
Abstract
Background
This study evaluated the long-term safety, tolerability and effectiveness of rimegepant, 75 mg orally disintegrating tablet, for the acute treatment of migraine in Chinese adults.
Methods
This phase 3, multicenter, open-label, single-arm study enrolled Chinese adults with a ≥1 year history of migraine (with or without aura), 6–18 moderate-to-severe migraine attacks/month within three months before a screening visit and at least six migraine days during a 30-day observation phase (OP). After the OP, eligible participants took rimegepant as needed (maximum one tablet per day) at the onset of mild-to-severe migraine attack for a long-term treatment (LTT) of 52 weeks.
Results
Overall, 240 participants were treated and 208 (86.3%) completed the study. During LTT, 203 (84.6%) participants reported ≥1 treatment-emergent adverse event (TEAE) and 46 (19.2%) reported ≥1 TEAE considered to be rimegepant-related. There were no rimegepant-related serious AEs or rimegepant-related TEAEs that led to treatment interruption or discontinuation. Mean reduction from the OP in monthly migraine days was observed as early as the first four weeks (−1.7; 95% confidence interval = −2.2 to −1.2), with an overall mean reduction of −4.4 (95% confidence interval = −4.9 to −3.9) days across LTT.
Conclusions
Rimegepant had a favorable long-term safety profile, was well tolerated in Chinese participants, and a reduction in the number of monthly migraine days was observed during the LLT.
This is a visual representation of the abstract.
Introduction
In China, an estimated 151.6 million people suffer from migraine, equating to an approximately 35% increase over the past 20 years (1). Almost all patients with migraine will require acute therapy; however, many oral migraine therapies were initially developed for other therapeutic indications and may be associated with low adherence due to limited efficacy and tolerability (2).
Rimegepant orally disintegrating tablet (ODT) is an oral small-molecule calcitonin gene-related peptide (CGRP) receptor antagonist for the acute treatment of migraine. Rimegepant is approved in the USA, European Union and UK for the acute treatment of migraine at a therapeutic dose of 75 mg, as needed (PRN; maximum 75 mg per 24 h) and the preventive treatment of migraine (every other day dosing). Rimegepant is also approved in China (January 2024) for the acute treatment of migraine with or without aura in adults.
Rimegepant for the acute treatment of migraine has been assessed in several clinical studies, which report the treatment of a single attack (3–8), and long-term safety and efficacy have been reported in a US population (9,10). Rimegepant demonstrated a favorable safety profile and was well tolerated in Chinese participants in a randomized, placebo-controlled phase 3 trial among adults living in China or South Korea; a single dose of rimegepant 75 mg was superior to placebo for pain freedom and freedom from the most bothersome symptom, with a safety and tolerability profile similar to placebo (7). The long-term safety and tolerability of rimegepant has not been reported in a Chinese population.
Here we report the results of a phase 3, multicenter, open-label, single-arm study for which the primary objective was to assess the long-term safety and tolerability of rimegepant 75 mg ODT PRN (maximum one tablet per calendar day) for the treatment of acute migraine in Chinese adults (ClinicalTrials.gov identifier: NCT05371652).
Methods
Participants
Eligible participants were aged ≥18 years with migraine onset before 50 years of age. Women of childbearing potential were required to have a negative pregnancy test at baseline visit before dispensing rimegepant. Participants were required to have ≥1-year history of migraine (with or without aura) consistent with a diagnosis according to the International Classification of Headache Disorders, a history of 6–18 moderate-to-severe migraine attacks per month within three months before the screening visit, six or more qualified migraine days during the Observation Phase (OP) and migraine attacks with a duration of 4–72 h if untreated. As the frequency of migraine attacks can fluctuate over time, such that episodic migraine (<15 headache days per month) and chronic migraine may transition into one another, our study allowed participants to have no more than 18 moderate-to-severe migraine attacks per month within three months before the screening visit.
Participants on prophylactic migraine medication could remain on therapy if the dose was stable for two or more months before baseline visit and the dose was not expected to change during the study. Triptans were permitted during the OP but were to be discontinued at baseline visit and not used during long-term treatment (LTT). Participants contraindicated for triptans were eligible, provided all other eligibility criteria were met.
Exclusion criteria included a history of basilar migraine with brainstem aura or hemiplegic migraine; history of HIV; poorly controlled/unstable cardiovascular disease, hypertension or diabetes; systolic blood pressure >150 mmHg or diastolic >100 mmHg after 10 min of rest; major depression/depressive episode within 12 months or other significant neurological disorders; history or diagnosis of Gilbert's syndrome or hepatic/biliary disorder; diagnosis of hematologic or solid malignancy within 5 years of screening; body mass index ≥35 kg/m2; use of St John's wort within 14 days of baseline visit; and use of opioids (e.g. morphine, codeine, oxycodone, hydrocodone) within 2 days of baseline visit. Participants were not eligible if they had taken part in a clinical trial with a non-biological investigational drug within 30 days of the screening visit, a biological drug within 90 days of the screening visit or were an active participant in any other investigational clinical study.
Study design
This was a multicenter, open-label, long-term safety study of rimegepant 75 mg ODT for the treatment of acute migraine in Chinese participants. The study design is summarized in Figure 1. Eligible participants entered the screening phase, which consisted of a screening visit and a 30-day baseline OP. After completing the screening visit, participants were issued an electronic diary (eDiary; eResearchTechnology, Inc.) to document each day whether a migraine occurred, the intensity of each attack, if it was treated and standard-of-care migraine treatment via a personal device application (Bring Your Own Device (BYOD); Shanghai Xincere Med Tech Inc.). After the OP, participants attended a baseline visit to review their continued eligibility. Participants with six or more migraine days during the OP and acceptable baseline laboratory assessments were eligible to enter LTT. Participants were instructed to take rimegepant 75 mg ODT (PRN, maximum 1 tablet/calendar day) at the onset of mild-to-severe migraine attacks. Onsite study visits were approximately every four weeks until week 16 and then every 12 weeks. Virtual visits via the BYOD application were conducted every four weeks in between every two onsite visits until week 52. Participants returned to the study site at the end of week 52 (±7 days) for the end of treatment visit, and a further follow-up visit 14 days (±2 days) after the week 52/early termination visit.

Study design. LTT, long-term treatment.
This study was conducted in accordance with Good Clinical Practice E6, as defined by the International Council for Harmonization and the local regulatory requirements by China National Medical Products Administration. Institutional review board/independent ethics committee approval was required for the protocol, consent form, recruitment materials/process and other written information. All participants provided written informed consent before any study activities.
Outcomes
The primary endpoints were adverse events (AEs), common AEs (incidence ≥5%), serious adverse events (SAEs) and AEs leading to rimegepant and/or study discontinuation, as well as physical examination, electrocardiogram, vital signs/physical measurements and clinical laboratory test data. The secondary endpoint was the number of migraine days and severity of migraine attacks every four weeks (monthly migraine days (MMDs)) during LTT with rimegepant 75 mg ODT compared to the OP. Participants recorded migraine occurrence, severity, rimegepant doses and standard-of-care migraine treatments using an eDiary and the BYOD application. Rimegepant exposure was evaluated based on the average number of tablets taken per month.
Exploratory endpoints included quality of life as measured by the change from baseline using the Migraine-Specific Quality of Life Questionnaire (MSQoLQ, version 2.1) (11); migraine-related disability as measured by the change from baseline using the Migraine Disability Assessment (MIDAS) (12), and participant preference, satisfaction and overall therapeutic response as assessed using three single-item assessments. The MSQoLQ is a 14-item questionnaire that spans three domains: restrictive role function (RR), preventive role function (PR) and emotional function (EF). The raw score of each MSQoLQ domain was transformed into a 0–100 scale. MIDAS is a retrospective, patient-reported questionnaire that measures migraine-related disability on a scale of 0–5 (little/no disability), 6–10 (mild disability), 11–20 (moderate disability) and ≥21 (severe disability). The Migraine Preference of Medication, Satisfaction with Medication, and Clinical Global Impression–change (CGI-C) scale (13) were used to assess participant preference, satisfaction and overall therapeutic response, respectively.
Statistical analysis
Statistical analyses were descriptive and no formal hypothesis testing was performed. The study aimed to screen approximately 330 participants and enroll approximately 240. For 240 participants, if the AE rates were 5% and 1%, then the probabilities of observing at least one AE were >99.9% and 91.0%, respectively, thus providing sufficient information for safety and tolerability assessment.
Safety was evaluated for the treatment safety period (from start of treatment to week 52/end of treatment date +7 days) and the follow-up safety period (duration after week 52/end of treatment +8 days). Laboratory test results were graded using Common Terminology Criteria for Adverse Events (CTCAE) (14) and AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA), version 26.1 (15). For efficacy evaluation, the LTT phase was defined as the duration from baseline visit or study drug start date (whichever was earlier) to week 52/end of treatment date. The number of migraine days by total and severity of migraine attacks was analyzed every month (four weeks) and compared with the OP. The number of MMDs were prorated into 28 days for the OP and each month (four-week interval) in the LTT phase. The full analysis set (FAS) included all participants who were enrolled and received at least one dose of rimegepant. The FAS was used for the analysis of baseline characteristics as well as the analyses of exploratory endpoint measures. The safety analysis set (SS) included all participants who received at least one dose of rimegepant and was used for safety analyses during the treatment safety period. The efficacy analysis set (EAS) included all participants in the FAS with ≥14 eDiary days (not necessarily consecutive) in both the OP and ≥1 month (four-week interval) of the LTT period. The EAS was used to assess the secondary endpoint.
Two-sided 95% confidence intervals (CIs) based on normal distribution were calculated for continuous endpoints, including migraine days, MSQoLQ and MIDAS. Two-sided exact Clopper–Pearson CIs were calculated for Migraine Preference of Medication, Satisfaction with Medication, and CGI-C.
Results
The first participant attended the first visit on 22 May 2022 and the last participant completed the study on 6 February 2024. Of 330 screened adults, 241 were enrolled. Of the enrolled adults, 240 (99.6%) were treated and 208 (86.3%) completed the study. The 240 treated participants were included in the FAS, EAS and safety analysis set. One participant was enrolled but not treated due to no migraine occurring. Participant disposition is summarized in Figure 2.
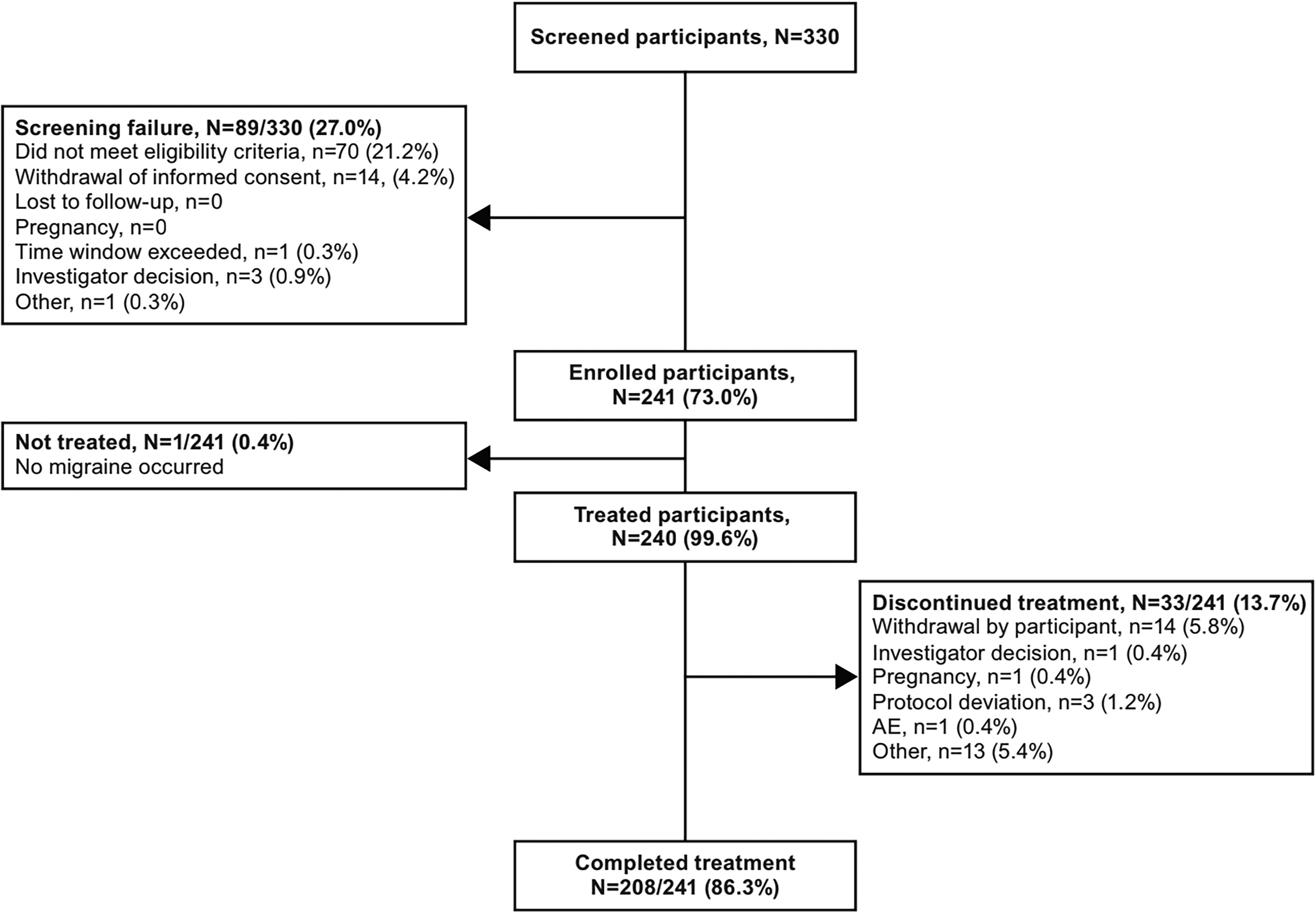
Participant disposition. AE, adverse event. Screening failure and enrolled participant percentages were based on the number of screened participants; all other percentages were based on the number of enrolled participants.
Baseline demographics, clinical characteristics, and migraine history are summarized in Table 1. Overall, participants had a mean (SD) age of 39.1 (11.0) years and 192 (80.0%) were female. Mean (SD) age at migraine onset was 27.3 (9.7) years and the mean (SD) duration of untreated migraine attacks was 17.9 (15.1) hours. Participants experienced a mean (SD) of 8.8 (2.2) moderate-to-severe migraine attacks per month within the last three months before the screening visit.
Baseline demographics, clinical characteristics and migraine history (full analysis set).

Data are the mean (SD), unless otherwise stated.
BMI = body mass index; MIDAS = Migraine Disability Assessment; MSQoLQ = Migraine-Specific Quality of Life Questionnaire.
The mean (SD) of eDiary daily compliance in the LTT phase was 94.6% (5.8%), with 206 participants (85.8%) demonstrating high compliance (≥90% in entering eDiary records) and 234 participants (97.5%) demonstrating compliance ≥80%.
Commonly (≥5% participants) taken classes of current non-study medications (i.e. those taken on or after informed consent and before rimegepant) included: anti-inflammatory and antirheumatics (ibuprofen, n = 144 (60.0%) and loxoprofen sodium, n = 12 (5.0%)) and analgesics (acetylsalicylic acid, caffeine, paracetamol (n = 21 (8.8%)) and caffeine, paracetamol, propyphenazone (n = 13 (5.4%)). Commonly (≥5% participants) taken concomitant non-study medications (i.e. those taken with rimegepant) included: anti-inflammatory and antirheumatics (ibuprofen, n = 129 (53.8%)); analgesics (acetylsalicylic acid, caffeine, paracetamol, n = 19 (7.9%); amantadine hydrochloride, caffeine, chlorphenamine maleate, cow bezoar, paracetamol, n = 13 (5.4%); paracetamol, n = 12 (5.0%)) and antivirals for systemic use (calcium sulfate dihydrate, dryopteris crassirhizoma rhizome, Ephedra spp. herb, forsythia suspensa, Glycyrrhiza spp. root with rhizome, houttuynia cordata herb, isatis tinctoria root, lonicera japonica flower, menthol, pogostemon cablin herb, Prunus spp. seed, Rheum spp. root with rhizome, rhodiola crenulata root with rhizome, n = 14 (5.8%)).
The proportion of participants who were taking triptans as an acute migraine medication (on or after informed consent and before rimegepant) were 3.3% (n = 8, zolmitriptan nasal spray and tablets) and 2.1% (n = 5, rizatriptan benzoate/rizatriptan). No participants had taken current or concomitant gepants as a non-study medication.
Primary endpoints: Safety
Of 240 treated participants, 203 (84.6%) experienced at least one TEAE and 46 (19.2%) experienced TEAEs that were considered by the investigator to be rimegepant-related (Table 2). Seven (2.9%) participants had an SAE: arteriosclerosis coronary artery, pneumonia, clavicle fracture, invertebrate disc protrusion, benign breast neoplasm, brain injury and varicose vein; none were considered by the investigator to be rimegepant-related. There were no rimegepant-related TEAEs that led to treatment interruption, discontinuation, or study discontinuation (Table 2). One participant had a serious TEAE (brain injury) that led to treatment and study discontinuation, and death. This was considered unrelated to rimegepant (car accident as pedestrian). One participant experienced an AE leading to study discontinuation and treatment interruption due to atrial septal defect, which was deemed definitely unrelated to rimegepant. During follow-up, 24/228 (10.5%) participants had an AE, with no SAEs or AEs leading to death.
Summary of TEAEs in participants treated with rimegepant 75 mg (safety analysis set).

Data are n (%).
CTCAE = Common Terminology Criteria for Adverse Events; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Common TEAEs occurring in ≥5% of participants were COVID-19 (n = 100 (41.7%)), upper respiratory tract infection (n = 28 (11.7%)), nasopharyngitis (n = 17 (7.1%)), hyperuricemia (n = 18 (7.5%)), hyperlipidemia (n = 16 (6.7%)) and pyrexia (n = 13 (5.4%)). The most frequently reported rimegepant-related TEAEs occurring in ≥2% of participants were sinus arrhythmia (n = 6 (2.5%)) and hepatic function abnormal (n = 6 (2.5%)). During the treatment safety period, TEAEs with maximum CTCAE Grade 1 were reported for 68 participants (28.3%), maximum Grade 2 for 120 participants (50.0%) and maximum Grade ≥3 for 15 participants (6.3%), of which most were Grade 3 (11 (4.6%)). Rimegepant-related TEAEs with maximum CTCAE Grade 1 were reported for 39 participants (16.3%) and maximum Grade 2 for 6 participants (2.5%). There was one participant with rimegepant-related CTCAEs Grade ≥3, TEAEs of aspartate aminotransferase (AST) increased (Grade 3) and blood creatine phosphokinase (CPK) increased (Grade 4). These TEAEs were not considered serious by the investigator, and the participant recovered (levels returned to normal/baseline).
The majority of abnormal laboratory parameters were of Grades 1–2 and are summarized in Table 3. Grades 3 and 4 laboratory abnormalities were reported for 11 participants: four (1.7%) CPK increased, two (0.8%) hypertriglyceridemia and one (0.4%) for each of AST increased, anemia, white blood cell decreased, neutrophil count decreased, lymphocyte count decreased and hypokalemia. Among them, five participants also reported TEAEs with CTCAE Grade ≥3, including: three (1.3%) CPK increased, one (0.4%) for each of AST increased, hypokalemia and white blood cell decreased (including the participant with AST increased (Grade 3) and blood CPK increased (Grade 4) described previously).
Summary of abnormal laboratory parameters according to CTCAE grade occurring in ≥5% of participants, or CTCAE grade ≥3, treated with rimegepant 75 mg (safety analysis set).

All data are n (%).
ALT = alanine aminotransferase; AST = aspartate aminotransferase; CPK = creatine phosphokinase; CTCAE = Common Terminology Criteria for Adverse Events; NR = not reported.
*Based on estimated glomerular filtration rate.
Secondary efficacy endpoint and tablet use
During the OP, the mean (SD) number of MMDs was 11.2 (4.1) and 9.3 (4.2) days for all migraine and for migraine of moderate-to-severe pain intensity, respectively. For all migraine attacks, a mean reduction from the OP in the number of MMDs was observed as early as the first four weeks (−1.7; 95% CI = −2.2 to −1.2) with a trend of continued reduction over time to weeks 41–44 (−5.7; 95% CI = −6.3 to −5.1), followed with a plateau through week 52 (Figure 3). A similar trend was observed for migraine attacks of moderate-to-severe intensity, with reductions of −1.9 (95% CI = −2.4 to −1.4) and −5.9 (95% CI = −6.5 to −5.2) after the first four weeks and at weeks 41–44, respectively. The mean monthly reduction in MMDs across LTT was −4.4 (95% CI = −4.9 to −3.9) days for migraine attacks of any intensity and −4.6 (95% CI = −5.1 to −4.1) days for migraine attacks of moderate-to-severe intensity.
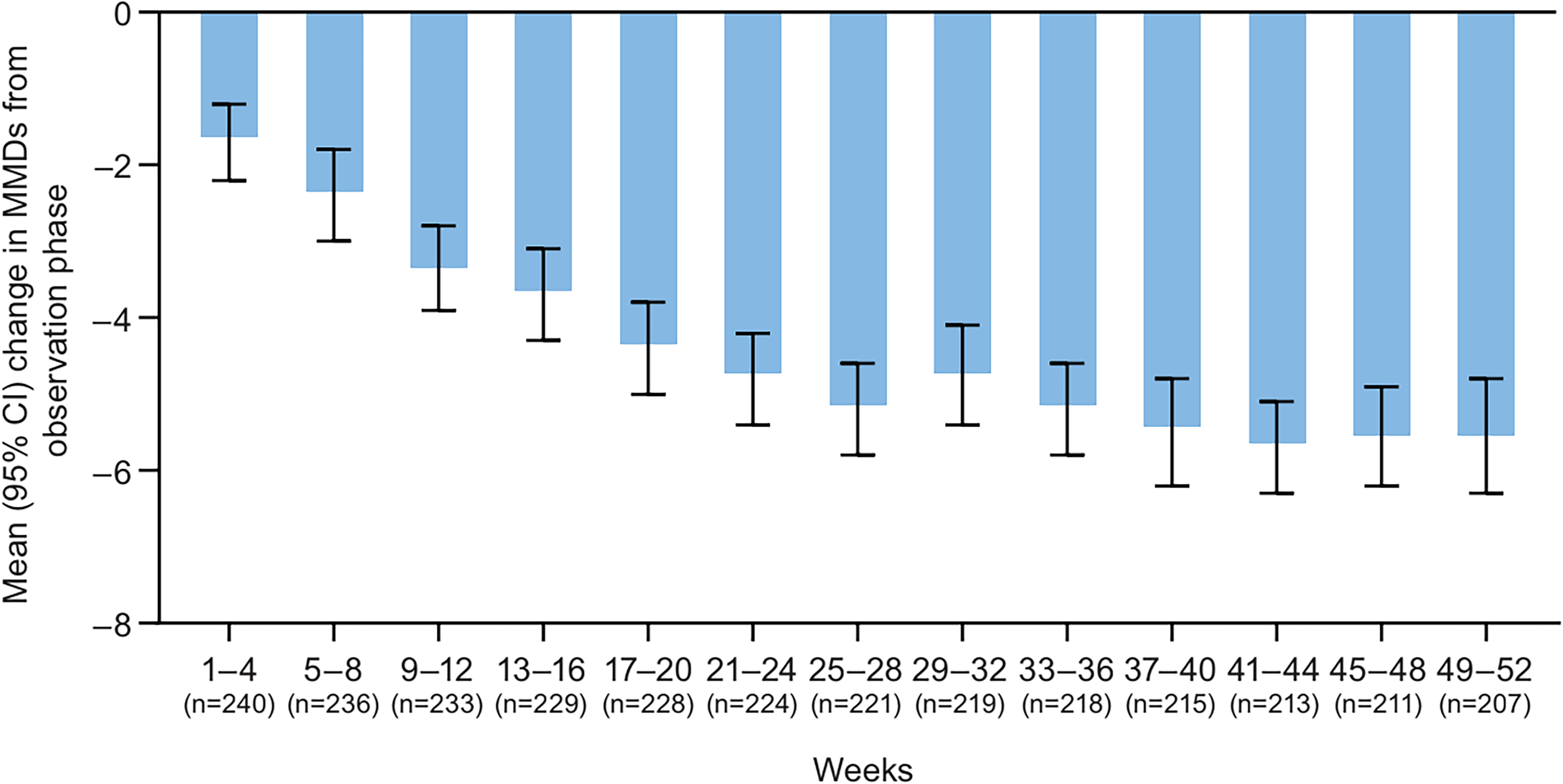
Mean (95% CI) change in MMD from the observation phase in participants treated with rimegepant 75 mg. CI, confidence interval; MMD, monthly migraine day (efficacy analysis set).
Overall, 173 (72.1%), 158 (65.8%), 106 (44.2%) and 70 (29.2%) participants had a ≥25%, ≥30%, ≥50% and ≥60% decrease from the OP in the number of migraine days per month for migraine attacks of any intensity, respectively. A nominally greater reduction was observed for migraine attacks of moderate-to-severe severity; 189 (79.4%), 178 (74.8%), 131 (55.0%) and 98 (41.2%) participants had a ≥25%, ≥30%, ≥50% and ≥60% decrease in the number of moderate-to-severe migraine days per month, respectively.
The number of rimegepant tablets administered each month during LTT is shown in Figure 4. The median (Q1, Q3) rimegepant dose was highest during month 2 (weeks 5–8) at 7 (4, 9) tablets per month and fell to 4 (3, 7) tablets per month by month 9 (weeks 33–36). The median (Q1, Q3) average rimegepant dose during LTT was 4.6 (3.1, 7.1) tablets per month. During LTT, 10.8% (26/240) of participants took less than two tablets per month, 70.8% (170/240) took ≥2 to <8 tablets per month, 12.9% (31/240) took ≥8 to <14 tablets per month and 5.4% (13/240) took ≥14 tablets per month.

Median (Q1, Q3) number of rimegepant tablets per month. Q, quartile (safety analysis set).
Exploratory endpoints
Total MIDAS score and all MSQoLQ domains improved by Week 12 and continued to improve throughout LTT (Figure 5). At week 52, the mean (95% CI) change in total MIDAS score was −38.4 (−44.4 to −32.5). At week 52, the mean (95% CI) change in RR function, PR function and EF was 24.3 (21.9–26.7), 22.7 (20.2–25.2) and 23.0 (20.3–25.8) respectively.

Mean (95% CI) change from baseline in MIDAS (a) and MSQoLQ domains (b) during LTT with rimegepant 75 mg. A decrease in MIDAS score indicates an improvement. An increase in MSQoLQ indicates an improvement. CI, confidence interval; LTT, long-term treatment; MIDAS, Migraine Disability Assessment; MSQoLQ, Migraine-Specific Quality of Life Questionnaire (full analysis set).
At Week 28, 89.3% (191/214) of participants indicated a preference for rimegepant 75 mg over their prior treatment. Preference was sustained throughout LTT and, at week 52, 91.3% (190/208) of participants indicated a preference for rimegepant 75 mg over their prior treatment (Figure 6(a)). Most participants (79.1%; 170/215) were “completely” or “very satisfied” with rimegepant treatment at week 28. Satisfaction was generally consistent through to week 52, when 82.2% (171/208) of participants were “completely” or “very satisfied” with rimegepant (Figure 6(b)). At week 28, treatment with rimegepant had led to a general improvement in 95.8% (206/215) of participants. This was maintained throughout LTT and, at week 52, 94.2% (196/208) of participants showed improvement on the CGI-C scale (Figure 6(c)).

Participant preference (a), satisfaction (b) and Clinical Global Impression–Change (c) during long-term treatment with rimegepant 75 mg (full analysis set).
Discussion
This phase 3, multicenter, open-label, long-term study demonstrated a favorable safety and tolerability profile for rimegepant 75 mg ODT (PRN up to one tablet per calendar day) for the treatment of acute migraine in Chinese adults over 52 weeks, and a two-week follow-up period. There were no rimegepant-related SAEs and no rimegepant-related TEAEs that led to treatment interruption, discontinuation or study discontinuation. The majority of TEAEs were mild-moderate and unrelated to rimegepant, and the most frequently reported TEAEs (COVID-19, upper respiratory tract infection, hyperuricemia, nasopharyngitis, hyperlipidemia and pyrexia) were predominantly due to the participants’ medical history and the COVID-19 pandemic. The safety and tolerability profile demonstrated in this study is reassuring as the dosing approach reflects the intended clinical use.
The safety and tolerability profile reported herein was consistent with that of previous phase 3, placebo-controlled trials for the acute treatment of a single migraine attack with rimegepant 75 mg (3,5,7,8,16). Rimegepant has demonstrated a safety profile comparable with placebo, with most AEs being mild-moderate in intensity and unrelated to rimegepant. Nausea was the only on-treatment AE reported in ≥1% of participants (US population) in either the rimegepant or placebo group in the pooled phase 3 studies (4–7).
In a supportive long-term, open-label safety study, rimegepant 75 mg (PRN) for up to 52 weeks (1514 participants) or every other day + PRN (maximum one tablet per calendar day) for up to 12 weeks (286 participants) demonstrated a favorable safety profile with no new safety signals (9). Consistently, most AEs were mild-moderate and were considered by the investigator to be unrelated to rimegepant. The most frequently (≥5% of participants) reported AEs (regardless of causality relationship) were upper respiratory tract infection (8.8%), nasopharyngitis (6.8%) and sinusitis (5.1%). No signal of drug-induced liver injury was identified, and there were no clinically relevant trends in laboratory test abnormalities (9). In our study, abnormal laboratory parameters and vital signs were generally within acceptable ranges, with a small percentage of participants experiencing elevations in liver function tests or blood pressure. No participant had drug induced liver injury meeting Hy's law, and no hepatic-related AE was reported as an SAE. As such, our study supports the long-term use of rimegepant for the acute treatment of migraine, with an acceptable liver safety profile.
In this study, rimegepant 75 mg (PRN) was associated with a reduction in the number of MMDs as early as the first four weeks, with a trend of continued reduction to weeks 41–44, followed by a sustained reduction through to week 52. This reduction was particularly pronounced among participants experiencing moderate-to-severe migraine attacks. Rimegepant's blockade of the key neuromodulator CGRP in the pathogenesis of migraine, and its established efficacy as both an acute (PRN) treatment and as a preventive (every other day) treatment, raises the possibility that PRN use may also have some preventive benefits by reducing a participant's sensitivity and reactivity to typical migraine triggers. These potential benefits cannot be studied in a typical acute treatment trial because, by design, relatively few attacks are treated, nor can this hypothesis be tested in a prevention trial, as, in that setting, treatment is taken in the absence of an attack. However, long-term safety studies where participants (with certain severity and frequency of migraine attacks) use the drug as needed for the acute treatment of migraine, over extended periods of time, offer an opportunity to explore this hypothesis.
Medication overuse headache (MOH) is common, occurring in approximately 1% of adults, and is often associated with drugs that target the 5HT-1 receptors (triptan-like drugs), opiates and combination products (17,18). In the event of MOH, abrupt withdrawal or tapering down of therapy is often recommended (19). Almost all of the therapeutic agents for the treatment of migraine currently in clinical use have been associated with MOH. The overuse of migraine therapeutics triggers MOH, increases the frequency of migraine attacks and is risk factor for the conversion of episodic migraine to chronic migraine, greatly increasing the additional burden of disease on patients and society (20). Extensive global clinical experience has demonstrated that antagonism of the CGRP signaling pathway (including small molecules and biologics) has no potential risk of producing MOH. The results of our study showed that rimegepant itself when used long-term as an acute treatment was associated with a reduction in MMD over time while the number of rimegepant tablets administered per month (four-week period) reached a median average value of four tablets per month at weeks 33–36. The pattern of monthly rimegepant exposure and MMDs suggests that MOH is unlikely to be associated with rimegepant. Rimegepant may be particularly suitable for people who have a higher frequency of headaches and are at risk of converting to medication overuse headache.
In China, the estimated years lived with disability due to headache disorders in 2017 was 6,609,000 person-years (1). People with migraine are at increased risk for comorbid conditions, including anxiety, depression, asthma, epilepsy and stroke (21). Disability due to migraine is associated with financial cost, including lost work hours and reduced productivity (21). Hence, there is a substantial medical need to reduce the burden of migraine. This study showed improvements in migraine-related disability and migraine-related quality-of-life domains at week 12, which continued to improve throughout LTT with rimegepant 75 mg.
People with migraine may also benefit from new acute treatments to meet unmet medical needs. A study of approximately 4000 respondents using oral acute migraine medication reported that >95% of respondents had at least one unmet treatment need, including rapid headache onset, inadequate two-hour pain freedom and headache recurrence within 24 hours (23). Many people with migraine express dissatisfaction with their acute treatment, leading to approximately 80% willing to try a new treatment (24). Participants in this study reported high levels of satisfaction with, and preference for, rimegepant compared to their prior migraine medication throughout 52 weeks. These high levels of satisfaction and preference are likely a result of the effectiveness and tolerability of rimegepant but may be partly attributable to participants’ current/concomitant medication. A retrospective analysis of antimigraine prescribing patterns in China (2018–2022) found that approximately 70% of outpatients were prescribed analgesics and approximately 26% were prescribed migraine-specific agents, the vast majority of which were triptans (25). In our study, anti-inflammatory/antirheumatics and analgesics were among the most common class of current and concomitant non-study medications. A relatively small proportion of participants were taking triptans (approximately 5%) at or after informed consent, and these were discontinued before starting rimegepant treatment. It is possible the high satisfaction for rimegepant may be partly due to placebo effects (i.e. a positive psychophysical response) following medication administration independent of pharmacological activity. Contextual, including placebo, effects have been reported to contribute to the benefit of CGRP antibodies in migraine treatment (26). However, the mechanisms driving placebo effects are multifaceted and subject to various psychological, physiological and contextual factors, and further research is warranted to discern the true impact in migraine trials. Satisfaction with rimegepant may also partly be attributed to PRN dosing that allows participants to customize their medication usage according to their specific needs. Furthermore, LTT with oral rimegepant (PRN and every other day +PRN dosing) is associated with reduced analgesic and antiemetic use (27). As such, rimegepant represents an important acute treatment option for migraine.
The strengths of the present study include the extended period of treatment and dosing approach, which reflect the intended clinical use of rimegepant, providing further reference for the acute treatment of migraine in Chinese participants. Limitations include the open-label nature of the study and lack of active comparator, which limits understanding of the clinical utility of rimegepant relative to other migraine treatments. Finally, a small number of participants dropped out over time and results were not imputed. If those participants were not experiencing treatment benefit, it may perpetuate positive results among the remaining participants; however, the completion rate for the study was high and the impact of dropout was likely minimal.
Conclusions
Rimegepant demonstrated a favorable safety profile and was well tolerated in Chinese participants during the long-term acute treatment of migraine. A reduction in the number of MMDs was observed as early as the first four weeks and continued to improve for 11 months with a maintained reduction through to the end of LTT. The LTT with rimegepant was associated with improvements in migraine-related disability and quality of life, and participants expressed high levels of preference and satisfaction with rimegepant treatment.
Clinical implications
A multicenter, open-label, long-term safety study of rimegepant 75 mg ODT for the treatment of acute migraine in Chinese participants was conducted.
Rimegepant 75 mg administered PRN for up to 52 weeks demonstrated a favorable safety profile and was well tolerated.
Rimegepant treatment was associated with a reduction in MMDs as early as four weeks that was maintained through week 52.
Footnotes
Acknowledgements
Medical writing support was provided by Leon Adams, PhD, of Engage Scientific Solutions and was funded by Pfizer.
Data availability
Declaration of conflicting interest
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Mingjie Zhang, Aihong Guo, Jin Wu, Hebo Wang, Yongbo Zhang, Hongjuan Dong, Jianguang Liu, Bei Zhang, Huailian Guo, Tingmin Yu and Shengyuan Yu have nothing to declare. Zhihong Lu was employed by Pfizer Inc. at the time of the study. Liheng Ma, Robert J. Fountaine, Qi Zhong and Xiaoran Han are employed by and own stock/stock options in Pfizer Inc. Glenn C. Pixton owns stock in AbbVie and is employed by and owns stock/stock options in Pfizer Inc.
Ethical statement
All participants provided written informed consent before any study activities.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Biohaven, which was acquired by Pfizer in October 2022.