
Abstract
Background
A subset of individuals with migraine are unsuitable for triptans due to intolerance, lack of efficacy, or contraindications. This phase 4 study assessed the efficacy and tolerability of a single 75-mg dose of rimegepant orally disintegrating tablet (ODT) for acute treatment of migraine in adults with documented triptan unsuitability.
Methods
Participants (aged ≥18 years with 4–14 migraine days per month) with documented history of (A) intolerance and/or lack of efficacy to ≥2 triptans or (B) contraindication to triptans were randomized (1:1) to rimegepant 75 mg ODT or placebo to treat a single migraine attack of moderate or severe pain intensity. Randomization was stratified by history of clinically relevant cardiovascular disease. The primary endpoint was the percentage of participants with migraine pain relief (no or mild pain) at 2 h post dose. Key secondary endpoints, tested using a hierarchal approach to control type 1 error, included the percentage of participants with migraine pain freedom at 2 h, rescue medication use within 24 h, return to normal function at 2 h, sustained return to normal function from 2–24 h and from 2–48 h, sustained migraine pain relief from 2–24 h and from 2–48 h, sustained migraine pain freedom from 2–24 h and from 2–48 h, and most bothersome symptom freedom at 2 h. Safety was assessed via adverse events (AEs) and laboratory tests.
Results
Overall, 585 participants (89.1% were female, mean age was 42.9 years) received study medication (rimegepant, n = 295; placebo, n = 290). Participants analyzed for efficacy (rimegepant, n = 286; placebo, n = 284) had documented failure to ≥2 triptans with ≥1 reason due to prior intolerance (30.5%) and/or ≥1 reason due to lack of efficacy (84.9%); 9.1% had a contraindication. Rimegepant demonstrated superiority over placebo for the primary endpoint of migraine pain relief at 2 h (55.9% vs 32.7%; difference [95% CI]: 23.2% [15.3–31.1%]; p < 0.0001) and all 10 alpha-protected key secondary endpoints including pain freedom at 2 h (all p ≤ 0.0005). AE rates were similar across treatments (12.5% vs 12.1%), with no severe AEs, serious AEs, or clinically significant laboratory test abnormalities reported in the rimegepant group.
Conclusions
A single 75-mg dose of rimegepant ODT was efficacious and well tolerated for acute treatment of migraine in adults unsuitable for triptans. This first prospective trial of a gepant in this population supports calcitonin gene-related peptide antagonism as a valuable option when triptans are unsuitable.
Trial Registration
Clinicaltrials.gov NCT05509400.
This is a visual representation of the abstract.
Introduction
Triptans have been the cornerstone of acute migraine therapy (1), but some patients may not respond well to, or cannot take, triptans. Insufficient response to at least two triptans occurs in 7–13% of patients (2–5). Additionally, 14–29% of triptan users discontinue because of adverse events (2,6–8). Moreover, 10–17% have formal contraindications to triptan use, primarily cardiovascular comorbidities (2,6,9–11). Collectively, these data underscore a critical unmet treatment need in individuals who are unsuitable for triptans due to lack of efficacy, intolerance, or contraindication.
Calcitonin gene-related peptide (CGRP) is a key mediator in migraine pathophysiology and small-molecule CGRP receptor antagonists (gepants) offer a mechanistically distinct approach without vasoconstrictive effects (12). Rimegepant orally disintegrating tablet (ODT), for example, is approved for both acute treatment of migraine (with or without aura) and preventive treatment of episodic migraine in adults (13). In pivotal phase 3 trials for the acute treatment of migraine, a single 75 mg dose of rimegepant ODT demonstrated significantly higher rates of freedom from pain and other migraine symptoms at 2 h post dose compared with placebo, and a favorable tolerability with no signal for hepatotoxicity (14–18). Prior gepant studies, however, either excluded or did not specifically evaluate patients with documented triptan unsuitability and no randomized trials designed to address the use of a CGRP antagonist for the acute treatment of migraine in a population unsuitable for triptans have been published to date. A post-hoc pooled analysis of three phase 3 rimegepant trials suggested efficacy in participants with self-reported triptan failure (19) but these findings required confirmation in a dedicated randomized controlled trial. Therefore, we conducted the first phase 4 randomized double-blind placebo-controlled study of single-dose rimegepant 75 mg ODT for the acute treatment of migraine in adults with documented unsuitability to triptans (NCT05509400).
Methods
Study design and oversight
This was a phase 4, multinational, randomized, double-blind, placebo-controlled study (Figure 1). Eligible participants who completed the double-blind treatment (DBT) phase could enter a twelve-week open-label extension (OLE) phase. The DBT phase was conducted from 18 October 2022, to 10 March 2025, at 93 sites in Australia, Austria, Belgium, Canada, Denmark, Finland, France, Germany, Italy, Mexico, Poland, Spain, Sweden, the United States, and the United Kingdom. This study reports findings from the DBT phase; findings from the OLE phase will be presented separately in a subsequent publication.
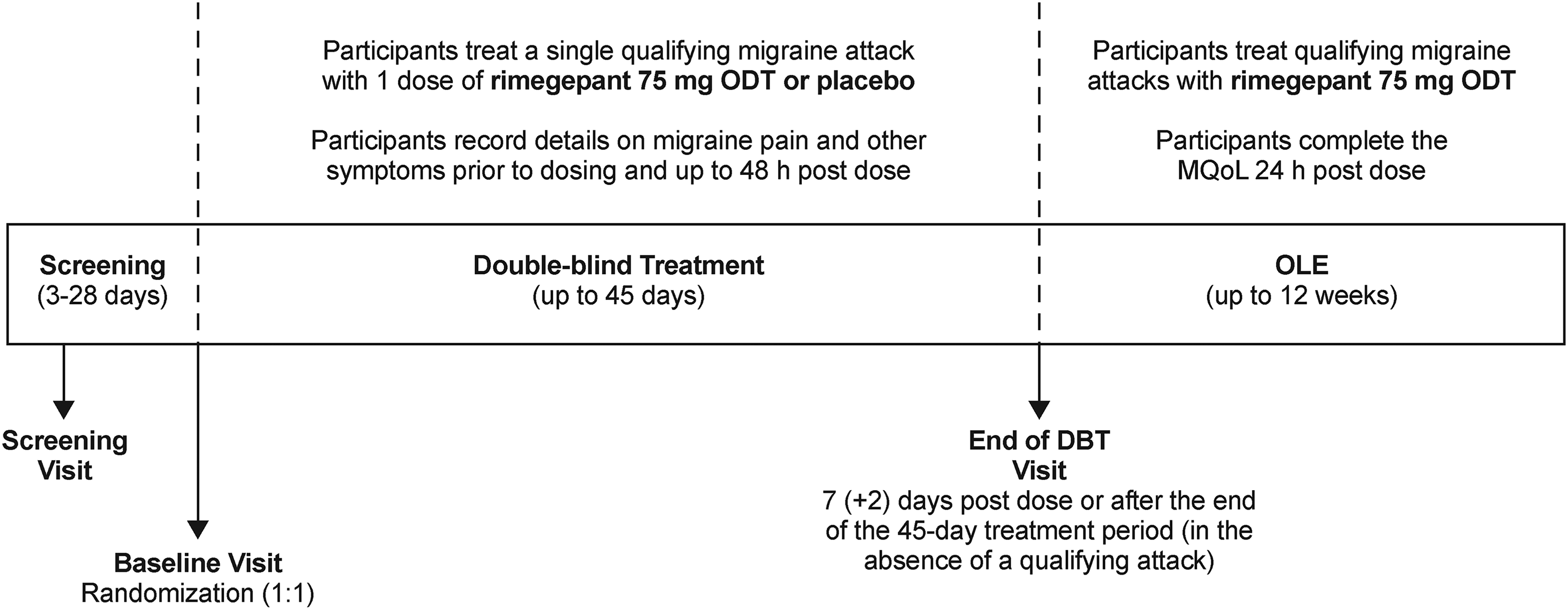
Study design. DBT = double-blind treatment; MQoL = Migraine Quality of Life questionnaire; ODT = orally disintegrating tablet; OLE = open-label extension.
The study protocol was approved by an Institutional Review Board/Independent Ethics Committee for each clinical site (Online Supplemental Table 1). All participants provided written informed consent. The study was conducted in compliance with ethical principles of the Declaration of Helsinki and all International Council on Harmonization Good Clinical Practice Guidelines. This manuscript follows CONSORT guidelines.
Participants
Males and females ≥18 years of age with at least a one-year history of migraine (with or without aura) according to the International Classification of Headache Disorders 3rd Edition (20), migraine onset prior to 50 years of age, an average of 4–14 migraine days per month in the three months prior to screening, untreated migraine attacks lasting an average of 4–72 h, and the ability to distinguish migraine attacks from tension headaches were eligible. Participants were also required to be unsuitable for triptan use, defined as a documented history of intolerance and/or lack of efficacy to at least two triptans or the documented presence of a contraindication. Documentation of triptan unsuitability was based on the medical/pharmacy record — complemented by participant interview, if needed — or via principal investigator interview of the treating physician.
Key exclusion criteria included a history of cluster headache, basilar migraine, or hemiplegic migraine; current medication overuse headache; headache attacks (migraine or non-migraine) for ≥15 days per month in any of the three months prior to screening; and an average of at least seven non-migraine headache days per month across the three months prior to screening. A full list of exclusion criteria is shown in Online Supplemental Table 2.
Treatment
Using the sponsor's centralized interactive response technology, eligible participants were randomized in a 1:1 double-blind manner to rimegepant 75 mg ODT or matching placebo. Randomization was stratified by history of clinically relevant cardiovascular disease (yes or no), with the number of participants in the “yes” stratum capped at 15%. Participants received a single dose of study medication and were instructed to take it when they have a qualifying migraine attack. A qualifying attack was defined as a migraine attack of moderate or severe pain intensity that is first treated with study medication, not with non-study acute migraine medication (e.g., nonsteroidal anti-inflammatory drugs [NSAIDs]). If a participant did not have a qualifying migraine attack within 45 days of the baseline visit, they were to return to the clinic for the end of DBT visit and return unused study medication.
Concomitant medications
Participants recorded use of concomitant medications throughout using a paper diary. The following non-CGRP preventive migraine medications were permitted if the dose was stable for at least three months prior to screening and not expected to change during the study: angiotensin-converting enzyme inhibitors/angiotensin receptor blockers, alpha-adrenergic agonists, antidepressants, antiepileptics, beta-blockers, calcium channel blockers, vitamins, and other recognized standard of care medications used for migraine prevention unless otherwise specified.
Participants could use permitted non-study medications for acute treatment of migraine (NSAIDs, acetaminophen [≤2000 mg per day for up to two consecutive days], antiemetics, baclofen, other recognized standard of care medications unless otherwise specified) as needed for the treatment of migraine attacks of mild or no pain intensity. No acute migraine medications were allowed for 2 h following dosing with study medication. After 2 h, participants could use permitted acute migraine medications for the purposes of rescue (for migraine symptoms that were not relieved by study medication at 2 h or for migraine symptoms that were relieved at 2 h but returned to moderate or severe pain intensity between 2 and 48 h post dose) as needed in accordance with standard of care.
The following medications were prohibited from the screening visit through the DBT phase: acetaminophen and acetaminophen-containing products for non-headache indications, non-narcotic analgesics taken at least fifteen days per month for non-headache indications, triptans, CGRP antagonists (oral or injectable), botulinum toxin injection, ergotamine, lamotrigine, muscle relaxants (except baclofen), marijuana and all forms of ingested or inhaled cannabidiol- or tetrahydrocannabinol-containing products, narcotics, and any investigational agent other than rimegepant. Strong or moderate inhibitors and inducers of cytochrome P450 family 3 subfamily A member 4 and strong inhibitors of P-glycoprotein transport were prohibited during the study. All devices and invasive interventions for the acute or preventive treatment of migraine were prohibited within three months of the screening visit and throughout the study.
Assessments
Participants used an electronic diary (eDiary) to record migraine headache pain intensity (none, mild, moderate, or severe), the status (present or absent) and intensity (mild, moderate, or severe) of migraine symptoms (nausea, phonophobia, or photophobia), and level of functional disability (normal, mild impairment, severe impairment, or requires bedrest) prior to dosing with study medication to treat a qualifying migraine attack and at 15 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, 6 h, 8 h, 24 h, and 48 h post dose. Participants also identified their current most bothersome symptom (MBS; among nausea, phonophobia, or photophobia) and aura status (present or absent) prior to taking study medication. The Migraine Quality-of-Life Questionnaire (MQoL) (21) was completed 24 h post dose.
Physical examinations, 12-lead electrocardiograms (ECG), clinical laboratory tests (hematology and serum chemistry), and liver function tests (LFTs) were assessed at screening and at the end of DBT visit. Vital signs and the Columbia-Suicide Severity Rating Scale (C-SSRS) were assessed at screening, baseline, and the end of DBT visit. Adverse events (AEs) were reported from the time of signed informed consent through the end of DBT visit. AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) Version 27.1 with severity and relationship to study medication determined by site investigators.
Endpoints
The primary efficacy endpoint was the percentage of participants with migraine pain relief (headache pain intensity of none or mild) at 2 h post dose. The ten key secondary efficacy endpoints included the percentage of participants with: (i) migraine pain freedom (headache pain intensity of none) at 2 h post dose, (ii) rescue medication use within 24 h post dose, (iii) return to normal function (functional disability score of normal) at 2 h post dose (among participants with functional disability at time of dosing), sustained return to normal function from (iv) 2–24 h and from (v) 2–48 h post dose (among participants with functional disability at time of dosing), sustained migraine pain relief from (vi) 2–24 h and from (vii) 2–48 h post dose, sustained migraine pain freedom from (viii) 2–24 h and from (ix) 2–48 h post dose, and (x) MBS freedom (absence of symptom) at 2 h post dose.
An exploratory outcomes research endpoint was the percentage of participants with a response of “moderately better” or “very much better” on the Patient Global Impression of Change (PGIC) question of the MQoL at 24 h post dose. The PGIC question asks participants about their overall change in migraine symptoms since taking study medication, with responses categorized on a 7-point scale from “very much worse” to “very much better”.
Safety-related secondary endpoints included the number and percentage of participants with on-treatment AEs, serious AEs, and grade 3 or 4 laboratory test abnormalities. Safety-related exploratory endpoints included the number and percentage of participants with on-treatment hepatic-related AEs and LFT elevations (e.g., alanine transaminase [ALT] or aspartate transaminase [AST] level >3x the upper limit of normal [ULN], total bilirubin level (TBL) > 1.5x ULN).
Analysis
Demographics and clinical characteristics were summarized descriptively for all participants who took double-blind study medication (safety population). On-treatment safety findings during the DBT phase were also summarized descriptively in the safety population. On-treatment AEs during the DBT phase were defined as those with onset date on or after the double-blind study medication dosing date through the earlier of the double-blind study medication dosing date plus nine days and the open-label rimegepant start date minus one day. On-treatment safety findings (e.g., laboratory tests, vital signs, ECGs) during the DBT phase were defined as those measured after the double-blind study medication dosing date through the earlier of the double-blind study medication dosing date plus nine days and the open-label rimegepant start date.
Efficacy was analyzed in all participants who were randomized once, had a qualifying migraine attack at time of dosing, took double-blind study medication, and had post dose efficacy data in the DBT phase (efficacy population). Given few participants had history of clinically relevant cardiovascular disease (i.e., “yes” randomization stratum), treatment groups were compared using unstratified crude risk estimation based on the normal approximation to the binomial distribution. Participants taking rescue medication at or before the time point of interest were imputed as failures. For single time point endpoints (e.g., 2 h post dose), participants with missing data at that time point were imputed as failures. For multiple time point endpoints (e.g., from 2–48 h post dose), participants with missing data at 2, 24, or 48 h post dose, or at more than one time point from 3–8 h post dose, were imputed as failures. Type I error was controlled using hierarchical testing whereby the primary endpoint was evaluated at a two-sided alpha level of 0.05. If the primary endpoint was significant, key secondary endpoints were each tested at a two-sided alpha level of 0.05 in the prespecified order. If an endpoint was not significant, nominal p values for all subsequent endpoints in the hierarchy were provided. Prespecified exploratory analyses of the primary and key secondary endpoints were also conducted in a subgroup of the efficacy population who failed at least two triptans with one or more reasons due to lack of efficacy. Hierarchical testing was not conducted for these exploratory analyses and all p values are nominal. The MQoL-based endpoint was summarized descriptively for the efficacy population, with non-missing MQoL data at 24 h post dose.
It was anticipated that about 90% of 300 participants randomized to each treatment group would have a migraine during the 45-day DBT phase, resulting in approximately 270 participants evaluable for efficacy in each treatment group. This was based on pooled results from three previous single-dose studies of rimegepant (14–16); migraine pain relief at 2 h post dose was 57.9% for rimegepant and 43.9% for placebo. A total sample size of 540 evaluable participants (270 per treatment group) provided approximately 90% power for the primary endpoint of migraine pain relief at 2 h post dose. This was based on a chi-square test with a two-sided alpha level of 0.05 and assumes that the true response rates in participants unsuitable for triptan use would be equivalent to the overall pooled response rates above.
Results
Participants
A total of 633 participants were randomized and 585 participants took double-blind study medication (safety population: rimegepant, n = 295; placebo, n = 290; Figure 2). Participants in the safety population were mostly female (89.1%), had a mean age of 42.9 years, had primarily migraine without aura (76.8%), and experienced a mean of 6.6 migraine days with moderate or severe pain intensity per month in the three months before screening. Demographics and baseline clinical characteristics were similar between treatment groups (Table 1). Concomitant preventive migraine medications were used by 21.9% of participants (rimegepant, 21.7%, placebo, 22.1%; Online Supplemental Table 3).

Participant disposition.
Summary of demographics and baseline clinical characteristics in the safety population. a

The safety population was defined as participants who took double-blind study medication.
Based on self-reported history of cardiovascular events, conditions, procedures and other risk factors.
Based on self-reported migraine history.
SD = standard deviation.
Overall, 570 participants were analyzed for efficacy (efficacy population: rimegepant, n = 286; placebo, n = 284). Reasons for triptan unsuitability in the efficacy population are shown in Table 2. Previous triptan medications used by participants in the efficacy population included sumatriptan (79.1%), zolmitriptan (45.4%), almotriptan (34.9%), rizatriptan (31.4%), eletriptan (18.8%), naratriptan (8.8%), and frovatriptan (5.6%).
Reasons for triptan unsuitability in the efficacy population. a

The efficacy population was defined as participants who were randomized only once, had a qualifying migraine attack at the time of dosing, took double-blind study medication, and had post dose efficacy data in the double-bind treatment phase.
Percentages do not add up to 100% because participants could be in more than one reason category.
Efficacy
Rimegepant was superior to placebo for the primary endpoint of migraine pain relief at 2 h post dose (Figure 3). Response rates were 55.9% for rimegepant and 32.7% for placebo, with a difference (95% confidence interval [CI]) of 23.2% (15.3%, 31.1%) between treatment groups (p < 0.0001). Hierarchical testing of all ten alpha-protected key secondary endpoints also demonstrated superiority of rimegepant over placebo (all p ≤ 0.0005; Figure 4). These included migraine pain freedom at 2 h post dose (difference [95% CI]: 15.3% [9.6%, 21.1%]), rescue medication use within 24 h post dose (difference [95% CI]: −28.2% [–35.6%, −20.8%]), return to normal function at 2 h post dose (difference [95% CI]: 16.2% [9.0%, 23.4%]), sustained return to normal function at 2–24 h post dose (difference [95% CI]: 11.4% [5.5%, 17.2%]) and 2–48 h post dose (difference [95% CI]: 11.7% [6.4%, 17.1%]), sustained migraine pain relief at 2–24 h post dose (difference [95% CI]: 24.4% [17.4%, 31.3%]) and 2–48 h post dose (difference [95% CI]: 23.4% [16.8%, 29.9%]), sustained migraine pain freedom at 2–24 h post dose (difference [95% CI]: 9.1% [4.3%, 13.8%]) and 2–48 h post dose (difference [95% CI]: 9.4% [5.2%, 13.7%]), and MBS freedom at 2 h post dose (difference [95% CI]: 12.5% [5.4%, 19.5%]).
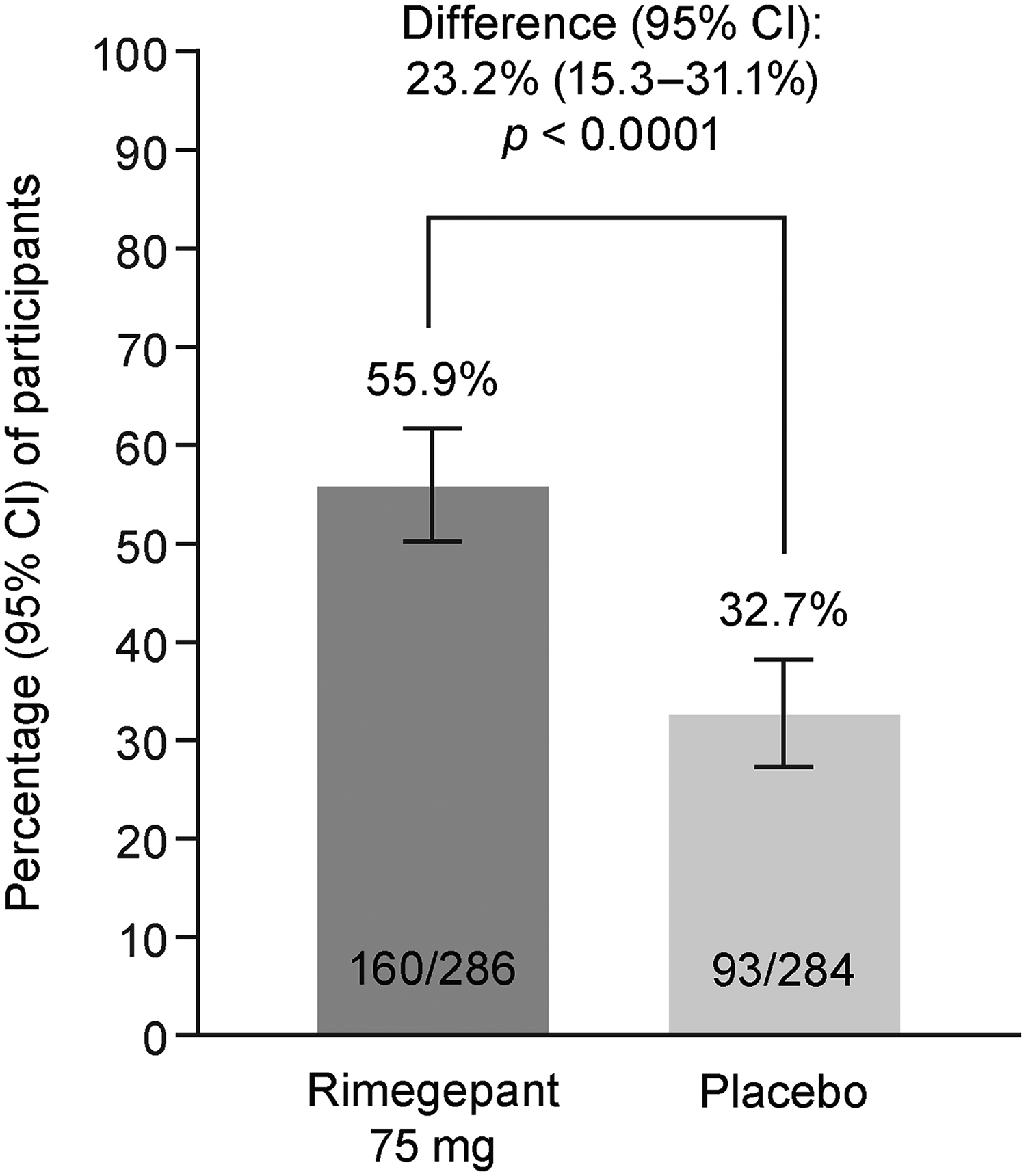
Migraine pain relief at 2 h post dose (primary endpoint) in the efficacy population.a

Key secondary endpoints in the efficacy population.a
The percentage of participants with a response of “moderately better” or “very much better” on the PGIC question of the MQoL (overall change in migraine symptoms since taking study medication) at 24 h post dose was higher in the rimegepant group (156 of 243 participants; 64.2%) than in the placebo group (75 of 250 participants; 30.0%).
Exploratory analyses in a subgroup of participants who were unsuitable for triptans with at least one reason due to lack of efficacy (rimegepant, n = 240; placebo, n = 244) favored rimegepant over placebo (nominal p < 0.05) for all primary and key secondary efficacy endpoints (Figure 5).
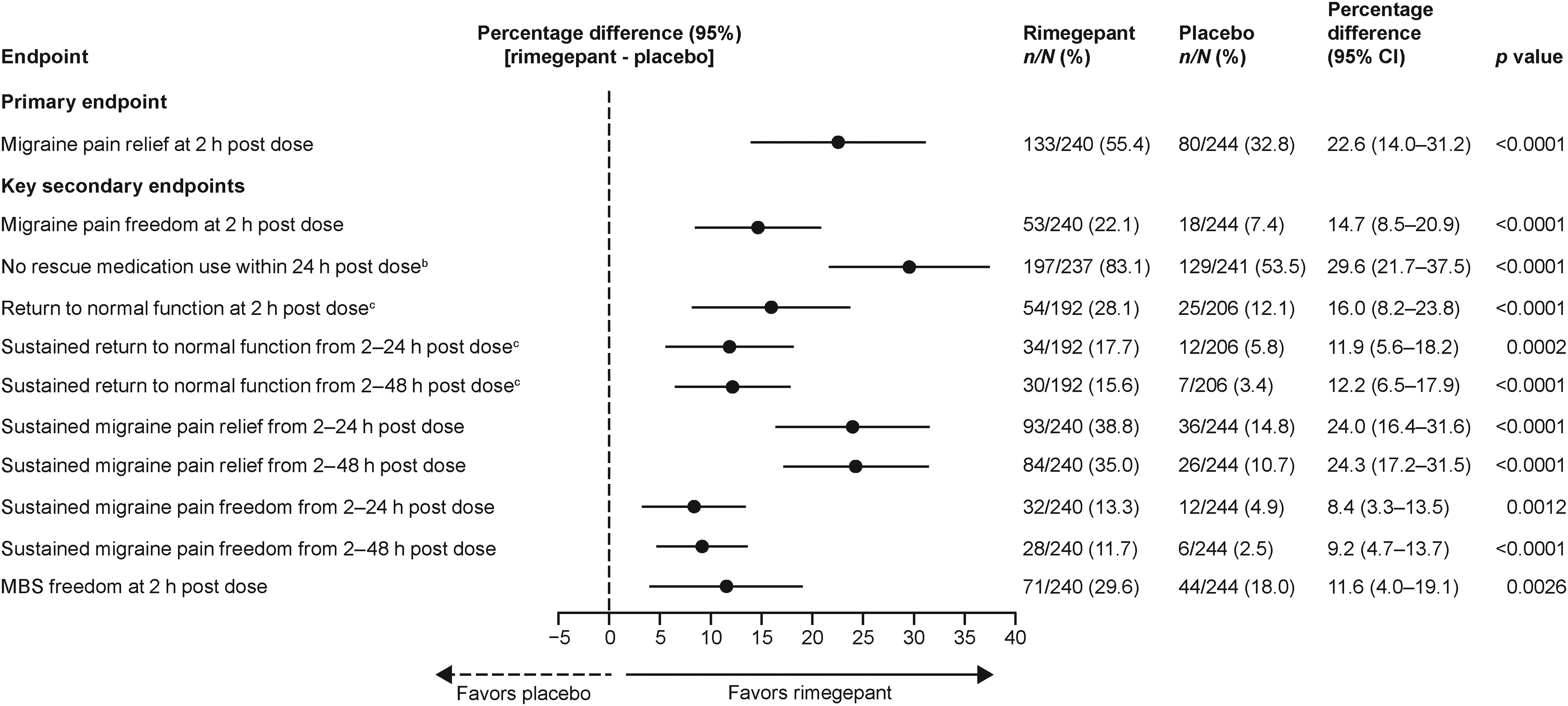
Primary and key secondary endpoints in the subgroup of participants in the efficacy populationa who failed at least two triptans with one or more reason due to lack of efficacy.
Safety
The frequency of on-treatment AEs during the DBT phase was 12.5% and 12.1% in the rimegepant and placebo groups, respectively (Table 3). Most on-treatment AEs were mild and there were no severe or serious AEs reported in the rimegepant group. The only on-treatment AEs with a frequency of ≥1% in either treatment group were nasopharyngitis (rimegepant, 1.7%; placebo, 1.0%), blood bilirubin increased (rimegepant, 0%; placebo, 1.0%), and migraine (rimegepant, 0%; placebo, 1.4%). An on-treatment AE of hypertension (mild in intensity) occurred in one (0.3%) participant in the rimegepant group; this participant did not have a prior diagnosis of hypertension. An on-treatment AE of Raynaud's phenomenon (moderate in intensity) occurred in one (0.3%) participant in the rimegepant group; this participant had a medical history of Raynaud's phenomenon. A full list of on-treatment AEs reported during the DBT phase is shown in Online Supplemental Table 4.
Summary of on-treatment adverse events in the safety population. a

The safety population was defined as participants who took double-blind study medication.
Based on AE preferred term “worst intensity” during the double-blind treatment phase.
AE = adverse event.
No participants in the rimegepant group had an on-treatment grade 3 or 4 laboratory test abnormality (CTCAE v5.0/DAIDS v2.1 toxicity grading scale); one (0.4%) participant in the placebo group had a grade 3 test abnormality of high potassium. No participants in either group had on-treatment ALT or AST level >3x ULN. No participants in the rimegepant group had TBL >1.5x ULN; two (0.8%) participants in the placebo group had TBL >1.5x ULN.
No clinically relevant findings related to physical examinations, vital signs, or 12-lead ECG were observed. No participants reported suicidal ideation, suicidal behavior, or non-suicidal self-injurious behavior on the C-SSRS.
Discussion
In this study, rimegepant 75 mg ODT produced clear and clinically meaningful benefits for the acute treatment of a single migraine attack of moderate or severe pain intensity in adults unsuitable for triptans. Treatment response favored rimegepant over placebo for the primary endpoint (migraine pain relief at 2 h post dose) and all 10 key secondary endpoints (including migraine pain freedom and MBS freedom at 2 h post dose). Importantly, participants in this study were generally representative of the broader triptan-unsuitable population, with documented reasons due to lack of efficacy, intolerance, or contraindication. Within the subgroup of participants who had failed at least two triptans with one or more reason due to lack of efficacy (representing 85% of the study population), analyses favored rimegepant over placebo (nominal p < 0.05) across the primary and all key secondary endpoints. The consistency of rimegepant effect across this subgroup underscores its potential as a broadly applicable acute treatment in individuals for whom 5-HT 1B/1D receptor agonists are not viable. This is supported by the observation that nearly two-thirds of rimegepant-treated participants reported that their migraine symptoms were moderately or very much better at 24 h post dose compared with fewer than one-third of placebo-treated participants.
International Headache Society (IHS) guidelines recommend co-primary endpoints of migraine pain freedom and MBS freedom at 2 h in acute migraine trials (22). Pain freedom is a stringent efficacy benchmark in clinical trials. In clinical practice, however, reduction to no or mild pain (typically termed “pain relief”) is a common and meaningful measure of benefit that supports return to normal functioning (23). Thus, to reflect real-world measures of treatment effectiveness, pain relief at 2 h post dose was chosen as the primary endpoint in this phase 4 non-registrational study. This aligns with IHS guidelines (22), emphasizing treatment at moderate to severe pain intensity to increase diagnostic specificity, reduce placebo-response, and better discriminate between treatment groups. To maintain alignment with IHS standards and ensure comparability with prior trials, both pain freedom and MBS freedom at 2 h were evaluated as key alpha-protected secondary endpoints. These endpoints were tested hierarchically and met statistical significance. This strategy of assessing both guideline-based (pain freedom and MBS freedom) and real-world outcomes (pain relief) provides a comprehensive evaluation of efficacy. While this approach differs from early intervention strategies often applied in clinical practice, it is consistent with regulatory standards for migraine trials.
Though direct comparisons are not possible, efficacy findings in this population of adults unsuitable for triptans are consistent with previous studies of rimegepant for the acute treatment of migraine in general migraine populations, which demonstrated sustained rimegepant-mediated improvements in pain, function, and rescue medication use (14–17). For example, differences between rimegepant and placebo for migraine pain relief at 2 h (primary endpoint in current study) were 23.2% in the current study and 15.2% in a pooled analysis of four previous studies (24). Differences for migraine pain freedom at 2 h (coprimary endpoint in previous studies) were 15.3% and 8.2% in the current and previous studies, respectively. Finally, differences for MBS freedom at 2 h (co-primary endpoint in previous studies) were 12.5% and 11.0% in the current and previous studies, respectively.
Findings from this dedicated prospective randomized controlled trial also confirm and expand upon preliminary findings from a pooled, post-hoc, subgroup analysis of phase 3 trials of rimegepant for the acute treatment of migraine in participants with a self-reported history of insufficient response to triptan therapy (including lack of efficacy and intolerance) (19). In the previous post-hoc analysis, rimegepant was more effective than placebo on the co-primary efficacy endpoints (pain freedom at 2 h post dose and MBS freedom at 2 h post dose) and the key secondary endpoint (pain relief at 2 h post dose) in individuals who had a history of insufficient response to one triptan and in those who had insufficient response to at least two triptans (19).
The reduced use of acute rescue medication in the rimegepant group compared with the placebo group observed in the current study is notable since chronic use of other classes of medications commonly used for acute treatment of migraine (e.g., triptans, NSAIDs, caffeine-containing compounds) can be associated with a risk of medication overuse headache (MOH) (20,25–27). Gepants have not been associated with a risk of MOH in preclinical, clinical, or real-world settings (28–30). Further, studies have shown that use of rimegepant reduces the requirement of agents that can cause MOH (31–33) and leads to a reduced prevalence of MOH (30).
European Headache Federation (EHF) consensus criteria define triptan non-responders as individuals who do not achieve effective treatment with a single triptan in at least three out of four migraine attacks. Individuals who fail (i.e., do not achieve effective treatment in at least three of four attacks) at least two and at least three triptans are classified as triptan-resistant and triptan-refractory, respectively (23). EHF criteria define effective treatment as achievement, within 2 h of administration and maintained for at least 24 h, of A) improvement of headache from severe or moderate to mild or absent; B) absent or minimal disturbances due to migraine-related non-pain symptoms; and C) no meaningful drug-related AEs (23). In the current study, triptan unsuitability was defined as documented failure to at least two triptan medications due to lack of efficacy or prior intolerance, or a contraindication to triptan use. Lack of efficacy was characterized by inadequate therapeutic response despite appropriate dosing on at least two separate occasions, with indicators including persistent or recurrent migraine symptoms at 2 h post-dose or lack of sustained relief through 24 h. Intolerance was defined as discontinuation of a triptan due to AEs considered intolerable by the treating investigator.
Determination of lack of efficacy and intolerance in this study was based on medical/pharmacy records; if the record was missing or there was a lack of adequate documentation of lack of efficacy or intolerance in the record, structured physician or participant interviews that were targeted in approach and formally documented were acceptable. We acknowledge limitations associated with reliance on medical/pharmacy records or physician/participant interviews, particularly with potential variability in subjective reporting. Nonetheless, the study methodology is consistent with EHF consensus criteria for effective treatment of an acute migraine attack (23), despite being initiated before publication of the consensus, and is comparable to methods used in other migraine studies assessing prior treatment failure (34–36).
The safety profile of gepants, including rimegepant, is a key strength of this medication class. As in previous studies in general migraine populations (14–17), the safety profile of rimegepant was similar to placebo in this population of adults unsuitable for triptans. This was evidenced by the low overall number of participants with an on-treatment AE, and the absence of any severe on-treatment AEs, serious on-treatment AEs, on-treatment grade 3 or 4 laboratory test abnormalities, or signs of hepatotoxicity reported among rimegepant-treated participants. The only on-treatment AE with a frequency ≥1% in the rimegepant group was nasopharyngitis. In the current study, AEs of hypertension and Raynaud's phenomenon occurred in one (0.3%) participant each in the rimegepant group. The AE of Raynaud's phenomenon occurred in a participant with a medical history of Raynaud's phenomenon. Although the AE of hypertension occurred in a participant without a prior diagnosis of hypertension, it was considered mild in intensity and was not deemed to be related to study drug by the principal investigator. Overall, no new safety concerns were identified in this population of adults unsuitable for triptans.
The American Headache Society and the IHS recommend a trial of two or more triptans (in the absence of contraindication or intolerability) before initiating use of gepants or other agents (e.g., lasmiditan) for moderate to severe migraine (1,37). In real-world practice, however, certain clinical circumstances such as severe intolerance, MOH, or high cardiovascular risk may justify earlier consideration of non-triptan options such as gepants or lasmiditan. Post-hoc analyses of rimegepant indicate similar efficacy regardless of whether patients have failed one or multiple triptans (19). Pooled post-hoc analyses of ubrogepant clinical trial data echo these findings (38). However, these exploratory results are hypothesis generating and until prospective trials evaluate use of gepants after only one triptan failure, the established standard of trying two distinct triptans prior to other agents should remain the recommended treatment approach.
Findings from this study suggest that rimegepant may be an appropriate option for the acute treatment of migraine in individuals who are unsuitable for triptans. However, conclusions on the use of rimegepant in this population are limited since this study only evaluated the effects of one dose of rimegepant to treat a single migraine attack. Findings from the 12-week OLE phase of this trial will allow for evaluation of the effectiveness and safety of repeated dosing of rimegepant (as needed up to once per day) in participants unsuitable for triptans. Timing of symptom onset relative to administration of study medication was not collected, which may be perceived as a study limitation since earlier versus later treatment may influence outcomes. However, the randomized, double-blind design mitigates potential differences between groups (i.e., time from symptom onset to treatment administration would be expected to be balanced across groups). Moreover, choosing a timing for study drug intake based on migraine pain severity instead of time since symptom onset increases the specificity of the migraine diagnosis (22).
The key strength of this study is that it is the first, to our knowledge, prospectively designed trial of a gepant in individuals with a documented history of unsuitability to triptans. Previous analyses of individuals unsuitable to triptans were based on subgroup analyses and not prospectively designed (19,38). The large sample size is also a strength of the study, with 585 participants receiving study medication. Finally, the statistical analysis plan was robust, with a hierarchical gate-keeping approach used to control type 1 error across the primary endpoint and ten key secondary endpoints.
Conclusions
A single dose of rimegepant 75 mg ODT demonstrated efficacy superior to placebo and a favorable safety profile that was similar to placebo for the acute treatment of migraine in adults with documented unsuitability for triptans due to previous lack of efficacy, intolerability, or contraindication. This is the first prospective study to demonstrate efficacy of a gepant for acute treatment of migraine in individuals unsuitable for triptans and suggests rimegepant may be an appropriate option for this population.
Article highlights
Single dose rimegepant 75 mg demonstrated efficacy and a favorable safety profile for acute treatment of migraine in adults with documented unsuitability to triptans.
This is the first randomized study to demonstrate efficacy of a gepant for acute treatment of migraine in individuals unsuitable to triptans and provides evidence that rimegepant may be an appropriate therapeutic option in this population.
Supplemental Material
sj-docx-1-cep-10.1177_03331024251395298 - Supplemental material for Rimegepant for acute treatment of migraine in triptan-unsuitable adults: A randomized, double-blind, placebo-controlled phase 4 trial
Supplemental material, sj-docx-1-cep-10.1177_03331024251395298 for Rimegepant for acute treatment of migraine in triptan-unsuitable adults: A randomized, double-blind, placebo-controlled phase 4 trial by Messoud Ashina, Peter McAllister, Charly Gaul, Adolfo Leyva-Rendon, Luz M. Ramirez, Catherine Nalpas, Alexandra Thiry, Lucy Abraham, Robert J. Fountaine and Terence Fullerton in Cephalalgia
Supplemental Material
sj-docx-2-cep-10.1177_03331024251395298 - Supplemental material for Rimegepant for acute treatment of migraine in triptan-unsuitable adults: A randomized, double-blind, placebo-controlled phase 4 trial
Supplemental material, sj-docx-2-cep-10.1177_03331024251395298 for Rimegepant for acute treatment of migraine in triptan-unsuitable adults: A randomized, double-blind, placebo-controlled phase 4 trial by Messoud Ashina, Peter McAllister, Charly Gaul, Adolfo Leyva-Rendon, Luz M. Ramirez, Catherine Nalpas, Alexandra Thiry, Lucy Abraham, Robert J. Fountaine and Terence Fullerton in Cephalalgia
Supplemental Material
sj-docx-3-cep-10.1177_03331024251395298 - Supplemental material for Rimegepant for acute treatment of migraine in triptan-unsuitable adults: A randomized, double-blind, placebo-controlled phase 4 trial
Supplemental material, sj-docx-3-cep-10.1177_03331024251395298 for Rimegepant for acute treatment of migraine in triptan-unsuitable adults: A randomized, double-blind, placebo-controlled phase 4 trial by Messoud Ashina, Peter McAllister, Charly Gaul, Adolfo Leyva-Rendon, Luz M. Ramirez, Catherine Nalpas, Alexandra Thiry, Lucy Abraham, Robert J. Fountaine and Terence Fullerton in Cephalalgia
Supplemental Material
sj-docx-4-cep-10.1177_03331024251395298 - Supplemental material for Rimegepant for acute treatment of migraine in triptan-unsuitable adults: A randomized, double-blind, placebo-controlled phase 4 trial
Supplemental material, sj-docx-4-cep-10.1177_03331024251395298 for Rimegepant for acute treatment of migraine in triptan-unsuitable adults: A randomized, double-blind, placebo-controlled phase 4 trial by Messoud Ashina, Peter McAllister, Charly Gaul, Adolfo Leyva-Rendon, Luz M. Ramirez, Catherine Nalpas, Alexandra Thiry, Lucy Abraham, Robert J. Fountaine and Terence Fullerton in Cephalalgia
Footnotes
Acknowledgments
The authors thank the study participants, their families, site investigators, and staff who took part in the study. Medical writing support was provided by Matt Soulsby, PhD, CMPP, and was funded by Pfizer.
Author contributions
M.A.: Investigation; Writing – review & editing. P.M: Investigation; Writing – review & editing. C.G.: Investigation; Writing – review & editing. A.L.-R.: Investigation; Writing – review & editing. L.M.R.: Methodology; Writing – review & editing. C.N.: Methodology; Writing – review & editing. A.T.: Methodology; Formal analysis; Writing – review & editing. L.A.: Methodology; Writing – review & editing. R.J.F.: Methodology; Writing – review & editing. T.F.: Conceptualization; Methodology; Writing – review & editing.
Data availability statement
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: M.A. has served as an advisory board/consultant/speaker for AbbVie, Astra Zeneca, Eli Lilly, GlaxoSmithKline, Lundbeck, Pfizer, and Teva; has received institutional research grants from Danish National Research Foundation, Lundbeck Foundation, Novo Nordisk Foundation, Novartis, and Lundbeck; is an associate editor for Journal of Headache and Pain and for Brain, and serves on the editorial board of Neurotorium and has received honoraria. P.M. has served as an advisory board member/speaker/consultant for AbbVie, ANI, BrightMind.AI, Dompe, Lilly, Lundbeck, and Pfizer. C.G. has received honoraria for consulting and lectures within the past three years from AbbVie, Chordate, Dr Reddys, Hormosan Pharma, Lilly, Lundbeck, Merz, Novartis Pharma, Perfood, Orion, Organon, Pfizer, Reckitt-Benckiser, Sanofi-Aventis, TEVA, and Vectura Fertin Pharma. His research is supported by a grant from the German Research Foundation (DFG). A.L.-R. has served as advisory board/ speaker for Farmasa-Schwabe, Grünenthal, Lundbeck, Merz, Pfizer, Sunpharma, and Torrent. L.M.R., C.N., L.A., R.J.F., and T.F. are, or were, employees of and hold stock/options in Pfizer. A.T. is a former employee of Biohaven Pharmaceuticals, owns stock in Biohaven Ltd, is an employee of Pfizer, and owns stock/options in Pfizer.
Data presented in this article were presented in part at the 67th Annual Scientific meeting of the American Headache Society in June 2025 (Minneapolis, MN, USA), the 22nd International Headache Congress in September 2025 (São Paulo, Brazil), and the forthcoming 19th European Headache Congress in December 2025 (Lisbon, Portugal).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Pfizer.
ORCID iDs
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.