Atogepant significantly reduced migraine burden over 12 weeks in a real-world setting. Overall, the timing of atogepant administration did not affect its effectiveness or tolerability. However, a greater reduction in MIDAS scores was observed among evening users. Whether this reflects a pharmacological advantage or a ceiling effect remains unclear. Taking atogepant without food was associated with a significantly greater reduction in MHDs, whereas changes in MMDs and MIDAS scores did not differ between groups. Long-term and dedicated studies are needed to evaluate and confirm these findings.
Trial Registration
The main study (STAR) was preregistered on clinicaltrial.gov, NCT06414044.
This is a visual representation of the abstract.
Keywords
Introduction
Migraine is a cyclical neurological disorder characterized by recurring attacks with different individual frequencies. 1 Chronobiological rhythms can influence the recurrence and periodicity of migraine attacks. 2 Indeed, several studies reported morning hours (between 6 a.m. and 2 p.m.) as the peak time of onset for migraine attacks, suggesting that circadian rhythms, rather than infradian rhythms, may play a role in the recurrence of attacks.2,3 This is further supported by evidence of hypothalamic activation in the days preceding migraine onset, 4 suggesting a possible common denominator between attack initiation and circadian periodicity.
Interestingly, most metabolic processes in mammals are under circadian control, including those involved in the pharmacokinetics of drugs. 5 Studies have shown daily rhythmicity in the expression of drug-metabolizing enzymes and transporters.6,7 Then, the timing of drug administration may have a role in maximizing effectiveness and minimizing side effects. 8 For example, the pharmacokinetics of paracetamol, a common drug used in different conditions, is influenced by the timing of administration, with differences in absorption between the morning and evening. 9 This has led to an increasing interest in the study of chronotherapy, which aims to optimize medical treatments by considering the circadian rhythms. 10 Although chronobiological rhythms may play a direct role in the recurrence of migraine attacks, 2 chronotherapy and its effect on drugs have rarely been considered in migraine treatment.
Atogepant, a novel oral calcitonin gene-related peptide receptor antagonist (gepant), has been recently approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the preventive treatment of both episodic (EM) and chronic (CM) migraine. 11 Several randomized controlled trials (RCTs) have shown its efficacy, safety, and tolerability12–16; however, no studies have investigated whether the timing of administration influences its effectiveness or side effects, resulting in a lack of recommendations for the optimal timing of administration.
Furthermore, the timing of administration is intrinsically related to meals, implicating whether the intake of atogepant would be better with or without meal, a question typically asked by patients during clinical practice. A dedicated phase 1 study evaluated the impact of a high-fat meal on the exposure of atogepant in healthy adults. The results showed that food intake reduced atogepant exposure by approximately 18% for the area under the curve (AUC) and 22% for maximum plasma concentration (Cmax). However, these changes were not deemed clinically relevant, 17 but no data on correlation with effectiveness and adverse events in migraine patients have been reported to date.
In this post-hoc analysis of the real-world STAR study, 18 we aimed to evaluate whether the timing of atogepant administration or its intake with meals can influence its effectiveness and tolerability over 12 weeks in patients with migraine.
Methods
Study design
This post-hoc analysis was derived from the STAR study, a real-world, prospective, multicenter, investigator-initiated, and independent study performed across several Italian headache centers. The study was pre-registered on clinicaltrials.gov (NCT06414044) and has a two-year pre-planned follow-up. The study design, as well as its primary and secondary outcomes, have been previously reported. 18 This analysis was conducted on all available patients within the first 12 weeks of follow-up, including all consecutive patients treated with atogepant 60 mg orally for migraine prevention who had taken at least one tablet, regardless of discontinuation, from June to December 2024. Data collection has been conducted using the open online database Research Electronic Data Capture (REDCap) and the Empedocle electronic platform (developed for the RICe study). The study was approved by the local Ethics committee as part of the Registro Italiano Cefalee (RICe) study (Studio RICe, 14591_oss CEAVC and subsequent amendments). Further details regarding the RICe study can be found elsewhere.19,20 The study was conducted in accordance with the Declaration of Helsinki. All patients signed a written informed consent form before enrolment. At the time the study started, atogepant was not subsidized by the Italian National Health Service. Consequently, all patients received the drug without cost thanks to an agreement between the Italian Medicines Regulatory Agency (AIFA), regional healthcare systems, and the manufacturing company.
Participants
Eligible individuals were adults aged 18 years or older who had a diagnosis of EM, with or without aura, or CM in accordance with the diagnostic criteria of the International Classification of Headache Disorders, 3rd edition (ICHD-3). 21
Inclusion criteria were: i) availability of headache diaries over at least one month before the enrolment, ii) at least 4 monthly migraine days (MMDs) in the 3 months preceding the enrolment, iii) a clinical indication for prescription of atogepant 60 mg, iv) information about the timing of atogepant administration.
The main exclusion criteria were: i) any contraindications to gepants, ii) any additional diagnoses of medical conditions and/or comorbidities that could have influenced the aims of the study according to clinicians, iii) pregnancy and breastfeeding.
Participants were enrolled regardless of the previous number of preventive treatments interrupted for ineffectiveness or tolerability issues. According to the European Headache Federation (EHF) criteria, ineffectiveness was defined as no meaningful improvement in migraine-related variables after the administration of drugs for at least 6 weeks at the appropriate dose. 22
Collected variables
Clinicians verified the headache diagnosis and collected demographic features before enrolment. Clinical variables were collected at baseline (T0) and after 12 weeks of therapy (T3). A paper-and-pencil diary was used to collect data.
The timing of atogepant administration over the 12-week treatment period was collected at T3. Patients were subdivided into morning (from 6:01 a.m. to 6:00 p.m.) and evening (from 6:01 p.m. to 6:00 a.m.) drug takers depending on the timing of atogepant administration. For this post-hoc analysis, we pre-specified a binary contrast to preserve statistical power and ensure inclusion of all eligible patients, with the aim of generating preliminary effect-size estimates for our pre-planned 12-month study. Although the labels “morning” and “evening” may appear simplified, this dichotomy was selected for pharmacological reasons. Atogepant reaches peak plasma concentration approximately 1–2 h after intake and has a terminal half-life of about 11 h; under these conditions, a 12-h subdivision represents a reasonable framework for exploring whether aligning the main exposure with the early-morning peak of migraine attacks influences clinical outcomes.
There were no explicit recommendations within the STAR protocol regarding atogepant timing, and dosing was left entirely to patient/clinicians preference. Patients with irregular administration timing (less than 85% of adherence of the same timing of intake) or with missing data were excluded. Furthermore, we collected information on whether the drug was taken with or without food. No strict definition was mandated. For the STAR study, “with food” was defined as intake during a meal or within approximately 30 min after it, whereas “without food” was defined as intake on an empty stomach (i.e., before any meal in a fasted state).
Effectiveness was evaluated through the change in MMDs and monthly headache days (MHDs) between T0 and T3. Responders were defined as patients who achieved a reduction of MMDs by at least 50% [RR50%] compared to baseline. A headache day was defined as a calendar day on which the patient reported any headache. In contrast, a migraine day was defined as a calendar day on which the patient reported a headache with the characteristics of migraine or used a triptan to treat it.
Medication overuse (MO) was defined as a regular overuse of acute symptomatic treatments (10 or more days/month for triptans and combination analgesics or 15 or more days/month for non-opioid analgesics) for more than 3 months in patients with CM. 21
Migraine-related disability was assessed using the Migraine Disability Assessment (MIDAS) questionnaire, 23 which was administered at both T0 and T3. Other clinical variables, including comorbidities, were reported according to patients’ charts and outpatient interviews during clinical practice.
Adverse events (AEs) were recorded throughout the study period without the use of a checklist or active solicitation, reflecting real-world clinical practice. Additionally, gastrointestinal-related (GI) adverse events were also categorized (yes/no) to create a composite gastrointestinal adverse event (including nausea and constipation) and also categorized (yes/no) including patients with nausea or inappetence.
Statistical analysis
This is a post-hoc analysis of the STAR study; we did not perform a structured sample size calculation and based the analysis on a convenience sample. The Shapiro-Wilk test was used to assess normality and revealed a non-normal distribution of several variables. Thus, statistical analysis was conducted with non-parametric tests. We reported mean [95% confidence interval or interquartile range (IQr) or mean plus standard deviation (SD) as appropriate] for continuous variables and number (percentage) for categorical data. No imputation was made for missing data, which are reported in tables and text. Pre- and post-treatment differences within groups for quantitative variables were compared using the Wilcoxon signed-rank test, while the exact McNemar's test was applied for proportions in paired samples. A Mann-Whitney U test was conducted to assess differences between two independent groups for continuous variables.
To examine changes in MMD over time and assess the impact of the timing of drug intake, we employed a linear mixed-effects model (LMM) for repeated measures. The dataset was structured with each participant contributing two observations (one at baseline and one at follow-up). The number of predictors included in the model was limited to ensure the appropriate model fit given the small sample size and variables selected based on clinical availability, missing data, and predicted impact.
The model included a within-subject factor of time (T0 vs. T3), a between-subject factor representing timing of drug intake (coded as morning vs. evening), the interaction between time and administration-timing group, and a random intercept for each subject to account for within-individual correlation over time. The covariance structure was specified as unstructured. Parameter estimation was performed using restricted maximum likelihood (REML). The same model was applied for MHDs as dependent variables. Finally, we assessed changes in MIDAS scores over time, considering the effects of atogepant administration timing (morning vs. evening), MHDs, and their interactions. The model accounted for repeated measures and included MHDs as a covariate. The same LMMs were also applied using a between-subject factor representing food intake (food yes/food no).
A two-tailed p-value <0.05 was considered significant for all variables, with a Bonferroni's correction where appropriate. All data were analysed using SPSS software version 29.0 (IBM Corp. SPSS Statistics, Armonk, NY, USA), and graphs were designed using GraphPad Prism version 10.00 (La Jolla, USA).
Results
The final study population included 81 participants with the available time intake data, out of 106 patients completing the 12-week treatment. Among them, 86.4% were females, with a mean age of 50.8 ± 13.7 [SD] years, and an age at disease onset of 18.8 ± 10.5 years. Forty-six (56.8%) participants had CM; among them, 36/46 (78.3%) had a concomitant diagnosis of MOH. Figure 1 reports the study flowchart.
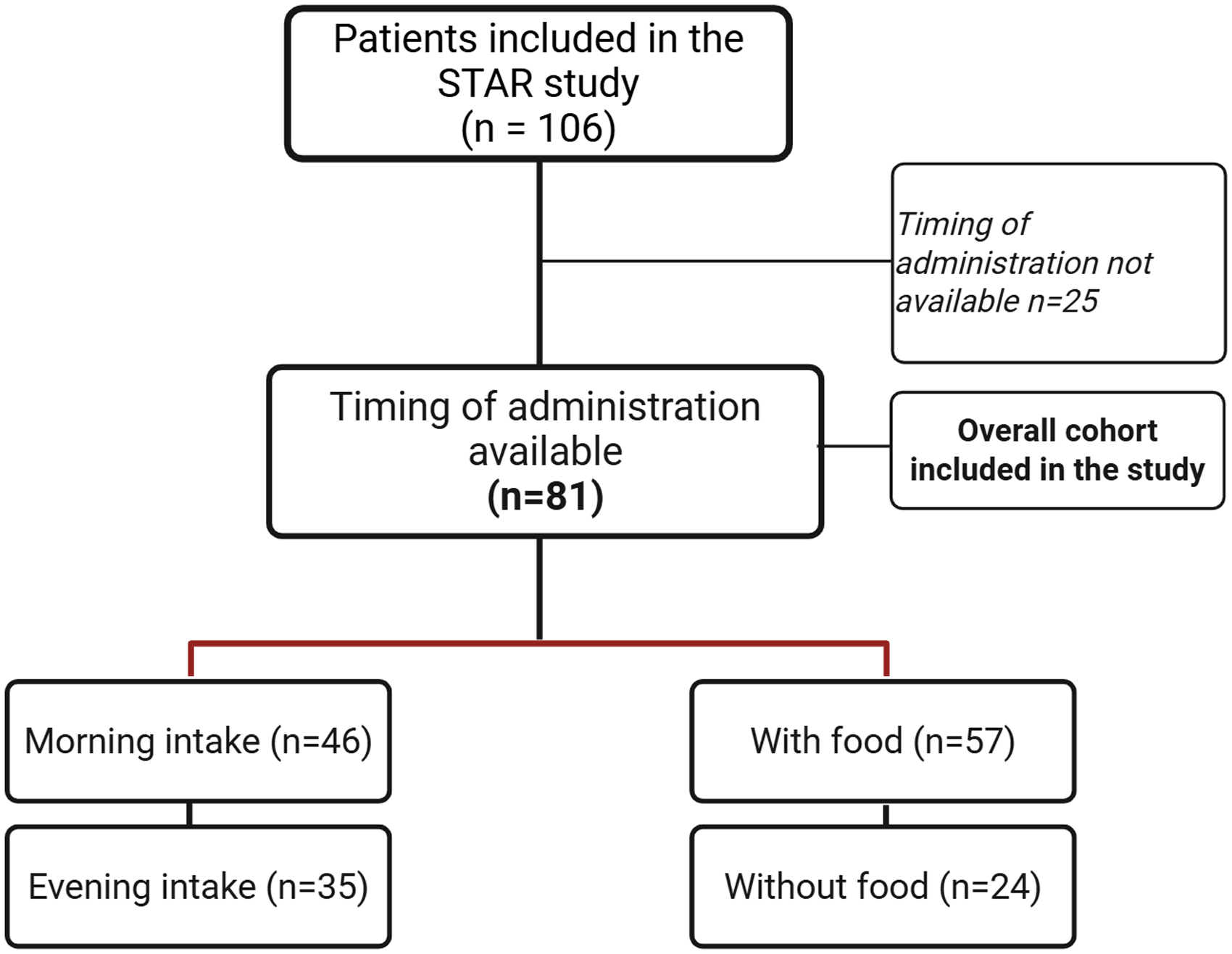
Study flowchart of patients included in the STAR study and stratification by atogepant intake (morning vs evening and with or without food)
At baseline (T0), patients reported a mean of 16.6 ± 8.3 MMDs and 19.2 ± 8.3 MHDs. Regarding medication intake, among the 63 patients with available data, the mean number of analgesics per month (AMNs) was 20.0 ± 18.4, while the acute medication days (AMDs) (n = 60) had a mean of 14.7 ± 9.5. Fifty-three percent of the participants had at least one clinically relevant comorbidity, of whom 14.8% and 16.0% reported psychiatric and gastroenteric comorbidities, respectively. Clinical, demographic features, and disability questionnaire scores (MIDAS and HIT-6) are fully detailed in Table 1. The mean number of failed preventive classes in the study population was 4.46 ± 2.21 (range: 0–9), with a median of 4. Among them, the ineffectiveness of anti-CGRP monoclonal antibodies (mAbs) and OnabotulinumtoxinA was reported by 50.6% and 39.5% of patients, respectively. Previous preventive treatments are detailed in Table S1.
Clinical and demographic features of the overall population and subdivided in morning/evening administration.

Calculated in patients with CM. Percentages are expressed on column total. AMNs, number analgesics per month; AMD; days with at least one analgesics use per month;BMI, body mass index; CM, chronic migraine; HIT-6, headache impact test; MIDAS, Migraine Disability Assessment questionnaire; MOH, medication-overuse headache; MHDs, monthly headache days; SD, Standard deviation.
Atogepant was taken in the morning (from 6:01 a.m. to 12:00 a.m.) by 41 patients (50.6%), in the afternoon (from 12.01 p.m. to 6:00 p.m.) by 5 individuals (6.2%), in the evening (from 6:01 p.m. to 00:00 a.m.) by 34 individuals (42.0%), and at night (from 00:01 a.m. to 6:00 a.m.) by only 1 patient (1.2%). Considering the variable dichotomously categorized in morning/evening, 56.8% of patients (46/81) took the study drug in the morning and 43.2% (35/81) in the evening (Figure 1). Overall, 70.4% (57/81) of patients took atogepant with food. The clinical, demographic features and disability (MIDAS and HIT-6) questionnaire scores subdivided for the timing of intake and with/without food are reported in Table 1 and Table 2, respectively.
Clinical and demographic features of the population subdivided in with/without food administration.

Calculated in patients with CM. Percentages are expressed on column total. AMNs, number analgesics per month; AMD; days with at least one analgesics use per month; BMI, body mass index; CM, chronic migraine; HIT-6, headache impact test; MHDs, monthly headache days; MOH, medication overuse headache; MIDAS, Migraine Disability Assessment questionnaire; SD, Standard deviation. p values are calculated with the Mann-Whitney U test.
According to the timing of intake, only the MIDAS score differed between the groups, with a mean of 55.43 (SD = 42.36) in the morning group and 78.54 (SD = 53.70) in the evening group (p = 0.034). For food intake, age when migraine became chronic (for CM patients), pain intensity, and number of AMNs, were significantly different between groups (
Only four subjects (4.9%) dropped out of the treatment with atogepant due to ineffectiveness (3/81, 3.7%) or adverse events (i.e., nausea and constipation [1/81, 1.2%]).
Effectiveness analysis
A significant reduction was observed in all collected variables of migraine frequency, acute symptomatic use and disability after 12 weeks of treatment with atogepant, regardless of the timing of atogepant administration. The number of MMDs decreased from a mean of 16.6 (SD = 8.4) at T0 to 9.7 (SD = 9.4) at T3 (Wilcoxon Z = –5.894, p < 0.001). Similarly, MHDs were significantly reduced at T3 (Z = –6.129, p < 0.001). Among the 57 patients with available data, AMNs decreased significantly (Z = –5.275, p < 0.001). Further analyses confirmed reductions in migraine-related disability and impact: AMDs (Z = –5.266, p < 0.001), MIDAS scores (Z = –6.784, p < 0.001), and HIT-6 scores (Z = –5.494, p < 0.001) all significantly improved at 12 weeks (Table 3). Fifty-two patients (64.2%) reached at least a 30% MHD response rate (RR). Specifically, 46 (56.8%) achieved ≥50%, 18 (22.2%) reached ≥75%, and 1 patient (1.2%) achieved migraine freedom. Similarly, for MMD 56 patients (69.1%) achieved 30%RR, 48 (59.3%) reached ≥50%RR, 27 (33.3%) ≥ 75%RR, and 4 patients (4.9%) experienced a complete response (100%).
Migraine-related variables and disability questionnaires at baseline and after 12-week therapy with atogepant in the overall population (n = 81).

Percentages are expressed on column total. AMNs, number analgesics per month; AMD; days with at least one analgesics use per month; HIT6, headache impact test; MIDAS, Migraine Disability Assessment questionnaire; MHDs, monthly headache days; SD, Standard deviation.
Timing of atogepant administration and effectiveness
The first model evaluated changes in MMDs during treatment (i.e., three months of treatment [time]) and assessed the impact of drug intake timing (morning/evening). The model confirmed a statistically significant main effect of treatment on MMDs (F(1,79) = 47.23, p < 0.001), indicating a reduction in migraine frequency after 12 weeks. The main effect of timing of intake was not significant (F(1,79) = 0.463, p = 0.498), suggesting no baseline difference in MMDs between timing groups. Likewise, the interaction between treatment and group stratified on timing of intake was not statistically significant (F(1,79) = 0.121, p = 0.729), indicating that the observed treatment effect did not differ depending on the time of day the medication was taken. Estimated marginal means (EMMs) [95%CI] showed that patients taking the medication in the morning experienced a decrease in MMDs from 15.9 [13.4–18.4] to 9.3 [6.5–12.1], while those who took it in the evening experienced a reduction from 17.4 [14.6–20.3] to 10.1 [6.9–13.3] (Figure 2A).
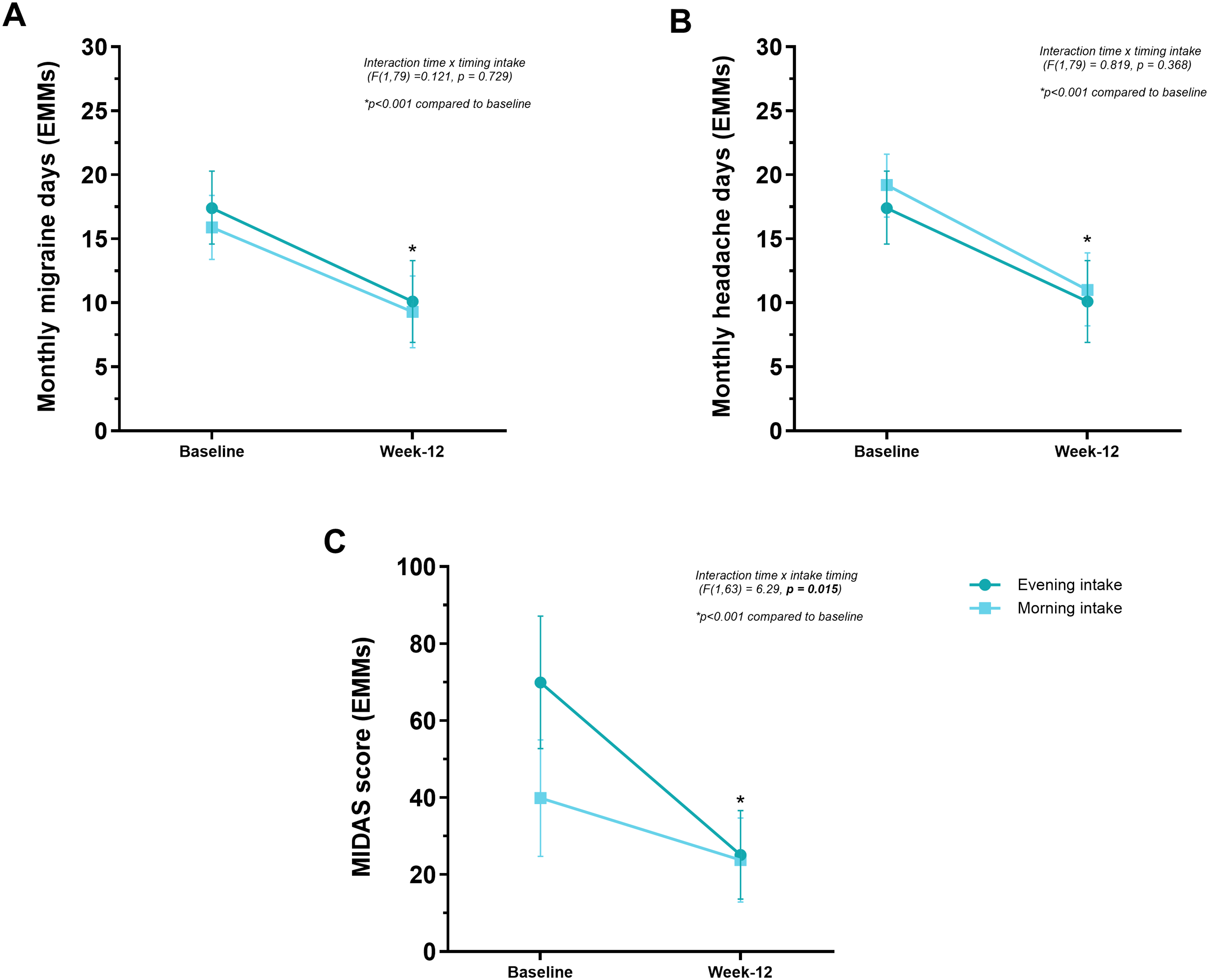
Effect of timing of atogepant intake (morning vs. evening) on clinical outcomes, MHDs (A), MMDs (B) and MIDAS score (C). Values are presented as estimated marginal means (EMMs) ± 95% Confidence interval, calculated using linear mixed-effects models. MIDAS score (panel C) is adjusted for MHDs.
Regarding MHDs, the second LLM revealed a significant main effect of treatment (F(1,79) = 57.76, p < 0.001), confirming that MHDs significantly decreased after 12 weeks. The main effect of intake timing group was not statistically significant (F(1,79) = 0.314, p = 0.577), suggesting that baseline MHD did not differ between morning and evening intake groups. Similarly, the treatment-by-timing of intake interaction was not significant (F(1,79) = 0.819, p = 0.368), indicating that the degree of improvement was not modified by the timing of drug intake. EMMs [95%CI] showed that patients taking the medication in the morning experienced a decrease in MHDs from 19.2 [16.7–21.6] to 11.0 [8.2–13.9] and those who took it in the evening from 19.3 [16.5–22.2] to 12.9 [9.6–16.2] (Figure 2B).
Finally, we evaluated whether the disability measure, the MIDAS score corrected for MHDs, could be a better variable to assess the morning-evening difference. In the LLM, there was a significant main effect of treatment (F(1,63) = 28.07, p < 0.001), indicating a reduction in MIDAS scores over the 12 weeks. MHD was also a strong predictor of MIDAS-related disability (F(26,57) = 4.07, p < 0.001), consistent with the expected association between headache burden and disability. The interaction between the timing of intake and MHD was not significant (p = 0.471). The main effect of timing of administration and MIDAS was not statistically significant (p = 0.139), suggesting that, on average, MIDAS scores did not differ significantly between patients taking the drug in the morning versus the evening at each follow-up. However, the interaction between treatment and timing of intake was statistically significant (F(1,63) = 6.29, p = 0.015), indicating that the degree of improvement in MIDAS scores during treatment varied according to the timing of intake. Specifically, evening users started from a substantially higher level of disability at baseline (EMMs [95%CI] = 69.9 [52.7–87.1]) compared to morning users (EMMs [95%CI] = 39.9 [24.7–55.0]), but both groups reached similar follow-up levels (EMMs [95%CI] = 25.1 [13.6–36.6] vs. 23.8 [12.9–34.7], respectively) (Figure 2C).
Food intake and effectiveness
As above, we also performed different LMMs to evaluate changes in MMDs, MHDs, and MIDAS in the first three months of treatment, assessing the impact of food intake (i.e., with or without meals).
For MMDs, a significant main effect of time was observed (F(1,79) = 51.435, p < 0.001), indicating a substantial reduction in MMDs from 17.0 at T0 to 9.3 at T3. The main effect of food intake was not statistically significant (F(1,79) = 0.001, p = 0.991), as well as the interaction of time and food intake (F(1,79) = 3.401, p = 0.069). EMMs [95%CI] show that patients who took the medication without food experienced a numerical reduction in MMD (from 18.0 [14.5–21.4] to 8.2 [4.4–12.1]) compared to those who took it with food (from 16.0 [13.8–18.2] to 10.2 [7.8–12.7]). However, this difference did not reach statistical significance (Figure 3A).

Effect of atogepant intake with or without food on clinical outcomes, MHDs (A), MMDs (B) and MIDAS score (C). Values are presented as estimated marginal means (EMMs) ± 95% Confidence interval, calculated using linear mixed-effects models. MIDAS score (panel C) is adjusted for MHDs.
For MHDs, the model showed a significant main effect of time (F(1,79) = 74.777, p < 0.001), reflecting a clear reduction in MHD from 19.9 at baseline to 11.3 at month 3. The main effect of food intake was not significant (F(1,79) = 0.001, p = 0.993), indicating no overall difference between groups. However, the interaction time per food intake was statistically significant (F(1,79) = 8.553, p = 0.005), suggesting that the reduction in MHDs over time differed depending on food intake. EMMs [95%CI] showed that patients taking the medication without food experienced a greater decrease in MHDs (from 21.3 [17.9–24.7] to 9.9 [5.8–13.8]) than those who took it with food (from 18.4 [16.2–20.6]to 12.7 [10.1–15.3]) (Figure 3B).
Finally, for the MIDAS score (adjusted also for MHDs), the model revealed a significant main effect of time (F(1,77.2) = 61.95, p < 0.001), indicating a substantial reduction in MIDAS scores over the treatment period. As expected, MHD was a strong independent predictor of MIDAS (F(26,57) = 4.07, p < 0.001). The main effect of food intake was not statistically significant on MIDAS (F(1,77.9) = 0.01, p = 0.919), suggesting that MIDAS scores did not differ meaningfully between patients who took the drug with or without food. Likewise, the interaction between time and food intake was not significant (F(1,77) = 1.110, p = 0.295), indicating that the degree of disability improvement over time was comparable between the two groups. After adjusting for MHD, estimated marginal means [95%CI] showed that MIDAS scores decreased from 69.0 [49.1–88.8] to 19.2 [9.0–29.5] in the group taking the drug without food, and from 63.9 [51.0–76.8] to 25.9 [19.2–32.6] in the group taking it with food. Still, this difference did not reach statistical significance (Figure 3C).
Overall tolerability and adverse events
Overall, 34 (41.9%) of patients reported at least one AE, while 12 patients reported more than one AE, with a total of 50 single AEs. The most reported event was constipation (29.6%, 24/81), followed by loss of appetite (14.8%, 12/81) and nausea (9.9%, 8/81). A total of 11.1% (9/81) of patients reported subjective weight loss, while fatigue and somnolence were infrequent, occurring in only 4.9% (4/81) and 2.5% (2/81) of patients, respectively. A composite GI adverse event category (including nausea and constipation, with both present) was observed in 33.3% of patients. Additionally, 21.0% of participants experienced either nausea or appetite loss. Finally, the objective loss of weight (i.e., all patients with ≤ 2 kilograms loss at 12 weeks) was shown in 26 patients (32.1%). Only one patient discontinued treatment due to AEs (constipation and nausea) (Table 4).
Adverse events in the overall cohort and subdivided for timing of administration and food intake.

Percentages are expressed on column total if not otherwise specified.
Timing of atogepant administration and adverse events
We compared the frequency of AEs between patients who took atogepant in the morning (n = 46) versus those who took it in the evening (n = 35), without any significant difference between the two groups for any evaluated adverse event (Table 4 and Figure 4A).

Effect of timing of atogepant intake (morning vs evening) (A and B) or with or without food (C and D) and on adverse events profile. Values are presented as means ± SD or percentage of patients. Ns. Not significant
Composite GI adverse events occurred in 13/46 (28.3%) of the morning group and 14/35 (40.0%) of the evening group (p = 0.343), while the combined occurrence of nausea or appetite loss was 9/46 (19.6%) vs 8/35 (22.9%) (p = 0.787).
Additionally, we analysed changes in body weight. The average weight reduction in the entire cohort was −0.89 ± 3.2 kg (ranging from −12 to +10 kg). When stratified by timing of administration, patients in the morning group (n = 46) experienced a mean reduction of −0.55 ± 2.9 kg, while those in the evening group (n = 35) had a numerical greater mean reduction of −1.34 ± 3.5 kg. However, this difference was not statistically significant (Mann–Whitney U = 801.5, p = 0.973) (Figure 4B). In a categorical analysis (≥2 kg reduction), proportions were similar across groups (32.6% morning vs. 31.4% evening; p = 0.910).
Food intake and adverse events
Finally, we assessed whether GI-related and other AEs differed based on whether atogepant was taken with or without food (
Discussion
In this study, we showed that atogepant significantly reduced both MHDs and MMDs and improved migraine-related disability over 12 weeks of treatment. Consistent with evidence from RCTs 24 and pharmacovigilance studies, 25 we found that GI-related symptoms, particularly constipation and nausea, are among the most common AEs in patients treated with atogepant.
However, to the best of our knowledge, this is the first study demonstrating that neither the timing of administration nor the atogepant intake with or without food influenced overall its effectiveness and tolerability. However, the reduction in MHDs was significantly greater in individuals taking the drug without food (p = 0.005), suggesting a modest but measurable better effect of dosing in the fasted state. Although the clinical magnitude of this difference appears small, it may reflect a minor pharmacokinetic interaction during routine treatment, optimizing effectiveness.
Indeed, although RCTs have shown that atogepant is effective in migraine prevention for both EM13,14 and CM, 16 none of them have investigated the possible effect of the timing of administration on its efficacy and tolerability, resulting in a lack of recommendations for the optimal time for its intake. This is mainly because the main RCTs on atogepant, phase 2b/3 trial, 12 ADVANCE, 13 ELEVATE, 14 PROGRESS, 16 and long-term phase 3 trial, 15 were not designed to investigate differences among different timings of administration. Indeed, in the ADVANCE trial, which investigated the safety and efficacy of atogepant in patients with EM, patients were randomized 1:1:1:1 to receive a once-daily dose of oral atogepant (10 mg, 30 mg, or 60 mg) or placebo for 12 weeks, without any specification or preference for the timing of administration. 13 A similar study design was applied for the ELEVATE trial, which investigated the safety and efficacy of atogepant in patients with EM and a previous failure of at least two classes of conventional oral migraine preventions. In this study, participants were randomized 1:1 to receive oral atogepant 60 mg once daily or placebo for 12 weeks, administered at approximately the same time of day, without preference for morning or evening. 14 A similar approach, but with a different randomization ratio (5:2), was applied in a Phase 3 trial assessing the long-term safety, tolerability, and efficacy of atogepant over 52 weeks. 15
Interestingly, the PROGRESS trial, which evaluated atogepant efficacy, safety, and tolerability for the preventive treatment of CM, was designed with a slightly different design. In this study, participants were instructed to take two tablets a day, in the morning and in the evening, for 12 weeks, receiving either a placebo twice daily, or atogepant 30 mg twice daily, or a morning dose of atogepant 60 mg with an evening dose of placebo. 16 A similar protocol was also employed in the first 2b/3 phase trial, in which patients were instructed to take subsequent doses twice daily at approximately the same times each day, with the following possible morning–evening administration combinations: placebo–placebo, atogepant 10 mg–placebo, atogepant 30 mg–placebo, atogepant 60 mg–placebo, atogepant 30 mg–atogepant 30 mg, and atogepant 60 mg–atogepant 60 mg. 12 Then, the latter two studies were the only RCTs that involved administering atogepant both in the morning and evening. However, none of them had a combination in which atogepant was administered alone in the evening with a placebo in the morning, precluding a possible comparison of efficacy and tolerability between morning and evening administration. Table 5 summarizes the study design and possible combinations of study drugs for each RCT.
Study design with ratio of randomization and possible combinations of study drugs for each randomized control trial conducted on atogepant for migraine prevention.

Independently from the study intervention, all the patients were instructed to take their study drug orally at approximately the same time. AM, morning dose; PM, evening dose.
Over the last decade, chronotherapy has gained the interest of numerous researchers across various fields, 10 with evidence suggesting that the timing of administration of several drugs may influence their effectiveness and AEs. 8 However, our results suggest that chronotherapy does not affect the effectiveness and tolerability of atogepant. Indeed, we found that both MMDs and MHDs were reduced after three months of treatment, independently of the timing of administration.
No significant differences emerged in the frequency and specific type of AEs, including GI-related symptoms and weight loss, between morning and evening users. Additionally, GI-related AEs were not alleviated by administering the drug with or without food. Taken together, these results suggest that GI AEs may be due to the pharmacodynamics of the drug rather than its pharmacokinetics.
The CGRP is ubiquitous in the human body, and its beta isoform is mainly localized in the nerves of the enteric nervous system. 26 Animal studies have shown that blocking the CGRP receptor may influence GI motility.27–29 A human provocation study showed that CGRP infusion causes symptoms related to GI hyperactivity in most participants, including abdominal pain, nausea, diarrhoea, and urge to defecate. These results support the idea that blocking CGRP can cause the opposite effect (i.e., GI hypomotility and consequent constipation). 30 Interestingly, this effect appears to be primarily caused by the CGRP receptor being blocked, rather than by targeting the CGRP itself. Indeed, constipation was mainly reported by patients treated with erenumab, 31 a monoclonal antibody that targets the CGRP receptor, and less frequently in patients treated with mAbs targeting the peptide directly, such as fremanezumab or galcanezumab. 32
It was hypothesized that blocking the CGRP receptor leaves the CGRP peptide free to bind the AMY1 receptor, 33 which inhibits gastric emptying34,35 and causes gastroparesis, contributing to both constipation 30 and nausea. 32 In contrast, as the stimulation of the CGRP receptor is excitatory on the GI motility, mAbs targeting the peptide may leave a small amount of free CGRP that can bind and stimulate preferentially the CGRP receptor, promoting the motility of the GI and reducing constipation. 30 As atogepant blocks the CGRP receptor, its GI-related AEs could be hypothesized to be due to a similar biological effect to that explicated by mAbs targeting the CGRP receptor. 36 Interestingly, RCTs showed a similar incidence of AEs across the different doses (10–60 mg daily), suggesting that the dose does not influence the rate of AEs.13,37
While gepants can block both the CGRP and AMY1 receptors, they bind the AMY receptor with up to 100-fold lower affinity than to the CGRP receptor, 38 leaving this receptor free to be bound by the circulating CGRP. The activation of AMY1 receptor by the circuulating CGRP could also be responsible for the nausea. Indeed, pramlintide, a synthetic analogue of human amylin, may frequently induce nausea as an adverse event. 39 The area postrema, an area located outside the blood-brain barrier, is a pivotal area for nausea and expresses amylin-specific receptors. 40 Then, similarly to the mechanism proposed for constipation, the free CGRP can bind the AMY1 receptor in the area postrema, causing nausea. 41 This may explain why a “peripheral” factor, such as administering atogepant concurrently with food, cannot reduce the incidence of GI-related AEs.
Interestingly, we observed a modest yet statistically significant interaction effect between the timing of atogepant administration and the degree of reduction in the MIDAS score. Indeed, although evening users presented a higher disability burden at baseline, they achieved similar MIDAS scores to morning users after 12 weeks, suggesting a greater improvement in disability in the evening users. Considering the absence of any effects of the timing of administration on the main measures of effectiveness, different hypotheses can be proposed to explain these results. On the one hand, it could be hypothesised that disability improved but reached a ceiling effect after 12 weeks (i.e., atogepant is equally effective regardless of baseline disability) and further improvement is difficult to obtain within this limited timeframe. On the other hand, the greater reduction in the delta of MIDAS scores observed among evening users could represent a pharmacodynamic advantage. Atogepant has a terminal elimination half-life of approximately 11 h, with a peak plasma concentration occurring 1 to 2 h after administration.13,42 Then, it could be speculated that evening administration may cause higher plasma levels in the first hour of the morning, when there is an increased risk of migraine attacks.2,3 This increased peak plasma concentration may help relieve and reduce the disability of attacks that emerge during this period. Unfortunately, this hypothesis could not be solved from the only two RCTs that used doses of atogepant in the evening (
Taking atogepant with or without food did not appear to significantly impact the overall drug's effectiveness or adverse events profile, although some data may warrant further investigation. We observed a robust and statistically significant reduction in both MMDs and MHDs with no significant main effect of food intake, but interestingly, the interaction between time and food intake reached statistical significance for MHDs. Specifically, patients taking the drug on an empty stomach showed a greater reduction in MHDs compared to those who took the drug with food. These results support the clinical flexibility of atogepant administration, as its effectiveness and tolerability appear largely unaffected by concomitant food intake, as also reported by a pharmacokinetic study in healthy controls, 17 but taking it in the fasting state could maximize effectiveness for some patients. This finding is of clinical interest and could reflect a modest pharmacokinetic interaction during long-term treatment in clinical practice.
The strengths of this study include its prospective data collection and multicentric design, reflecting the common clinical practice of several Italian headache centers, and the link to a nationwide registry of patients with headache disorders. However, it is important to acknowledge that our study has some limitations. First, this study is a secondary analysis of the principal study, which was not designed to investigate the difference between timing intake. 18 This precluded us from obtaining homogeneous cohorts with similar MIDAS scores or other variables between the two groups, although the statistical models used account for these differences. Additionally, we did not collect the time of onset of migraine attacks, and the study lacked a control group with a placebo. Our analysis did not include chronobiological measures such as circadian biomarkers or pharmacokinetic sampling, which limits our ability to directly assess chrono pharmacological mechanisms. Moreover, the STAR protocol did not provide explicit recommendations regarding the timing of atogepant administration or structured collection of adverse events, introducing potential variability that could influence chronobiological comparisons and expectation bias. Finally, the limited sample size precluded further analysis.
Despite these limitations, we strongly believe that, although preliminary, our results could promote other studies investigating this phenomenon to optimize migraine care with atogepant. Considering the above-mentioned limitations, our data should be interpreted with caution. Long-term and dedicated studies with matched cohorts are needed to confirm our findings.
Conclusion
Atogepant significantly reduced migraine burden over 12 weeks in a real-world setting. Overall, the timing of atogepant administration (morning vs. evening) or its intake with or without food seems not to affect its overall effectiveness or tolerability, including GI-related AEs. However, taking the drug without food produced a significantly greater reduction in MHDs (but not in MMDs and MIDAS score) and despite late-day users had higher MIDAS scores at baseline, they showed a greater improvement during treatment, reaching the same score as morning users after 12 weeks. Whether this reflects a pharmacological advantage, possibly due to the chronobiology of migraine, or a ceiling effect remains unclear. Long-term and dedicated studies are needed to further evaluate and confirm these findings.
Clinical implications
Atogepant reduced migraine burden over 12 weeks, independent of morning or evening intake, and regardless of food. Both monthly migraine days and monthly headache days showed significant improvement.
Evening users started with higher disability but improved more, reaching similar final MIDAS scores as morning users.
Supplemental Material
sj-docx-1-cep-10.1177_03331024261416490 - Supplemental material for The chronopharmacology of atogepant in migraine prevention: A real-world evaluation of influence of timing of administration on effectiveness and tolerability
Supplemental material, sj-docx-1-cep-10.1177_03331024261416490 for The chronopharmacology of atogepant in migraine prevention: A real-world evaluation of influence of timing of administration on effectiveness and tolerability by Luigi Francesco Iannone, Gabriele Sebastianelli, Federico De Santis, Michele Corrado, Marilena Marcosano, Raffaele Ornello, Licia Grazzi, Danilo Antonio Montisano, Francesco De Cesaris, Antonio Munafò, Giulia Vigani, Gianluca Avino, Michele Trimboli, Maria Albanese, Antonio Russo, Giorgio Dalla Volta, Marina Romozzi, Paolo Calabresi, Flavia Lo Castro, Alberto Boccalini, Paola Merlo, Maria Pia Prudenzano, Maria Rosaria Valente, Innocenzo Rainero, Giada Giuliani, Marta Altieri, Luisa Fofi, Alberto Doretti, Gloria Vaghi, Francesca Pistoia, Claudia Lanni, Delfina Ferrandi, Alessandra Rufa, Stefania Battistini, Gianluca Coppola, Pierangelo Geppetti, Simona Sacco, Simona Guerzoni, Claudia Altamura, Fabrizio Vernieri and in Cephalalgia
Footnotes
Acknowledgments
The “Società Italiana per lo Studio delle Cefalee” (SISC) is acknowledged for the “Registro Italiano delle Cefalee (RICe)”.
Ethical considerations
The local Ethics committee approved the study as part of the Registro Italiano Cefalee (RICe) study (Studio RICe, 14591_oss CEAVC and subsequent amendments).
Consent to participate
All patients signed a written informed consent form before enrolment.
Consent for publishing
All authors agreed to submit and publish with Cephalalgia.
Author contributions
LFI and GS had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis, designed this post-hoc study, performed statistical analysis and drafted the manuscript. FV and CA supervised the post-hoc and designed and conducted the main STAR study. All Authors critically reviewed the manuscript, agreed to be fully accountable for ensuring the integrity and accuracy of the work, and read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: LFI received financial support, consulting fees for the participation in advisory boards and support for attending meetings from: Teva, Eli Lilly, Lundbeck, Pfizer and AbbVie; he is Associate Editor for Frontiers in Neurology Headache and Neurogenic Pain section. GS received honoraria from AbbVie.
FV has received financial support from Allergan-AbbVie, Angelini and Lundbeck for investigator-initiated trials; consulting fees for the participation in advisory boards from AbbVie, Angelini, Eli Lilly, Lundbeck, Organon, Novartis, Pfizer, and Teva; honoraria for scientific lectures and presentations from AbbVie, Eli Lilly, Lundbeck, Novartis, Organon, Pfizer, and Teva; support for attending meetings from Abbvie, Amgen, Eli Lilly, Lundbeck, Pfizer, and Teva; he has been Principal Investigator in clinical trials sponsored by AbbVie, Eli Lilly, Lundbeck, Pfizer, and Teva; he is Co-Specialty Editor for Frontiers in Neurology Headache and Neurogenic Pain section.
SG has received fees and honoraria for advisory boards, speaker panels, or clinical investigation studies from Novartis, Teva, Eli Lilly, Pfizer, Lundbeck, Angelini, and AbbVie.
CA is Associate Editor for Frontiers of Human Neuroscience and Frontiers in Neurology Headache and Neurogenic Pain section; she received travel grants and/or personal fees for advisory boards and speaker panels, from Novartis, Eli-Lilly, Lundbeck, Teva, Lusofarmaco, Laborest, Abbvie/Allergan, Almirall, and Pfizer.
GV received personal fees from Lundbeck and travel grants from TEVA.
GG received personal fees for participation in advisory boards from Abbie and honoraria for presentation from Eli Lilly.MA received travel grant, honoraria as a speaker, or for participating in advisory boards from Abbvie, Teva, Organon, Pfizer, Eli Lilly and Lundbeck.
Other authors declare no relevant financial or non-financial interests to disclose.
Data availability statement
Open practices
Not applicable.
Rice study group
Supplemental material
Supplemental material for this article is available online.