
Abstract
Aim
LY3451838 is a monoclonal antibody against pituitary adenylate cyclase-activating peptide (PACAP), a target in migraine research. The present study aimed to evaluate LY3451838 as a preventive treatment for participants with treatment-resistant migraine.
Methods
Following preclinical assessment of LY3451838, including pharmacokinetic and pharmacodynamic studies, safety was evaluated in a phase 1 study of LY3451838 (n = 33) versus placebo (n = 13) in healthy participants. A phase 2 trial was carried out in treatment-resistant participants with chronic migraine (CM) (n = 16) or episodic migraine (EM) (n = 22). In phase 2, participants received a single intravenous (IV) dose of 1500 mg LY3451838 (n = 19) or placebo (n = 19) and completed ePRO daily diaries. Participants were followed for safety for 140 days.
Results
In phase 2, at one-month post-dose, patients who had received a single IV dose of LY3451838 exhibited greater changes from baseline than placebo in mean monthly migraine headache days in both the CM and EM subgroups (CM: −4.7 days vs. −3.0 days; EM: −1.7 days vs. −1.2 days), but the treatment contrast was not statistically significant in either subgroup. Similar non-significant results were seen at the three-month time point. The percentage of participants reporting treatment-emergent adverse events was similar for LY3451838 and placebo, with one serious adverse event of B-cell lymphoma in an LY3451838-treated participant that led to study discontinuation.
Conclusions
LY3451838 did not demonstrate superior efficacy over placebo in patients with treatment-resistant CM or EM. However, the difference observed between LY3451838 and placebo among CM patients is similar to the statistically significant difference reported in the recent HOPE trial, which primarily consisted of CM patients. Further clinical research with larger sample sizes is needed to inform on the utility of blocking PACAP in various migraine populations.
Trial Registration
ClinicalTrials.gov: NCT03692949 (phase 1); NCT04498910 (phase 2).
This is a visual representation of the abstract.
Introduction
Migraine affects more than a billion people worldwide (1). Preventive treatments exist, including two recently introduced classes of drugs: small-molecule calcitonin gene-related peptide (CGRP) receptor antagonists and monoclonal antibodies (mAbs) against CGRP or its receptor (2). However, there remains room for therapies that provide relief to patients who do not respond to available treatment options (3).
Encoded by the adenylate cyclase-activating polypeptide 1 (ADCYAP1) gene, pituitary adenylate cyclase-activating peptide (PACAP) is a member of the glucagon/secretin/vasoactive intestinal peptide (VIP) family of neuropeptides, with an amino acid sequence most closely resembling that of VIP (3). PACAP is initially expressed as a preproprotein that later undergoes post-translational processing to either the PACAP27 or PACAP38 form (4). Expressed in both central and peripheral tissues, the PACAP38 form largely predominates (3). Although both VIP and PACAP are agonists of VIP receptors type 1/2 (VPAC1/2), PACAP receptor type-1 (PAC1) has higher affinity (approximately 0.5 n
PACAP has been implicated in multiple physiological and endocrine functions, including vasodilation and circadian rhythms (9–11), as well as primary headache conditions (3,12). It is assumed that in primary headache conditions, PACAP27/38 modulates synaptic activity in the trigeminovascular system via a small but significant release of PACAP from the trigeminal ganglia, potentially activating PAC1 receptors on the local satellite cells (13). As a vasodilatory peptide that is associated with the trigeminal system, PACAP shares many similarities with the key migraine mediator CGRP (14). PACAP and CGRP both exhibit elevated plasma levels during migraine attacks (although CGRP more so than PACAP (15,16), and, when administered, can trigger headache in healthy subjects and migraine in migraine patients (15,17–21). However, PACAP expression only partially overlaps with that of CGRP in key migraine-linked neuronal pathways. For example, PACAP, but not CGRP, is released by parasympathetic neurons in the sphenopalatine ganglia that innervate the dura mater (14,22); despite this, neither PACAP nor its receptors are typically seen in the nerve fibers, the dura mater or its vasculature (13). Furthermore, although the in vivo vasodilatory effect of PACAP on the middle meningeal artery (a major supply artery to the dura mater) has been reported as pronounced and sustained (19), in vitro work has described PACAP as less potent in human meningeal arteries compared to CGRP (23–24). Given the density of PACAP in the parasympathetic ganglia (16,25–27), PACAP may have a role in headache disorders via inflammation of the trigeminovascular system.
LY3451838 is a fully human immunoglobulin G4 (IgG4)-variant mAb designed to block PACAP activity as a potential treatment for migraine prevention. Akin to the reduced migraine burden following CGRP blockade, non-clinical data have suggested that LY3451838 could be effective for preventive treatment of migraine by blocking the PACAP pathway in humans. With a similar affinity to both PACAP27 and PACAP38, LY3451838 was evaluated in preclinical, phase 1 clinical and phase 2 clinical testing. In phase 2, LY3451838 was tested in participants with treatment-resistant migraine, who might benefit most from the novel mechanism of action.
Methods
Ethical statement
Protocols for all clinical studies included in this analysis were approved by the Institutional Review Board or Ethics Committee at each participating site. All studies included in this analysis were conducted in accordance with the ethical principles of the Declaration of Helsinki. Written informed consent was obtained from all eligible participants before undergoing study-related procedures. The phase 1 study protocol was reviewed by the Institutional Research Review Committee and the National Healthcare Group Domain Specific Review Board (NHG DSRB Reference: 2018/01021). Institutional Review Board approval for the phase 2 study was obtained from Advarra (Reference: Pro00041989). All animal studies were conducted in accordance with the policies and recommendations of the National Institutes of Health guidelines for laboratory animals and conform to the protocols approved by the Eli Lilly Institutional Animal Care and Use Committee. ARRIVE and CONSORT reporting guidelines were employed as appropriate (28,29).
The preclinical methodology is described in the supplementary material (Doc. S1).
Phase 1 study (NCT03692949)
The safety of a single intravenous (IV) dose of LY3451838 (25, 75, 250, 500, 1000 or 1500 mg) versus placebo was evaluated in a first-in-human, double-blind, randomized study in healthy human participants at one center in Singapore between 11 December 2018 and 26 February 2020. In total, 47 participants were enrolled and treated (LY3451838, n = 33; placebo, n = 14); all but one participant (placebo recipient) completed the study. Screening was conducted during the 28 days before dosing. Participants were required to be 21–65 years of age, with a body mass index (BMI) of 18–35 kg/m2 and an estimated glomerular filtration rate of ≥60 mL/min/1.73 m2. Exclusion criteria included a history or presence of certain medical illnesses that rendered the individual unsuitable for participation, such as significant autoimmune diseases (see supplementary material, Doc. S1).
Eligible participants were assigned sequentially into six dosing cohorts and randomized to LY3451838 or placebo in a 6:2 ratio within each cohort. Participants were admitted to the clinical research unit on Day −1, and eligibility was confirmed before dosing on Day 1. Participants were monitored for 140 days post-dose for safety and pharmacokinetic (PK) sample collection. Serum PK samples were analyzed by an internally designed and validated enzyme-linked immunosorbent assay to determine LY3451838 concentration.
The endpoints in this study included safety and PK in healthy participants. Summary safety statistics for each cohort are provided by dose level and for all placebo recipients combined. Serum LY3451838 PK (maximum drug concentration (Cmax) and area under the serum concentration–time curve (AUC) from time 0 to infinity (AUC0–∞)) were calculated using noncompartmental methods and summarized by dose level using descriptive statistics.
Phase 2 study (NCT04498910)
The safety and efficacy of 1500 mg LY3451838 versus placebo were evaluated in adult participants with treatment-resistant migraine across nine centers in the United States between 16 November 2020 and 9 November 2022. This dose was chosen to ensure adequate target engagement and to account for the uncertainty in translating the preclinical model to human efficacy.
In addition to being 18–75 years of age, key inclusion criteria were two-fold: a diagnosis of migraine as defined by the International Headache Society International Classification of Headache Disorders, 3rd edition, and documentation of treatment resistance, defined as failure to respond to two to four different migraine preventive medication categories, including anti-CGRP medications, in the past 10 years due to inadequate efficacy or safety/tolerability reasons. Additional inclusion criteria were a history of migraine headaches of at least one year prior to visit 1, and migraine onset prior to age 50 years. Exclusion criteria included concurrent use of other preventive migraine treatments, failure of more than four migraine preventive medication categories in the past ten years, and a history of cluster headaches or migraine subtypes. For additional information, see the supplementary material (Doc. S1).
This proof-of-concept study was conducted in four periods (Figure 1). In the screening period, informed consent was obtained, a full clinical assessment was performed and participants discontinued excluded medications or treatments. In the baseline period, study eligibility and baseline data were established, which included daily logging in an electronic participant-reported outcomes (ePRO) diary. Data logged included headache occurrence, duration, features and severity, and also whether any acute headache medication was taken; the name, dose and date of any acute headache medication taken was logged and returned to site staff at every visit. In the double-blind treatment period, eligible participants were stratified by migraine type (chronic migraine (CM) or episodic migraine (EM)) and randomly assigned 1:1 within each stratum to receive a single dose of LY3451838 (1500 mg) or placebo through an IV infusion over at least 60 min. EM was defined as 4–14 migraine headache days and fewer than 15 headache days per one-month period. CM was defined as at least 15 headache days per one-month period, of which at least eight were migraine days. Participants continued completion of daily ePRO diary entries and were permitted to continue taking allowed acute migraine headache medication while recording this use. In the follow-up period, participants were followed for safety for 140 days post dose and continued to complete the ePRO diary and record their use of acute headache medications.

Study design of the phase 2 trial evaluating LY3451838 in patients with treatment-resistant CM or EM. *Treatment with LY3451838 (n = 60) or placebo (n = 60). CM = chronic migraine; EM = episodic migraine; ePRO = electronic participant-reported outcomes; HD = headache days; IV = intravenous; MHD = migraine headache days; Mth = month.
The primary endpoint was the mean change from baseline in the number of monthly migraine headache days during the one-month treatment phase. Secondary endpoints included the proportion of participants with a ≥50%, ≥75% or 100% reduction from baseline in monthly migraine headache days, treatment-emergent adverse events (TEAEs), serious adverse events (SAEs) and AUC and Cmax for LY3451838. Bayesian analysis was performed for the primary efficacy endpoint and corresponding frequentist analyses were conducted as a sensitivity analysis. All other statistical inference was performed using frequentist methods.
Results
Preclinical studies: In vitro
In vitro work determined that LY3451838 blocks PACAP27- and PACAP38-based activation of human PAC1, VPAC1 and VPAC2 on Chinese hamster ovary (CHO) cells (mean potencies: 77–605 p
Preclinical studies: In vivo
LY3451838 pharmacology and PK were evaluated in Sprague–Dawley rats (see supplementary material, Doc. S1). After IV administration of a single 10 mg/kg dose of LY3451838 in rats, the mean clearance and half-life (t1/2) were 0.46 ml/h/kg and 138 hours, respectively (see supplementary material, Table S1). LY3451838 pharmacodynamics were evaluated in CD-1 mice and Sprague–Dawley rats. In mice, a subcutaneous 3-day pretreatment injection of 0.3 to 10 mg/kg LY3451838 dose-dependently blocked PACAP-induced increases in plasma cyclic adenosine monophosphate (cAMP) levels when samples were collected 10 minutes after IV administration of 13 nmol/kg PACAP38 (Figure 2(a)). In contrast, the IgG4 PAA control mAb (LSN2835015) failed to prevent a PACAP-induced increase in plasma cAMP levels (Figure 2(a)). These data demonstrate an in vivo pharmacodynamic response in mice with LY3451838 treatment. In rats, LY3451838 at doses of 10 and 30 mg/kg, but not 3 mg/kg, inhibited plasma protein extravasation in the dura following electrical stimulation of the trigeminal ganglion. In contrast, LSN2835015 did not inhibit plasma protein extravasation. This effect is illustrated at 10 minutes after IV administration in Figure 2(b).

Preclinical LY3451838 pharmacodynamic assays. (a) In vivo inhibition of PACAP38-induced increase in mouse plasma cAMP levels with LY3451838 treatment. CD-1 mice (male) were injected subcutaneously with 0.3 to 10 mg/kg LY3451838 or the control IgG4 PAA antibody LSN2835015 as a three-day pretreatment. Rolipram (100 µg/kg) was included in the vehicle to inhibit rapid degradation of plasma cAMP. Plasma was then collected 10 minutes after injection of either vehicle or PACAP38 (13 nmol/kg) as indicated. Plasma cAMP levels are graphed as the mean (standard error), n = 5 to n = 6. Percentages represent the percent inhibition. #p < 0.001 vs. vehicle, **/***p < 0.01/0.001 vs. PACAP (using an ANOVA model followed by Dunnett's post-hoc test). (b) Inhibition of trigeminal stimulation-induced PPE in the dura of rats following pretreatment with LY3451838 or the control IgG4 PAA antibody LSN2835015. LY3451838 or LSN2835015 was administered 10 minutes before electrical stimulation of the trigeminal ganglion. Data are expressed as the extravasation ratio (stimulated/unstimulated). Data are represented as the mean (SEM). *p < 0.05 vs. control antibody (using an ANOVA model followed by Dunnett's post-hoc test, mean (standard error), n = 3). ANOVA = analysis of variance; cAMP = cyclic adenosine monophosphate; IgG = immunoglobulin G; IV = intravenous; PACAP = pituitary adenylate cyclase-activating polypeptide; PPE = plasma protein extravasation; SC = subcutaneous; SEM = standard error of the mean.
Phase 1 study
In total, 47 participants were randomized and treated (LY3451838, n = 33; placebo, n = 14), of whom 46 completed the study (LY3451838, n = 33; placebo, n = 13) (see supplementary material, Figures S1 and S2 and Table S2). Phase 1 participants had a mean age of 42.8 years (range 22–63 years), mean (standard deviation) BMI of 25.1 (3.6) kg/m2, and were primarily male (84.9%) and all Asian. There were no deaths, SAEs or discontinuations due to TEAEs reported for these healthy participants. The proportion of participants who experienced TEAEs was comparable between groups (LY3451838, 82.1%; placebo, 85.7%). The most common TEAEs considered related to LY3451838 included headache, abdominal discomfort and diarrhea. No clinically significant changes in vital signs or orthostatic changes in blood pressure or heart rate were observed. For additional information, see supplementary material (Table S3).
The Cmax, AUC from time 0 to the last measurable serum concentration and AUC0–∞ of LY3451838 all increased in an apparent dose-proportional manner (see supplementary material, Table S4). Median time to Cmax consistently occurred at early time points and ranged from 0.57 to 4.54 hours after the end of IV infusion. Calculated geometric mean t1/2 was generally consistent across dose groups, ranging from 213 to 340 hours. Geometric mean total body clearance ranged from 10.0 to 15.8 ml/h (for serum concentration-time profiles, see supplementary material, Figure S3), with geometric mean volume of distribution at steady state (VSS) ranging from 3.54 to 5.21 litres, reflecting low distribution to peripheral tissues.
Phase 2 study
This study did not meet its originally planned enrollment goals, with slower-than-expected recruitment resulting in changes to the planned analyses (for additional information, see supplementary material, Doc. S1). In total, 38 participants were randomized and treated (LY3451838, n = 19; placebo, n = 19) (for the time course of serum LY3451838 concentration, see supplementary material, Figure S4), of whom 33 completed the study (LY3451838, n = 16; placebo, n = 17) (see CONSORT diagram in Figure 3). Overall, demographic and baseline characteristics were balanced across treatment groups (Table 1). Participants had a mean age of 48.3 years, a mean (standard deviation) BMI of 30.0 (6.2) kg/m2 and were primarily female and white. At baseline, participants experienced a mean of 13.5 monthly migraine headache days (see supplementary material, Table S5).
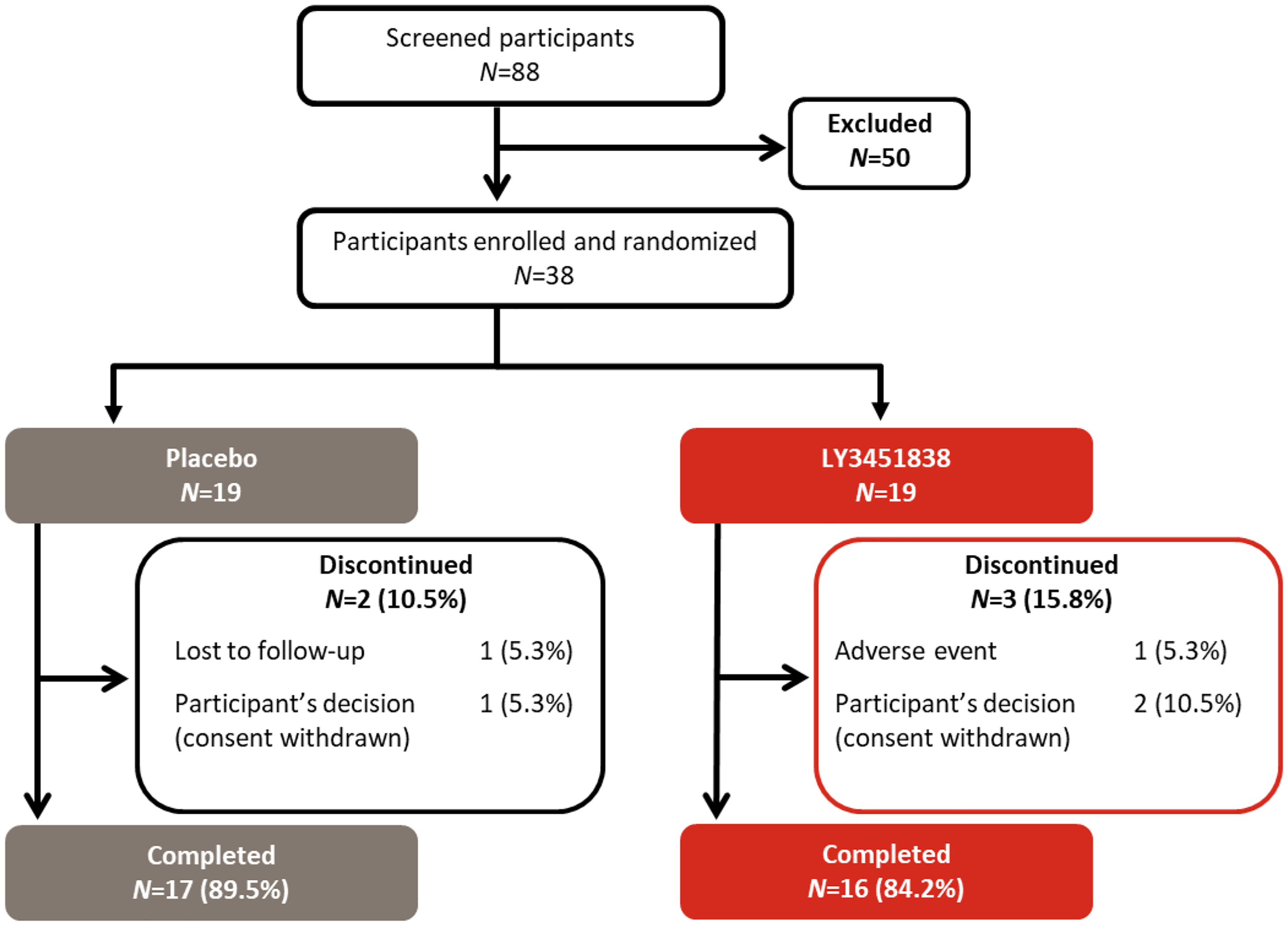
CONSORT diagram for the phase 2 proof-of-concept study. CONSORT = Consolidated Standards of Reporting Trials; n = number of participants.
Baseline demographics and disease characteristics of adults with treatment-resistant migraine in the phase 2 proof-of-concept study.

Data are presented as the mean (standard deviation) or n (%). Prior migraine preventive medications included any migraine preventive medications that were discontinued in the past 10 years due to safety or tolerability concerns, an inadequate response, or no response.
BMI = body mass index; MHD = migraine headache days; n = number of participants.
Although participants were meant to have failed between two to four previous treatments, the final sample may have included participants who had failed between one and nine treatments (for prior medications administered to more than one participant, see supplementary material, Table S6) because we have insufficient documentation to determine otherwise (for additional information, see Discussion). All phase 2 participants were administered at least one acute concomitant medication, with oral analgesic combinations containing paracetamol (n = 20; 52.6%) and triptans (n = 19; 50.0%) being the most common (see supplementary material, Table S7).
At one month post-dose, participants who had received LY3451838 exhibited greater changes from baseline in mean monthly migraine headache days than those who received placebo, for both the CM (−4.7 days vs. −3.0 days) and EM (−1.7 vs. −1.2) subgroups (Figure 4(a)). The difference from placebo was not statistically significant for either subgroup; the 95% confidence intervals (CIs) associated with LY3451838 – placebo were −5.2 to 2.0 days for CM and −3.8 to 2.6 days for EM. This pattern persisted to three months post-dose, with greater changes from baseline with LY3451838 than placebo (CM: −5.3 days vs. −3.0 days; EM: −1.9 days vs. −1.2 days) (Figure 4(b)) that were not statistically significant; the 95% CIs for LY3451838 – placebo were −6.1 to 1.8 days for CM and −4.3 to 2.7 days for EM. Similarly, exploratory analyses did not reveal differences between treatment groups in the proportion of participants with ≥50% (n = 4 LY3451838; n = 5 placebo), ≥75% (n = 0; n = 1), or 100% (n = 0; n = 1) reduction from baseline in monthly migraine headache days (see supplementary material, Table S8).

Overall mean change in monthly migraine headache days. Differences in the LS mean change from baseline (error bars depict the standard error) are represented within the figure on top of the bars. LY3451838 was dosed at 1500 mg via intravenous administration. Baseline, monthly number of migraine headache days at baseline. LS = least-squares; n = number of participants.
One SAE was reported during the study, in a participant who received LY3451838: primary diffuse large B-cell lymphoma of the brain, which was assessed as not related to the study drug (see supplementary material, Table S9). This SAE led to the participant's discontinuation and death after study completion. No clinically meaningful changes in blood pressure or heart rate, nor any AEs from such changes, were observed; however, this study was not designed to look at such changes, only AEs in a broad sense.
The incidence of TEAEs was similar between those receiving LY3451838 (n = 11; 57.9% with ≥1 TEAE) and placebo (n = 13; 68.4%); the same was true for mean changes in clinical laboratory values and vital signs. The most common TEAE was upper respiratory tract infection (see supplementary material, Table S10).
Similar PK were observed in participants with treatment-resistant migraines as in healthy participants (Table 2), with a geometric mean Cmax of 613 μg/ml, a geometric mean t1/2 of 352 hours, AUC0–∞ of 178,000 μg·h/ml, geometric mean total body clearance of 8.53 ml/h, and geometric mean VSS of 3.81 litres. At 30 days post dose, the geometric mean LY3451838 concentration was 143 μg/ml (range 89–354 μg/ml). The geometric mean LY3451838 concentration at 90 days post-dose was 9.56 μg/ml (range 2.85–30.9 μg/ml).
Summary of LY3451838 PK parameters after a single intravenous infusion of 1500 mg LY3451838 the phase 2 proof-of-concept study.

AUC0–∞ = area under the serum concentration-time curve from time 0 to infinity; AUC0–t = area under the serum concentration-time curve from time 0 to the last measurable serum concentration; CL = total serum clearance; Clast = last measurable concentration; Cmax = maximum observed serum drug concentration; CV = coefficient of variation; n = number of participants; NA = not applicable; NC = not calculated; PK = pharmacokinetic; t1/2, terminal elimination phase half-life; tlast = time to the last measurable concentration; tmax = time to reach the maximum observed serum drug concentration; Vss = volume of distribution at steady state.
Discussion
In the phase 2 trial evaluating the efficacy and safety of LY3451838 in participants with treatment-resistant migraine, a single IV dose of 1500 mg LY3451838 led to a reduction in mean monthly migraine headache days, particularly for those with CM, although this reduction did not statistically differ from that with placebo. In both phase 1 and phase 2 clinical testing, LY3451838 and placebo participants exhibited TEAEs at similar frequencies, with one LY3451838-treated participant discontinuing phase 2 testing due to an SAE of primary diffuse large B-cell lymphoma of the brain. Serum concentrations of LY3451838 increased with increasing IV dose (25, 75, 250, 500, 1000 or 1500 mg) in the phase 1 evaluation of LY3451838 in healthy human participants, with PK parameters suggesting linear PK over the administered dose range.
Although not statistically significant, the observed phase 2 reductions in mean monthly migraine headache days were numerically greater with LY3451838 than with placebo and similar to those seen in previous studies such as the CONQUER trial (30). Participants in the CONQUER trial who received the anti-CGRP mAb galcanezumab (120 mg) during a three-month treatment phase displayed a least-squares mean reduction in monthly migraine headache days of −4.1 (vs. −1.0 with placebo) (30), while those who received LY3451838 exhibited a mean three-month reduction of −3.6 days (vs. −2.2 with placebo). Furthermore, the observed reductions with LY3451838 were greater than those seen in a recent trial of the PAC1 antagonist AMG 301, although neither trial demonstrated a statistical difference versus placebo on any efficacy measure (31). Unlike AMG 301, LY3451838 inhibits PACAP directly and was given as a single infusion rather than multiple administrations; however, participants in both trials were evaluated at the same three-month time point. The least-squares mean (standard error) three-month reduction in monthly migraine days in the trial of AMG 301 (n = 343 enrolled) was −2.2 (0.5) days for both AMG 301 treatment groups and −2.5 (0.4) days for placebo (31), while the current trial (n = 38) found a mean (standard error) reduction of −3.6 (1.2) days with LY3451838 and −2.2 (1.3) days with placebo.
More recently and more relevantly to the LY3451838 mechanism of action, the HOPE proof-of-concept trial reported superior efficacy of the anti-PACAP mAb Lu AG09222 versus placebo in patients with treatment-resistant migraine (32). When comparing the HOPE trial results to this trial, it is important to recognize that, although both sets of inclusion criteria required treatment failure with two to four previous preventive treatments, the HOPE trial was designed to have a 70% CM population and only reported results for the combined CM and EM population. The baseline migraine days for each trial reflect this difference: the HOPE trial had a baseline value of 16.7 days, while this trial had baseline values of 19.6 and 9.0 days in the CM and EM populations, respectively. As such, a comparison of the CM segment of this trial to the overall population in the HOPE trial where 70% of the population is CM, is perhaps the most appropriate. The HOPE trial placebo group (n = 94) showed a mean change from baseline in monthly migraine days of −4.2, while the high dose of the anti-PACAP mAb Lu AG09222 (n = 97; 750 mg IV infusion) resulted in a reduction of −6.2 days over the four-week treatment span (32). This reported difference from placebo of −2.0 days in the HOPE trial is not substantially different from that observed in the CM segment of this much smaller trial: −1.7 and −2.3 days versus placebo at one and three months, respectively. Ultimately, further clinical research with larger sample sizes is needed to inform on the utility of blocking PACAP with a mAb treatment in various migraine populations.
Inhibition of PACAP has become an attractive target in migraine treatment; however, it remains an ongoing area of research as the physiology is not yet fully understood (33–34). PACAP signaling is inherently complex because both PACAP27 and PACAP38 bind with similar affinity to four receptor types that have widespread distributions. As a result, PACAP is a highly multifunctional peptide, involved in the regulation of neurotransmission, vasodilation, intestinal motility, cell proliferation and differentiation, neuroprotection, reproduction, and immunity, and may even serve as a potent antimicrobial peptide (3). This complexity is exacerbated by the fact that it remains unclear which receptor subtypes are specifically linked to migraine, and clinical trials evaluating antagonism of PAC1 (e.g. AMG 301) have not demonstrated efficacy (31). The complex signaling environment, together with the limited number of specific agonists and antagonists available for PACAP receptors, has led to the development of anti-PACAP molecules instead (3). Given the diverse functions of PACAP, a clearer understanding of the underlying physiology and identification of a more specific drug target are warranted (3).
The present study has several limitations. First, the study was limited by sampling from a treatment-resistant population, which precludes drawing conclusions with respect to the broader population suffering from migraine. Second, this trial was powered based on the large treatment effect size seen in the CONQUER trial with galcanezumab (30) (a −3.1 treatment effect) and was insufficiently powered for detecting statistical significance akin to the more modest −2.0 treatment effect as seen in the CM population in this trial and reported in the HOPE trial with Lu AG09222 (32). Third, we were limited by our lack of detailed documentation on prior treatment failures in our global database (i.e. beyond site-level data); we relied on site staff to confirm all eligibility parameters and only required documentation of confirmation, which precludes us from confirming that all participants had previously failed two to four preventive medication categories as intended. Importantly, no protocol deviations were reported by any site. Finally, given the low sample size, efficacy analyses were limited to the primary endpoint and select secondary and exploratory endpoints. It is possible that a larger sample size would have revealed a statistically significant effect on the primary endpoint, similar to that seen in the HOPE trial (32).
Conclusions
In conclusion, LY3451838 blocked PACAP activity and showed promise as a potential treatment for migraine prevention during preclinical analyses and had a similar safety profile to placebo during phase 1 testing. In the phase 2 proof-of-concept study reported here, the anti-PACAP mAb LY3451838 did not demonstrate superior efficacy over placebo in an adult, treatment-resistant population of participants with CM and EM. However, the study was powered to detect a large effect size and thus the sample size might have been insufficient to detect a more modest effect. Further research into PACAP mechanisms is needed to determine whether a subgroup of patients suffering from migraine can have clinically meaningful benefit from treatment with an anti-PACAP mAb.
In both phase 1 and phase 2 testing, the incidence of TEAEs, as well as the mean changes in clinical laboratory values and vital signs, were similar between groups, with no treatment-related SAEs. The phase 2, randomized, double-blind, placebo-controlled, proof-of-concept study in adults with treatment-resistant migraine did not meet its primary endpoint. LY3451838 did not demonstrate superior efficacy over placebo in treatment-resistant episodic or chronic migraine. Efficacy of LY3451838 and placebo was assessed at one and three months. The study was carried out in a treatment-resistant episodic and chronic migraine population.
Supplemental Material
sj-docx-1-cep-10.1177_03331024251368757 - Supplemental material for Preclinical and clinical evaluation of LY3451838, a PACAP-neutralizing monoclonal antibody, in randomized, double-blind, placebo-controlled phase 1 and phase 2 studies involving healthy adults and adults with treatment-resistant migraine
Supplemental material, sj-docx-1-cep-10.1177_03331024251368757 for Preclinical and clinical evaluation of LY3451838, a PACAP-neutralizing monoclonal antibody, in randomized, double-blind, placebo-controlled phase 1 and phase 2 studies involving healthy adults and adults with treatment-resistant migraine by Michael P. Johnson, Judith Krikke-Workel, Chetan N. Patel, S. Michelle Morin, P. Kellie Turner, Kristie A. Clark, David Donley, Yan Jin, Kirk W. Johnson, Maurice Vincent, John R. Stille, Lisa M. Broad and Ashok Patel in Cephalalgia
Footnotes
Acknowledgments
Eli Lilly and Company thank the clinical trial participants and their caregivers, without whom this work would not be possible. Further details regarding the trial investigators and study sites associated with this study are provided in the supplemental material. The authors also thank Daniel Girard, BSc, for substantial contributions to the preclinical work, as well as Emma Raftis, PhD, Philiswa Mbandlwa, PhD, and Dominika Kennedy, PhD, for their project management support and expertise in medical writing and strategic communication.
Data availability
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request six months after the indication studied has been approved in the USA and European Union and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report and blank, or annotated case report forms, will be provided in a secure data sharing environment upon request. For details on submitting a request, see the instructions provided at ![]() .
.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/orpublication of this article: Michael P. Johnson, Judith Krikke-Workel, Chetan N. Patel, S. Michelle Morin, P. Kellie Turner, Kristie A. Clark, Yan Jin, John R. Stille and Lisa M. Broad are employees and minor stake/shareholders of Eli Lilly and Company. David Donley is a contractor at Eli Lilly and Company via EMB Statistical Solutions. Maurice Vincent and Kirk W. Johnson were employees and minor stake/shareholders of Eli Lilly during the time of the study but are not current employees. Dr Chetan N. Patel has received personal fees and clinical grants from Eli Lilly. Daniel Girard, Emma Raftis, Philiswa Mbandlwa and Dominika Kennedy are employees of Eli Lilly and Company. No other disclosures were reported.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is sponsored by Eli Lilly and Company. Role of the Funder/Sponsor: Eli Lilly and Company was responsible for design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review or approval of the manuscript; and the decision to submit the manuscript for publication.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.