
Abstract
Substance P, a neuropeptide associated with pain and inflammation, has been implicated in migraine pathophysiology through its action within the trigeminovascular system. This narrative review summarizes current evidence on the synthesis, release, receptor binding and downstream effects of substance P. It integrates preclinical and clinical findings to reassess its therapeutic relevance. Substance P is released from primary afferent neurons and acts on neurokinin-1 receptors, which are widely expressed in both peripheral tissues and central pain-processing regions. In the meninges, substance P contributes to vasodilation, plasma protein extravasation, mast cell degranulation and immune cell recruitment, all of which facilitate neurogenic inflammation and possibly lower the activation threshold of meningeal nociceptors. In the central nervous system, substance P promotes excitatory neurotransmission, potentiates glutaminergic activity and attenuates inhibitory GABAergic signaling, cumulatively amplifying pain transmission. Preclinical studies consistently demonstrate that neurokinin-1 receptors antagonists inhibit substance P-induced responses, such as neurogenic inflammation and neuronal activation, supporting their therapeutic potential. However, randomized controlled trials with neurokinin-1 receptors antagonists were not superior to placebo in treating migraine. A re-appraisal of these trials reveal that the disappointing results might be due to methodologic shortcomings, including underpowered samples and suboptimal efficacy endpoints. A recent randomized, double-blind, placebo-controlled, two-way crossover study showed that intravenous infusion of substance P is potent inducer of headache and arterial dilation in healthy adults. These findings align with the established biological functions of substance P and warrant renewed therapeutic interest. Advances in translational research, particularly those emphasizing direct measurement of meningeal nociceptor activity and refined clinical trial design, might overcome past limitations and clarify the role of substance P in migraine. The dismissal of substance P signaling as a therapeutic target might thus have been premature. Renewed efforts might uncover novel therapeutic strategies, offering hope for patients with migraine who remain unresponsive to existing treatment.
This is a visual representation of the abstract.
Introduction
Migraine is a disabling neurological disorder (1,2), characterized by recurrent attacks of moderate-to-severe headache and accompanying photophobia, phonophobia, nausea and vomiting (3). Fundamental to migraine pathogenesis is the activation of the trigeminovascular system (TVS), which comprises the trigeminal nerve and its axonal projections that innervate the meninges and its blood vessels (4,5). Activation of this system leads to the release of various neuropeptides from sensory nerve fibers, including but not limited to substance P and calcitonin gene-related peptide (CGRP) (6,7).
Substance P is an 11-amino-acid neuropeptide of the tachykinin family that has been extensively studied for its role in pain transmission and neurogenic inflammation (8,9). Early preclinical experiments implicated substance P in migraine mechanisms, suggesting that it contributes to vasodilation, plasma protein extravasation (PPE), mast cell degranulation and immune cell recruitment within the meninges (10). These insights led to the development of neurokinin-1 receptor (NK1R) antagonists as potential therapeutic agents in migraine (11). In addition to its role in pain transmission, substance P has also been implicated in mood regulation, likely due to its expression in limbic brain regions such as the amygdala and hippocampus (12–14). These regions are associated with the affective-emotional components of pain and psychiatric comorbidities, including anxiety and depression (15,16), which are common in people with migraine (17,18).
Despite these mechanistic insights, the translation into effective therapies has proven challenging. A considerable proportion of individuals with migraine continue to experience inadequate relief from existing treatments, underscoring a persistent unmet need (19). The failure of clinical trials involving NK1-R antagonists, despite promising preclinical data (20–28), has contributed to a shift in focus toward other neuropeptide systems (29). However, this abandonment might have been premature, as trial design limitations and evolving understanding of migraine pathogenesis warrant renewed investigation of substance P signaling.
Randomized controlled trials of NK1R antagonists, dapitant, lanepitant, GR205171 and L-758,298, did not prove superior to placebo in treating migraine (30–34). This disconnect between robust preclinical findings and negative clinical trials illustrates the challenges in translating molecular insights into effective migraine treatments. Importantly, recent human experimental data might shift this perspective. A randomized, double-blind, placebo-controlled, two-way crossover study demonstrated that intravenous infusion of substance P reliably induced headache and arterial dilation in healthy adults (35). Notably, this represents the first study to investigate the role of substance P in headache generation in humans directly because previous studies with substance P had only reported headache as a side effect rather than examining it as a primary outcome (36–38). These findings provide direct evidence of the ability of substance P to trigger headache in humans and align with its known pro-nociceptive and vasodilatory properties.
Together, this emerging evidence supports a renewed appraisal of substance P. It appears to be a mechanistically and clinically relevant drug target in migraine. This narrative review summarizes current evidence on the synthesis, release, receptor binding and downstream effects of substance P. It integrates preclinical and clinical findings to reassess its therapeutic relevance.
Methods
We conducted a literature search using the MEDLINE database (via PubMed) for articles published up until April 1, 2025. The search terms included combinations of: “Substance P”, “Neurokinin-1 Receptor”, “Migraine”, “Trigeminovascular System” and “Neurogenic Inflammation”. The reference lists of selected articles were also manually searched for additional relevant studies. No language restrictions were imposed during the literature search.
Substance P in the trigeminovascular system
The TVS is widely accepted as the anatomical and physiological substate from which nociceptive messaging originates and ultimately yields the perception of migraine headache (4). In migraine, nociceptive signals are posited to arise from pain-sensitive intracranial structures, such as the meninges and its blood vessels (39,40). These signals are detected by the peripheral processes of first-order sensory neurons, which include thinly myelinated Aδ fibers and unmyelinated C fibers (41). The cell bodies of these pseudounipolar neurons reside in the trigeminal ganglion (TG) (42).
The central axonal processes of these neurons enter the brain stem at the level of the pons and descend ipsilaterally within the spinal tract of the trigeminal nerve (43). They terminate in the spinal trigeminal nucleus, particularly its caudal subdivision known as the trigeminal nucleus caudalis (TNC) (44). The second-order TNC neurons extend into the upper cervical spinal cord segments (C1-C3), where they interface with dorsal horn neurons (45). Together, TNC neurons and upper cervical dorsal horn neurons constitute the trigeminocervical complex (TCC), a functional integrated hub in migraine pathophysiology (46).
From the TCC, second-order neurons decussate and ascend contralaterally as the trigeminothalamic tract, carrying nociceptive messages to higher-order processing centers. These ascending fibers primarily target the thalamus, which serves as the major relay for trigeminal sensory input (47,48). The third-order thalamic neurons project to multiple cortical regions involved in sensory-discriminative and affective-emotional dimensions of pain, including the somatosensory cortices, anterior cingulate cortex and insula (49).
The integrated activity of these pain-processing networks underlies not only the sensory perception of migraine headache, but also its emotional salience. Within this framework, substance P acts at multiple levels to modulate nociceptive messaging, possibly contributing to the initiation, amplification and persistence of migraine attacks.
Synthesis and transport of substance P
Substance P is synthesized by a plethora of cell types, including sensory neurons and immune cells (50). In the TVS, its production is located in the cell bodies of first-order sensory neurons within the TG (51). The synthesis beings with transcription of the preprotachykinin A gene, which encodes the precursor protein preprotachykinin A (52). This precursor undergoes post-translational processing to generate substance P and neurokinin A (53).
Once synthesized, substance P is packaged into large dense-core vesicles, which are then transported via axonal transport mechanisms to both peripheral and central nerve terminals (54). This bidirectional enables the rapid and localized release of substance P in response to neuronal activation. The synthesis, localization and role of substance P in TVS are explained in Figure 1.

Role of substance P in the trigeminovascular system. Transcription of the TAC1 gene in pseudounipolar sensory neurons of the trigeminal ganglion leads to the production of preprotachykinin A, which is post-translationally processed into substance P. Once synthesized, substance P is stored in dense-core vesicles and transported to both peripheral and central nerve terminals. Upon release from peripheral terminals, it induces vasodilation, plasma protein extravasation and immune cell recruitment. These responses are considered to activate perivascular afferents of first-order trigeminal nociceptive neurons, which then relay signals to second-order sensory neurons in the brainstem.
Release mechanisms in the trigeminovascular system
The release of substance P within the TVS can be initiated by a range of noxious stimuli, including mechanical stress, chemical irritants, thermal inputs and inflammatory mediators (55,56). These triggers depolarize sensory neurons, leading to the opening of voltage-gated calcium channels and a subsequent influx of calcium ions (57–59). Elevated intracellular calcium concentrations serve as a key signal for exocytosis, prompting the fusion of substance P-containing dense-core vesicles with the neuronal membrane and the release of substance P into the extracellular space (60).
Importantly, substance P is not released in isolation. It acts in concert with CGRP (61), with which it is co-localized in the presynaptic terminals of primary afferent neurons (62,63). Immunohistochemical studies have indeed demonstrated this co-expression (64), suggesting a synergistic relationship wherein both substance P and CGRP contribute to migraine mechanisms.
The release of substance P is regulated by multiple modulating factors. These include certain ion channels, receptor-mediated signaling pathways and interactions with other neurotransmitters that fine-tune both the timing and extent of substance P release (55,65).
Binding to neurokinin-1 receptors
Substance P binds to the G-protein coupled receptors of the neurokinin receptor family, including neurokinin 1 (NK1), neurokinin 2 (NK2) and neurokinin 3 (NK3) receptors (66). Among these, substance P exhibits the highest affinity for the NK1 receptor (NK1R) (52), which constitutes its primary functional target. NK1R is widely expressed across both the central and peripheral nervous systems, as well as in various non-neuronal cells such as endothelial cells and vascular smooth muscle cells (VSMCs) (67,68). Within the TVS, NK1R expression has been found in meningeal arteries, the TG and TCC, underscoring its potential relevance to migraine mechanisms.
Upon binding, NK1R undergoes conformational activation and initiates intracellular signaling cascades, most notably through the activation of phospholipase C (69). This activation leads to the generation of second messengers, inositol triphosphate and diacylglycerol, which in turn promote intracellular calcium release and protein kinase C activation. These signaling events mediate a range of downstream effects, including vasodilation, PPE, neurogenic inflammation, and modulation of neuronal excitability (70).
Although substance P preferentially targets NK1R due to its highest binding affinity, it can also bind to NK2 and NK3 receptors with considerably lower affinity (71). NK2Rs and NK3Rs are primarily activated by neurokinin A and neurokinin B, respectively, and their contribution to migraine pathophysiology remains poorly understood (70). However, it warrants mention that these receptors are also expressed in TG neurons and might therefore play a role in pain and inflammation (67).
Emerging evidence has refined the traditional view of NK1R as a plasma membrane-restricted receptor (72). Following ligand binding and activation by substance P, NK1R can undergo agonist-induced internalization into endosomal compartments (73). This endosomal signaling might help sustain or alter intracellular pathways under certain conditions, potentially extending the effects of substance P beyond the initial cell surface receptor interaction. In migraine, such prolonged signaling could theoretically play a role in maintaining pain during attacks.
Regulation of the activity of substance P
The activity of substance P is regulated by several factors (74,75). One primary mechanism of regulation involves enzymatic degradation by widely expressed cell surface peptidases, including neutral endopeptidase and angiotensin-converting enzyme (74). These enzymes cleave substance P into non-functional fragments (76), thereby limiting the duration of the action of substance P.
Beyond enzymatic degradation, receptor-level desensitization also plays a critical role. Prolonged exposure to substance P activates G protein-coupled receptor kinases, which in turn facilitate the recruitment of β-arrestins to the NK1R (75). This process involves receptor internalization, effectively removing NK1R from the cell surface and attenuating further responsiveness to extracellular substance P. Importantly, internalized receptors can continue to signal from endosomal compartments or be targeted for degradation via β-arrestin-mediated pathways, contributing to both sustained intracellular signaling and signal termination. Moreover, genetic and epigenetic factors can influence the expression levels of substance P and its NK1R (77), further shaping individual variability.
Peripheral actions of substance P
Cephalic vasodilation
One of the principal peripheral actions of substance P is the induction of dilation in cerebral and dural blood vessels (78). This process begins with NK1R activation on endothelial cells, which stimulate endothelial nitric oxide synthase and leads to the generation of nitric oxide (NO), a potent vasodilator (79–82). NO then diffuses into adjacent VSMCs, where it activates soluble guanylate cyclase (83). This enzyme catalyzes the conversion of guanosine triphosphate into cyclic guanosine monophosphate, a second messenger that activates protein kinase G (PKG) (Figure 2(a)) (84).
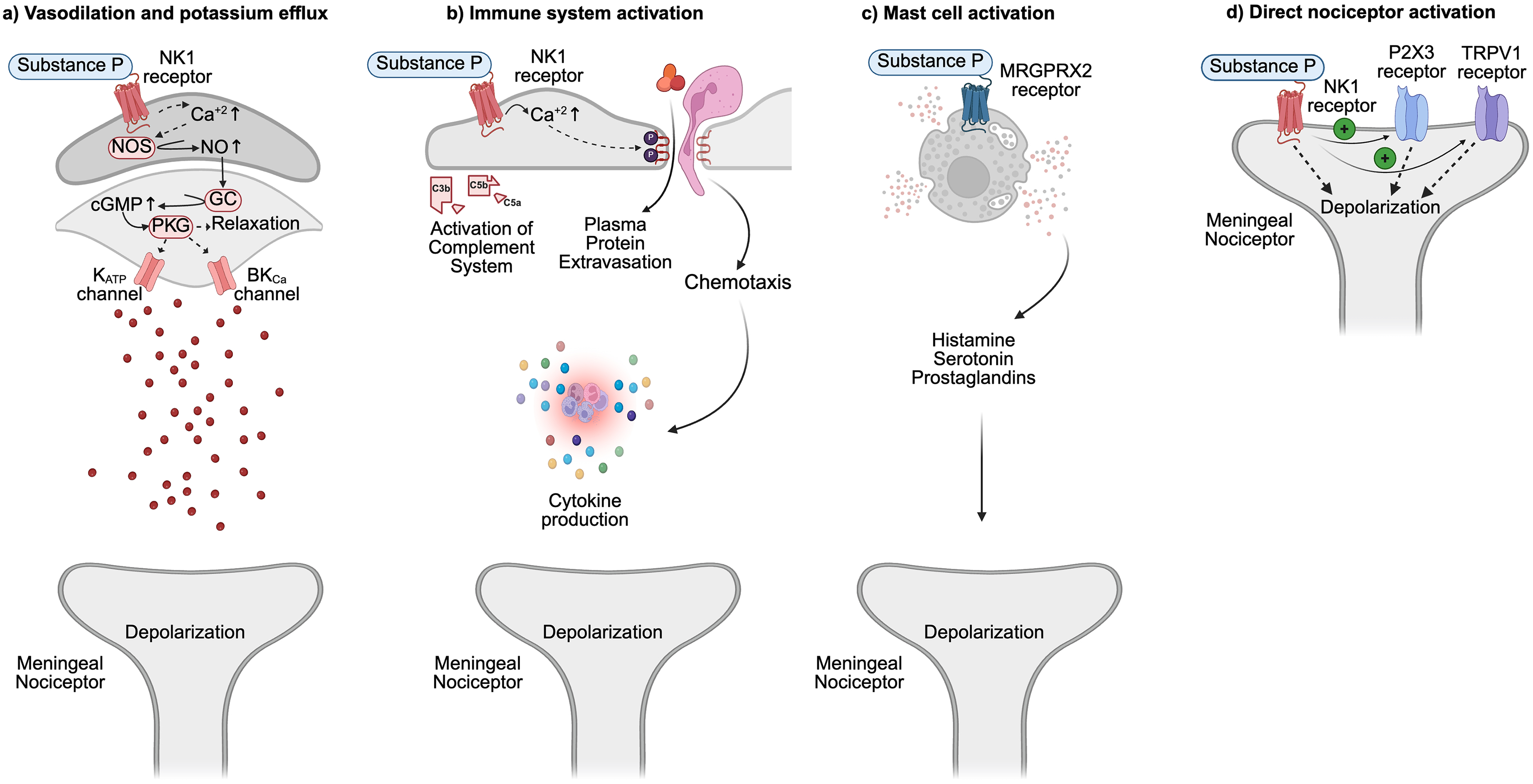
Hypothesized mechanisms of substance P action on meningeal nociceptors. (a) Substance P promotes cephalic vasodilation by activating neurokinin 1 (NK1) receptors on endothelial cells, stimulating nitric oxide (NO) production via endothelial nitric oxide synthase (NOS). NO diffuses to vascular smooth muscle cells, where it activates soluble guanylate cyclase (GC), elevating cyclic GMP (cGMP) levels and subsequently activating protein kinase G (PKG). This cascade triggers potassium efflux through ATPsensitive potassium (KATP) channels and large conductance calcium-activated potassium (BKCa) channels, leading to membrane hyperpolarization, reduced calcium influx, and smooth muscle relaxation. (b) Substance P increases vascular permeability by disrupting endothelial tight junctions, resulting in plasma protein extravasation (PPE), perivascular edema and neurogenic inflammation. It also enhances chemotaxis and stimulates the release of pro-inflammatory cytokines from macrophages, neutrophils and lymphocytes, contributing to immune activation and sensitization of meningeal nociceptors. (c) Activation of mast cells via Mas-related G-protein coupled receptor member X2 (MRGPRX2) receptors by substance P leads to the release of histamine, serotonin, and prostaglandins. These mediators promote vasodilation, PPE, and immune-driven sensitization, processes implicated in migraine pathophysiology. (d) Substance P could also directly modulate neuronal excitability by facilitating ion flux through P2X3 and transient receptor potential vanilloid 1 (TRPV1) channels in trigeminal neurons, potentially enhancing nociceptor responsiveness. Although these effects are mainly observed at supraphysiological concentrations, they suggest a possible direct mechanism of nociceptor activation.
PKG exerts its vasorelaxant effects by phosphorylating multiple downstream targets, including ATP-sensitive potassium (KATP) channels and large-conductance calcium-activated potassium (BKCa) channels (85). Phosphorylation of these channels facilitate their opening, allowing potassium ions to exit the cell, which hyperpolarizes the VSMC membrane and reduces the probability of voltage-gated calcium channel opening (86). As a result, intracellular calcium levels decrease, leading to diminished actin-myosin interactions and VSMC relaxation, thereby promoting vasodilation (87).
In the context of migraine, arterial dilation is hypothesized to activate perivascular meningeal nociceptors through both mechanical and chemical stimuli (4,88). The expansion of vessel diameter might stretch mechanosensitive nociceptors, potentially involving Piezo channels (89), whereas local increases in extracellular potassium might further depolarize neurons, lowering their activation threshold. Together, these processes might initiate or exacerbate migraine headache. Notably, NO donors, as well as pharmacological openers of KATP channels and BKCa channels, have been shown to induce migraine attacks in people with migraine but not in healthy adults (90,91). These human experimental findings underscore the pathophysiological relevance of this signaling cascade.
Increase in vascular permeability and plasma protein extravasation
In addition to promoting vasodilation, substance P increases vascular permeability by inducing cytoskeletal contraction and disrupting endothelial tight junctions (Figure 2(b)) (92). This leads to the formation of intercellular gaps, allowing plasma proteins and pro-inflammatory mediators to extravasate into the perivascular space (93). The resultant tissue edema and activation of complementary pathways contribute to neurogenic inflammation within the meninges (94). This inflammatory environment sensitizes nearby meningeal nociceptors (95), lowering their activation threshold and perpetuating cephalic pain. In this way, PPE serves as an important interface between vascular responses and sustained nociceptive messaging characteristics of migraine.
Activation of mast cells and immune responses
Substance P also plays a central role in immune modulation by directly activating mast cells via Mas-related G-protein coupled receptor member X2 (MRGPRX2) expressed on their surface (Figure 2(c)) (96). This activation results in the release of various pro-inflammatory mediators, including histamine, tryptase, prostaglandins, cytokines and chemokine (97). Among these, histamine is particularly relevant, as it promotes vasodilation and PPE (98), augmenting the actions initiated by substance P (99). In addition, histamine and certain prostaglandins are well-established molecular migraine triggers in humans with migraine (100,101), underscoring the likelihood of mast cell involvement in migraine pathogenesis.
Modulation of other immune cells
Beyond mast cells, substance P modulates the activity of various other immune cell types, including macrophages, neutrophils and lymphocytes (Figure 2(b)) (102). It promotes chemotaxis, enhancing the production of pro-inflammatory cytokines, and modulates immune cell function (103,104). This widespread impact on the immune system is thought to contribute to the maintenance of inflammation and sensitization within the TVS, perpetuating migraine attacks (47).
Direct activation of meningeal nociceptors
In addition to its well-established vascular and pro-inflammatory effects on non-neuronal cells, substance P might also exert direct excitatory actions on meningeal nociceptors (Figure 2(d)). Experimental work by Spiegelman et al. (105) demonstrated that application of substance P onto TG slices in vitro elicited neuronal depolarization. In support, preclinical data have shown that substance P, via NK1R-dependent mechanisms, enhances calcium influx through P2X3 receptors in trigeminal neurons (106). However, an important caveat is the concentration of substance P used in these mechanistic studies. The in vitro studies were conducted using micromolar doses, approximately one million times higher than the picomolar concentrations used in human provocation experiments that demonstrated headache induction (35). This disparity raises questions about the physiological relevance of observed effects, such as neuronal depolarization and P2X3 activation (106), under experimental conditions. These supraphysiological doses might elicit non-specific cellular responses that do not accurately reflect endogenous signaling in human migraine. Consequently, although mechanistically informative, such findings must be interpreted with caution when extrapolating to clinical settings.
Emerging evidence indicates that substance P modulates nociceptive signaling through functional interactions with transient receptor potential (TRP) channels, which are key regulators of neurogenic inflammation and pain transduction (107). Substance P appears to sensitize TRPV1 and TRPA1 channels via NK1R-dependent mechanisms (108,109), enhancing their responsiveness to noxious stimuli and promoting peripheral sensitization. TRPV1 channels, non-selective cation channels predominantly expressed in nociceptive neurons (108), are co-localized with substance P and NK1 receptors in trigeminal sensory ganglia (110,111). Furthermore, TRPM8, a cold-sensitive channel also co-expressed with substance P in subsets of trigeminal neurons (112), has been linked to NO donor- and CGRP-induced migraine-like behaviors in rodents (113). These findings support a model in which TRP channels serve as downstream effectors of substance P signaling, amplifying trigeminal excitability and sustaining migraine-associated pain.
Taken together, the direct excitatory effects of substance P on nociceptive neurons might act synergistically with its indirect pro-nociceptive actions mediated vascular and immune mechanisms. This combined influence could activate meningeal nociceptors and trigger nociceptive messaging in ascending trigeminal pain pathways.
Interaction with other signaling molecules
Substance P does not exert its effects in isolation; rather, it operates within a broader network of neuropeptides and signaling molecules that collectively appear to shape migraine pathophysiology (29). Among these, CGRP is of particular importance due to its well-established role in the neurobiological basis of migraine. Substance P and CGRP are co-released from trigeminal sensory neurons (62,63), and both peptides contribute to vasodilation and neurogenic inflammation (61). However, these actions are mediated via distinct receptor systems and intracellular signaling pathways, with substance P acting primarily via NK1R and CGRP through its own G protein-coupled receptors (70,114). The combined effects of substance P and CGRP might therefore amplify vascular and nociceptive responses, particularly in the meninges. Thus, targeting a single mediator might be insufficient to fully disrupt the cascade of events that drive migraine attacks. A better understanding of how these signaling pathways converge is essential for the development of more effective and possibly multi-targeted therapeutic agents.
Central actions of substance P
Facilitating excitatory neurotransmission
NK1Rs are widely expressed within the central pain matrix, including the trigeminal nucleus caudalis, thalamus, cortex and brainstem nuclei involved in descending pain modulation (115–117). Interestingly, preclinical data have demonstrated that iontophoretic application of substance P increases spontaneous firing in second-order nociceptive neurons (118). This effect has been observed in both feline and rodent models (118,119). In addition to enhancing baseline neuronal excitability, substance P amplifies response to noxious stimuli (120), suggesting a possible role in central sensitization.
Substance P can also modulate glutaminergic signaling by facilitating NMDA receptor function (121,122). It promotes phosphorylation of NMDA receptor subunits, reducing the magnesium block and increasing calcium influx (123). This process amplifies excitatory synaptic transmission and might contribute to changes in gene expression associated with chronic pain.
Moreover, substance P can attenuate inhibitory transmission by reducing GABA release from interneurons (124). The combined effect of increased excitation and reduced inhibition shifts the balance towards hyperexcitable neurons, promoting the transmission of nociceptive messages within the central nervous system (CNS).
Interaction with descending modulatory pain pathways
Substance P also modulates descending pain pathways, which originate in key brainstem regions such as the periaqueductal gray and the rostroventromedial medulla (123). Findings from preclinical experiments indicate that substance P can augment local excitatory inputs and enhance endocannabinoid signaling within these neural circuits, potentially increasing analgesic tone (125). By contrast, direct administration of substance P to the rostroventromedial medulla has been shown to elicit thermal antinociception in rats, suggesting a context-dependent role in pain modulation (126). These insights highlight the bidirectional effects of substance P within CNS structures involved in descending pain modulation, which might depend on both anatomical site and physiological state.
Preclinical neurokinin-1 receptor antagonist testing
Building on the mechanistic evidence described earlier, researchers developed NK1R antagonists as potential migraine treatments (Table 1) (127). Initial preclinical studies in rodents showed that compounds such as RP67580 and GR82334 suppressed PPE in a dose-dependent manner following TG stimulation (20,21). These results supported the involvement of NK1R in neurogenic inflammation.
Summary of preclinical studies evaluating NK1R antagonists in experimental migraine models.

Abbreviations: PO = per oral; IV = intravenous; TG = trigeminal ganglion; TCC = trigeminocervical complex; CGRP = calcitonin gene-related peptide; nNOS = neuronal nitric oxide synthase; NK1 = neurokinin 1.
Subsequent experiments evaluated GR205171, a CNS-penetrant NK1R antagonist with high affinity for the human receptor (127). In rabbits, GR205171 inhibited substance P-induced vasodilation of the carotid artery (26). In the same study, it reduced dural PPE and attenuated c-Fos expression in second-order trigeminal neurons, comprising an established marker of neuronal activation, in rodents. Despite these promising effects in neurovascular and inflammatory models, Akerman et al. (128) demonstrated that GR205171 did not attenuate NO donor-induced hypersensitivity or spontaneous firing in neurons of the trigeminocervical complex in rats. This finding aligns with rodent data from Pedersen et al. (129), who reported that the NK1R antagonist L-733,060 failed to prevent mast cell degranulation following NO-donor infusion. Together, these studies seem to underscore a critical mechanistic distinction: NK1R signaling precedes NO production and is therefore unlikely to influence pathophysiological processes once NO-mediated cascades have been initiated.
Additional animal studies demonstrated similar effects with other NK1R antagonists. For example, GR203040 reduced vasodilation induced by NK1R agonism and inhibited dural PPE after trigeminal stimulation (23). Likewise, CP-99,994 decreased both PPE and c-Fos mRNA expression in second-order trigeminal neurons (24).
These findings were extended to lanepitant, which binds with high affinity to both human and guinea pig NK1Rs (130,131). In guinea pig models, lanepitant inhibited PPE following TG activation (25). Dapitant, a highly selective antagonist for human NK1Rs, further supported these results (132). It suppressed c-Fos expression in second-order sensory neurons after capsaicin, a TRPV1 agonist, was administered into the cisterna magna (22).
Collectively, these consistent preclinical findings demonstrate that NK1R antagonists inhibit key markers of migraine-related processes, including PPE and c-Fos expression. This robust body of evidence provided a strong rationale for advancing NK1R antagonists into clinical trials as candidate migraine treatments.
Clinical trials of neurokinin-1 receptor antagonists in migraine
NK1R antagonists have been investigated across a range of pain disorders, including osteoarthritis, postoperative pain, fibromyalgia and neuropathic pain (133). Despite promising preclinical data, these agents failed to demonstrate clinical efficacy by the late 1990s. Controlled trials of NK1R antagonists in migraine similarly produced negative or inconclusive results (Table 2). Pharmaceutical agents such as dapitant, lanepitant, GR205171 and L-758,298 were evaluated in randomized, double-blind, placebo-controlled studies, yet none were superior to placebo (30–34).
Clinical trials evaluating NK1R antagonists for migraine: design, dosing and efficacy outcomes.

Abbreviations: PO = oral; IV = intravenous.
The study by Goldstein et al. (33) contains an inconsistency in reporting of efficacy outcomes. Although some sections refer specifically to “migraine headache days”, others describe the outcome more broadly as “headache days”. It is unclear whether the analyses were limited to migraine-specific headaches or included all headache types.
Dapitant trial
Dapitant (RPR100893) is a non-peptide NK1R antagonist developed as a potential treatment for acute migraine attacks (22). Diener et al. (34) conducted a randomized, double-blind, placebo-controlled, parallel-group trial to assess the efficacy and tolerability of dapitant in 139 adults with migraine. Participants were randomized to treat a single migraine attack of moderate-to-severe pain intensity with one of three oral dapitant doses (1, 5 or 20 mg) or placebo. Headache intensity was rated using a four-point scale, ranging from 0 (no pain) to 3 (severe pain). The primary efficacy endpoint was headache relief, defined as a reduction in pain intensity from moderate or severe (scores of 2 or 3) to mild or none (scores of 1 or no) at two hours post-dose. However, none of the dapitant doses were superior to placebo. The most common adverse events were asthenia (3–8%) and somnolence (3–10%).
Lanepitant trials
Lanepitant (LY303870) is a non-peptide, competitive NK1R antagonist with high affinity for both the human and guinea pig receptors (25). Based on promising preclinical data, Goldstein et al. (30,33) performed two randomized clinical trials to assess lanepitant's efficacy in both acute and preventive migraine treatment.
The first clinical trial applied a randomized, double-blind, placebo-controlled, crossover design (30). Fifty-three participants with migraine were enrolled and received three oral doses of lanepitant (30, 80 and 240 mg) and placebo. Only 40 participants treated their attacks with all four interventions (i.e. three active doses and the placebo). The primary efficacy outcome was the responder rate, defined as the proportion of participants achieving headache relief at 120 minutes post-dose. However, no lanepitant dose showed superiority over placebo. Adverse events were similar across all interventions.
In a separate 12-week, double-blind, placebo-controlled, parallel-group trial, lanepitant was evaluated for migraine prevention (33). Eighty-four participants were randomized to receive either lanepitant 200 mg daily (n = 42) or placebo (n = 42). The primary efficacy endpoint was the proportion of participants achieving at least a 50% reduction in monthly migraine days from baseline through week 12. Although 41% of participants in the lanepitant group achieved at least a 50% reduction in monthly migraine days, compared to 22% in the placebo group, the difference was not statistically significant. Notably, by the third month, the lanepitant group exhibited a statistically significant reduction in monthly migraine days compared to placebo (40.6% vs. 17.4%).
GR205171 trial
GR205171 is a NK1R antagonist with high affinity for the human receptor. Connor et al. (31) conducted a randomized, double-blind, placebo-controlled trial to evaluate its efficacy in the acute treatment of migraine attacks. Sixty-three participants with migraine received an intravenous dose of GR205171 (25 mg; n = 31) or placebo (n = 32) to treat a single migraine attack. The primary endpoint was headache relief at two hours post-infusion, measured on a four-point scale ranging from 0 (no pain) to 3 (severe pain). GR205171 failed to provide superior headache relief, compared to placebo. The trial did not report any data on treatment-emergent adverse events.
L-758,298 trial
L-758,298 is a CNS-penetrant, selective NK1R antagonist (134). Its potential as an acute migraine treatment was evaluated in a double-blind, placebo-controlled, in-clinic trial involving 72 patients with migraine (32). Patients were randomly assigned to receive an intravenous dose of L-758,298 at 20 mg (n = 4), 40 mg (n = 4) or 60 mg (n = 39), or placebo (n = 25). The primary efficacy endpoint was headache relief at two hours post-dose. The 60-mg dose of L-758,298 showed no statistically significant difference from placebo in achieving this outcome (33% vs. 52%). The reported adverse events were generally mild and transient.
Analysis of clinical trial outcomes
Multiple design-related limitations likely contributed to the negative outcomes observed in clinical trials evaluating NK1R antagonists for migraine. In studies assessing acute treatment, the primary endpoint was typically headache relief at two hours post-dose (30–32,34), a measure now considered suboptimal relative to the more stringent standard of headache freedom recommended in current international guidelines (135). In addition, trial protocols frequently omitted key methodological details. Sample size calculations were either absent or inadequately justified, with insufficient documentation of expected treatment and placebo response rates. Many studies failed to specify whether missing data were imputed, if an intention-to-treat analysis was implemented, or whether statistical adjustments were applied for multiple comparisons, all of which could have substantially influenced the validity of the reported findings.
The only preventive trial, which assessed lanepitant (33), enrolled participants exclusively with episodic migraine, thereby limiting generalizability to individuals with chronic migraine. Although a statistically significant reduction in monthly migraine frequency was observed at three months, 40.6% with lanepitant compared to 17.4% with placebo, this result did not correspond to the trial's predefined primary endpoint, which was an average ≥50% reduction from baseline to months 1–3. The failure to meet this benchmark likely reflects limited statistical power due to a small sample size, which could have biased the efficacy estimates. Secondary outcomes and responder analyses were also inadequately powered and inconsistently reported, contributing further to interpretive uncertainty.
Taken together, these methodological shortcomings constrain the interpretability of trial outcomes and raise the possibility that observed inefficacy might reflect flawed study design rather than intrinsic pharmacologic failure. A rigorous re-evaluation of NK1R antagonists, using contemporary endpoints, transparent statistical frameworks and a more representative participant population, could therefore be justified.
Translational challenges
The failed NK1R antagonist trials in migraine might suggest limited translational value of commonly used preclinical markers. For example, most animal studies relied solely on dural PPE after TG stimulation as a surrogate for migraine headache (20,21,23,25). Although widely used, dural PPE is only an indirect marker of trigeminovascular activation and does not fully capture complex nature of headache or migraine pain experienced in humans. Inhibiting PPE might not reliably predict clinical efficacy, as evidenced by the discrepancy between preclinical and clinical trial outcomes (136–139). As an alternative, direct electrophysiological recordings of meningeal nociceptor activity could offer a more precise and translatable metric of trigeminovascular activation.
Likewise, some preclinical studies used c-Fos expression to indicate neuronal activation within the ascending trigeminal pain pathways (22,24). Although c-Fos levels rise with intense neuronal firing (140), their suppression does not consistently correlate with therapeutic benefit in animal models of migraine (141). Moreover, c-Fos expression is neither specific to meningeal afferents, nor to nociceptive neurons.
Moreover, most preclinical migraine studies have relied exclusively on male animals, despite the higher prevalence of migraine in females and evidence for sex-specific differences in nociceptive signaling (142). Incorporating both sexes in experimental designs will improve the generalizability of preclinical findings and better reflect the clinical population. Together, these issues underscore the need for more translatable preclinical approaches, such as electrophysiological recordings of meningeal nociceptor activity, that might better align with human migraine biology.
Future directions
Conclusions about the failure of NK1R antagonists in migraine appear somewhat premature. Most trials used small samples and outdated efficacy endpoints. Modest but inconsistent therapeutic benefits appeared in the lanepitant trial for migraine prevention (33), indicating a potential signal. New controlled trials should therefore consider using up-to-date efficacy metrics: two-hour pain freedom post-dose for acute treatments and ≥50% reduction in monthly migraine days for migraine prevention.
Novel evidence has also renewed interest in substance P signaling and its relationship to headache disorders. Our lab has recently demonstrated that intravenous infusion of substance P induces headache in 15 (71%) of 21 healthy adults compared to two (10%) after placebo. In addition, we found that substance P dilated the superficial temporal artery. Although intriguing, these experimental data warrant confirmation in placebo-controlled studies that enroll people with migraine (35,143).
Preclinical models must likewise evolve. Dural PPE and trigeminal c-Fos expression are indirect measures of cephalic pain and should only be considered complementary. Instead, researchers should record meningeal nociceptor firing, which directly maps trigeminovascular activation and offers a clearer mechanistic bridge to cephalic pain. Rigorous, mechanism-based endpoints at both bench and bedside will give NK1R antagonists a fairer test and might reveal a niche for this drug class in migraine treatment.
Conclusions
Accumulating preclinical and emerging human experimental evidence converges on substance P and NK1R signaling as a pathogenic contributor to trigeminovascular activation in migraine. Negative results from NK1R antagonist trials could reflect methodological shortcomings, including underpowered samples and outdated endpoints, rather than true pharmacological failure. Carefully designed studies are needed to determine whether targeting substance P or its NK1R offers therapeutic promise for migraine.
Article highlights
Substance P has recently been shown to trigger headache and arterial dilation in humans, reinforcing its role in migraine and its potential as a therapeutic target.
Despite supportive preclinical data, NK1R antagonists have failed in clinical trials—likely due to outdated designs, small sample sizes, and suboptimal endpoints.
Future research should adopt modern clinical efficacy endpoints and more translatable preclinical models, such as direct recordings from meningeal nociceptors.
Footnotes
Acknowledgments
Figures were created using BioRender.com.
Author contributions
HA and MA conceptualized the manuscript. BA conducted the article screening and drafted the main text. All authors reviewed and revised the manuscript for intellectual content.
Data availability
Upon reasonable request, the corresponding author will provide the necessary materials to interested researchers for the purpose of academic scrutiny, reproducibility and further scientific investigation.
Declaration of conflicting interests
RHC has received personal fees from Pfizer and Lundbeck, outside of the submitted work. HMA has received personal fees from Lundbeck and Pfizer, outside of the submitted work. MA has received personal fees from AbbVie, Astra Zeneca, Eli Lilly, GlaxoSmithKline, Lundbeck, Novartis, Pfizer and Teva, outside of the submitted work. MA also serves as an Associate Editor of Brain and The Journal of Headache and Pain. HA has received personal fees from AbbVie, Lundbeck, Pfizer and Teva, outside of the submitted work. HA also serves as an Editorial Board Member of The Journal of Headache and Pain. The remaining author (BA) reports no competing interest.
Funding
This manuscript received funding from the Lundbeck Foundation (R403-2022-1352 to HA). Lundbeck Foundation (grant number R403-2022-1352).