
Abstract
Background
Familial hemiplegic migraine (FHM) types 1–3 are associated with protein-altering genetic variants in CACNA1A, ATP1A2 and SCN1A, respectively. These genes have also been linked to epilepsy. Previous studies primarily focused on phenotypes, examining genetic variants in individuals with characteristic FHM symptoms. This study aimed to investigate the association of FHM genetic variation with migraine and epilepsy, utilizing a genotype-first approach.
Methods
Whole-exome sequence data from 454,706 individuals from the UK Biobank were examined for self-reported and inpatient-diagnosed migraine and epilepsy. Carriers were compared with non-carriers in a burden analysis using logistic regression while accounting for age, biological sex and UK Biobank assessment center. A machine learning-based approach was employed to predict whether variants resulted in gain-of-function (GoF), loss-of-function (LoF) or neutral effects.
Results
Heterozygous carriers of GoF CACNA1A variants, LoF ATP1A2 variants or neutral SCN1A variants were at increased risk of migraine. Homozygous carriers of neutral SCN1A variants were also associated with migraine but these carriers showed a reduced disease risk of epilepsy.
Conclusions
Heterozygous genotypes in all three FHM genes were associated with migraine but not epilepsy in this genotype-focused study. Homozygous SCN1A genotypes also showed increased disease risk of migraine, yet these carriers were protected against epilepsy.
This is a visual representation of the abstract.
Introduction
Familial hemiplegic migraine (FHM) represents a type of migraine with aura that follows an autosomal dominant inheritance pattern (1,2). Migraine attacks in FHM are characterized by distinctive aura symptoms, including unilateral muscle weakness followed by severe headache (1). Not all carriers of FHM-associated genetic variants exhibit the characteristic hemiplegic phenotype; instead, some individuals have more common aura symptoms, such as visual disturbances (3). This suggests a potential contribution of FHM-associated variants to more common types of migraine with aura (4). Probands with FHM and their first-degree relatives exhibit an increased risk of other types of migraine with aura, aside from hemiplegic aura, while showing no increased risk of migraine without aura (4). However, no conclusive evidence has yet established the role of FHM-associated genes in common forms of migraine (5). Despite its presumed rarity, with a prevalence of 0.01% (2,3), FHM has been a subject to extensive research because of its familial occurrence, offering unique insights into the genetics of migraine. This has been achieved through a large number of case reports and familial characterizations, which have resulted in the discovery of a triad of genes linked to the FHM phenotype: CACNA1A, ATP1A2 and SCN1A associated with FHM1, FHM2 and FHM3, respectively (1,2). These genetic associations have been instrumental in elucidating the hereditary basis of this complex neurological disorder. They have also enabled the investigation of pathophysiology of migraine with aura in preclinical studies (6–14).
FHM-associated genetic variants may have associations beyond migraine alone. Hence, several case reports have documented clusters of epilepsy cases within families affected by FHM2 (15–18). Similar co-morbidity with epilepsy has been reported in FHM1 (19–21) and FHM3 families (22,23). A recent systematic review based on data from 32 case series on FHM families suggests that 40% of family members affected by FHM also experienced seizures, with an equal division between those exhibiting focal and generalized epilepsy (24). The association between FHM-linked genetic variants and epilepsy finds further support in a study on a cohort of 318 epilepsy patients. The study found distinct CACNA1A variants in ten unrelated patients, with eight of the variants classified as pathogenic/likely pathogenic (25). Notably, none of these ten epilepsy patients experienced migraine, reinforcing the idea that CACNA1A mutations may contribute to an epilepsy phenotype in addition to their well-established role in FHM1. Heterozygous variants in the CACNA1A gene have also been linked to other severe neurological disorders including epileptic encephalopathies (26) and spinocerebellar ataxia-6 often associated with CAG repeat exon expansion (27). CACNA1A variants have also been reported in families with episodic ataxia type 2 (28). Notably, more than half of these patients also exhibit migraine, some with episodic hemiplegia. As for the other two FHM-associated genes, these have also been associated with neurological diseases other than migraine. Thus, a case series has characterized polymicrogyria syndrome linked to homozygous ATP1A2 variants (29) and developmental and epileptic encephalopathy with infantile onset, Dravet syndrome, is associated with SCN1A loss-of-function (LoF) variants (30).
Investigations thus far have predominantly centered on characterizing the genetic profile of individuals and families exhibiting the distinctive FHM phenotype or epilepsy. By contrast, the present study adopted a genotype-first approach to examine carriers of variants in the three FHM-associated genes. This approach revealed new insights into the genetic role of these genes and their correlation with the disease. It was hypothesized that missense and protein-truncating variants within the three FHM-associated genes were associated with migraine and epilepsy. The hypothesis was tested by examining the population-based UK Biobank (UKB) (31). The UKB has enrolled approximately half a million individuals who have been extensively characterized with genetic and electronic health records being available. As exome sequence data has become available for the UKB (32), this constitutes a unique resource to investigate the potential association between sequence variants within the three FHM-associated genes and self-reported and hospital diagnoses for migraine and epilepsy.
Methods
In this study, a genotype-first approach was employed, where carriers of missense and protein-truncating variants in CACNA1A, ATP1A2 and SCN1A were first identified, followed by an analysis of their association with migraine and epilepsy. This approach contrasts with the traditional phenotype-first method, in which individuals are selected based on clinical characteristics and subsequently their genetic profile is analyzed. Whole exome sequence data from the Original Quality Functionally Equivalent (OQFE) pipeline was used. Between 2006 and 2010, approximately 9.2 million people aged 40–69 years, who lived within a reasonable travel distance (up to 25 miles) of one of 22 assessment centres across the UK, were invited to participate in the UK Biobank. In total, 502,000 adults (5.5% of those invited) were recruited (33). Participants completed a wide range of baseline assessments, including touchscreen questionnaires covering sociodemographic factors, family history, lifestyle, medical history, cognitive function tests and environmental exposures. Also, different biofluids were collected including blood samples, which subsequently has been used for chip genotyping, whole exome and genome sequencing data (31,32,34). Because participation in the UK Biobank cohort was voluntary, it does not fully represent the current UK population in several ways (35). However, the significance of this depends on the specific research question being addressed. To ensure that findings can be generalized to a broader or future population, it may be more important to have a sufficiently large number of participants with varying levels of exposure and disease incidence (36).
A single variant-level filter was applied, requiring that at least 90% of all genotypes for a given variant had a read depth of at least 10, meaning each genetic position was read at least 10 times, aiming to identify variants within the FHM-associated genes (37). The predicted consequence of the identified sequence variants within the three FHM-genes was obtained using Variant Effect Predictor (VEP) (38). Only the canonical transcript of each of the three FHM-associated genes (CACNA1A [NM_001127222.2]; ATP1A2 [NM_000702.4]; SNC1A [NM_001165963.4]) was considered, meaning that any FHM-variants annotated to any of the other transcripts were not considered. Sequence variants and carrier status was obtained from the whole exome sequencing data by utilizing the R-package qgg (39,40).
The study focused on protein-altering variants (i.e. missense variants resulting in amino acid changes and protein-truncating variants, including variants resulting in premature stop (nonsense), canonical splice sites (splice-donor or splice-acceptor) or insertion/deletion variants that shifted frame (frameshift) (41). To test for association between disease outcomes and FHM sequence variants, a burden association test was applied. For each FHM gene, three different genetic scenarios were defined: (i) All missense and protein-truncating variants subdivided according to their predicted gain-of-function (GoF), LoF or neutral effects based on LoGoFunc, a machine learning approach developed to explicitly predict pathogenicity (42); (ii) missense or LoF variants that have been associated with FHM in ClinVar (43); and (iii) FHM-associated variants that were reported in ClinVar as pathogenic, likely pathogenic or pathogenic/likely pathogenic using the American College of Medical Genetics and Genomics (ACMG) classification. In Scenarios ii and iii, an assessment was made to determine whether ClinVar-reported FHM variants, regardless of pathogenicity, were associated with migraine and epilepsy, and whether the association was amplified when restricting the analysis to rare pathogenic/likely pathogenic variants (see supplemental material, Tables S1 and S2). For each of the three different genetic scenarios, we defined additional two groups: individuals having at least one heterozygous or at least one homozygous alternative sequence variants. Individuals that fulfilled both zygosity-criteria for multiple genetic variants were included in both comparative groups. Medical diagnoses were obtained from the hospital inpatient data (World Health Organization's international classification of diseases (ICD-10), UK Biobank data field: 41270). To maximize statistical power, the analyses of diagnostic subtypes were performed on all ICD-10 subcategories containing more ≥500 cases (corresponding to a cohort prevalence of 0.001; see supplemental material, Table S3). This threshold allows us to maintain statistical power without including categories where sample sizes are too small to yield meaningful insights. The following diagnoses were thus included in the subsequent genetic burden analysis: Migraine with aura (ICD-10: G43.1), migraine unspecified (G43.9), generalized idiopathic epilepsy and epileptic syndromes (G40.3) and epilepsy unspecified (G40.9). In addition to hospital diagnosis codes, self-reported cases of migraine and epilepsy (UK Biobank data field: 20002) were also included.
The burden analysis was performed by assigning each UKB participant with an indicator variable (0|1) of whether the individual was either homozygous or heterozygous for any of the identified FHM genetic variants in one of the three scenarios. For each FHM gene and scenario, a logistic regression was fitted with carrier status, defined for each gene with one of the three scenarios, as predictor and with the response variable being diagnosed or self-reported migraine or epilepsy while accounting for age at inclusion, biological sex and UKB assessment center. Note, in the analyses of being either heterozygote or homozygote carriers, individuals from the other contrast group were included among the non-carriers. The degree of association of the carrier status was determined using a likelihood ratio test,
Results
Among all UKB participants, 454,706 individuals had whole exome sequence data available. There were 38,466 more female participants than male participants with sequence data available and, on average, the female participants were slightly younger than male participants (Table 1). There were almost three times more female participants with self-reported migraine compared with male participants (Table 1). Consistently, we observed more inpatient female participants with migraine with aura or unspecified migraine. Slightly more females had reported epilepsy, whereas more males had an ICD-10 inpatient record for unspecified epilepsy (Table 1). In total, 6,065 individuals had at least one epilepsy ICD-10 code and, among them, 2,691 had reported they had epilepsy in the questionnaire. A much larger proportion of UKB participants had self-reported migraine compared with having an inpatient ICD-10 code of any migraine diagnosis (16,621 vs. 6,492, respectively). Approximately one-third of the UKB participants with a migraine diagnosis also reported migraine (i.e. 2,059 UKB participants with both diagnosed and self-reported migraine).
Descriptive statistics of the study population. Age is represented as the mean ± SD, and other numbers are represented as counts. Age comparison was tested with the Wilcoxon rank sum test, whereas the count comparisons were performed using chi-squared test. The subdiagnoses for epilepsy (G.40) and migraine (G.43) were included in further analysis based on a threshold prevalence of more than 500 cases per 500,000 individuals in the UK Biobank cohort. For details, see supplemental material, Table S3.

CACNA1A
Out of 4,128 identified sequence variants in CACNA1A, 1,423 missense variants and 125 protein-truncating variants were identified (see supplemental material, Table S4A). The heterozygous carriers accounted for 85% of the cohort and 18% were homozygous carriers of a CACNA1A missense or protein-truncating variant (Table 2). These heterozygous and homozygous carriers did not show an increased risk of migraine (Figure 1A; see aso supplemental material, Dataset S1). Variants were subdivided based on predicted consequence for protein function where an equal number of CACNA1A variants were classified as GoF and LoF, whereas most variants were classified as neutral (Table 2; see also supplemental material, Dataset S2). Carrier frequency differed between the groups, with 0.2% of the UKB cohort being heterozygous carriers of GoF CACNA1A variants, in contrast to the carrier frequencies of heterozygous LoF and neutral variants at 43.3 and 26.5%, respectively (Table 2). The mean minor allele frequency was also lower in GoF variants compared with neutral and LoF CACNA1A variants (see supplemental material, Table S5). There was an increased risk of self-reported migraine among heterozygous GoF carriers (Figure 1B), whereas no associations with migraine were observed among heterozygous carriers of LoF and neutral CACNA1A variants (Figure 1C,D). No carriers of homozygous GoF variants were detected, but homozygous carriers of LoF and neutral variants were observed in 3.2 and 2.1% of the UKB cohort, respectively. However, the association of these homozygous carrier groups with migraine with aura did not reach statistical significance after correcting for multiple testing (see supplemental material, Dataset S1). Twenty-two CACNA1A variants reported in ClinVar to be associated with FHM were detected (Table 2), but these were not associated with either migraine or epilepsy (see supplemental material, Dataset S1). Five of the FHM-associated variants reported in ClinVar were classified as pathogenic/likely pathogenic, and carriers of these variants showed an association with increased risk of diagnosed migraine with aura and diagnosed unspecified migraine (see supplemental material, Figure S1A). Given the limited number of carriers and cases, interpreting the results from these few carriers of pathogenic variants should, however, be approached cautiously. Some variants in ClinVar were also reported to be associated with diseases other than FHM (see supplemental material, Table S2). No association between CACNA1A variants and either self-reported or diagnosed epilepsy was detected independently of heterozygous/homozygous carrier status and pathogenicy and predicted functional impact of the variants (Figure 2; see also supplemental material, Figure S1B).
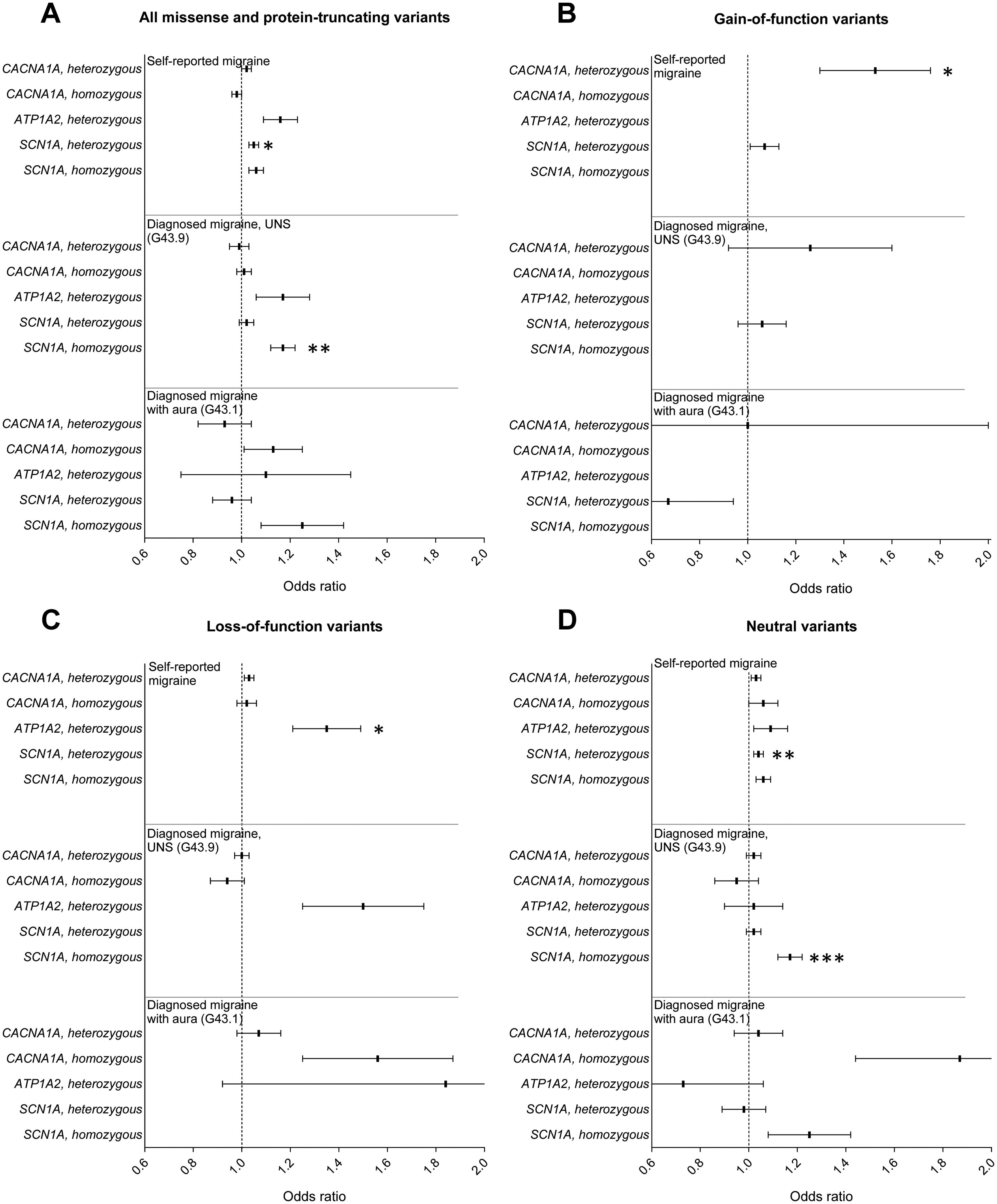
Association between migraine and variants within familial hemiplegic migraine (FHM)-associated genes; CACNA1A, ATP1A2 and SCN1A. Burden analysis of all detected missense and protein-truncating variants (A), predicted gain-of-function variants (B), predicted loss-of-function variants (C) and variants with predicted neutral effects on protein function (D). Outcomes were self-reported migraine (upper), diagnosed migraine unspecified (UNS; ICD-10 diagnosis G43.9; middle) and diagnosed migraine with aura (ICD-10 diagnosis code G43.1; lower). For detailed statistics, see supplemental material, Dataset S1. Error bars, standard error. *p < 0.05, **p < 0.01 and *p < 0.001, burden analysis with Benjamini-Hochberg adjustment for multiple testing.

Homozygous carriers of SCN1A variants showed a reduced disease risk of epilepsy. Examination of the burden of epilepsy linked with familial hemiplegic migraine (FHM)-associated CACNA1A, ATP1A2 and SCN1A gene variants for all identified missense and protein-truncating variants (A), predicted gain-of-function (B), predicted loss-of-function (C) and predicted neutral variants (D). For detailed statistical analysis including number of carriers and cases, see supplemental material, Dataset S1. Error bars, standard error. *p < 0.01 in burden analysis after Benjamini-Hochberg adjustment for multiple testing.
Number of heterozygous and homozygous carriers of missense and protein-truncating variants (PTVs) in the three familial hemiplegic migraine-associated genes. Functional impact of variants was predicted to be gain-of-function (GoF), loss-of-function (LoF), or neutral based on a machine learning-based method (30). Variants categorized as pathogenic/likely pathogenic were based on ClinVar classifications. The number of carriers is represented as a percentage of the UK Biobank cohort within parentheses. In groups with five or less carriers, the number of carriers is not disclosed due to General Data Protection Regulation.

ATP1A2
In the FHM2-associated ATP1A2 gene, 2,221 sequence variants were identified with 424 being missense variants and 50 protein-truncating variants (see supplemental material, Table S4B). In total, 8,197 individuals (1.8% of the cohort) were identified being heterozygous carriers of at least one missense or protein-truncating variant (Table 2). Only three of the variants were predicted to be GoF, 205 were predicted to be LoF, and 216 were predicted to have neutral effects (see supplemental material, Dataset S2). Although a similar number of LoF and neutral variants were detected, the carrier frequency of predicted neutral variants was more than three times higher than that of LoF variants (Table 2) because of a higher minor allele frequency for the neutral variants than for LoF variants (see supplemental material, Table S5). Carriers of LoF variants showed an increased risk of self-reported migraine (Figure 1C), whereas GoF and neutral ATP1A2 variants were not associated with migraine (Figure 1B,D). Twelve homozygous carriers of neutral variants and less than five carriers of homozygous LoF variants were identified (Table 2), but these genotypes were not associated with migraine or epilepsy (Figure 2A,D). In total, 7,391 heterozygous carriers of 135 missense and protein-truncating variants that have been reported in ClinVar to be associated with FHM were detected. No association with self-reported or diagnosed migraine or epilepsy was found for these carriers (see supplemental material, Dataset S1). When only including pathogenic/likely pathogenic missense and LoF ATP1A2 variants (13 variants and 31 heterozygous carriers), an increased disease risk of self-reported migraine was detected (see supplemental material, Figure S1A). No association with epilepsy was observed among carriers of these pathogenic/likely pathogenic variants (see supplemental material, Figure S1B).
SCN1A
Within the SCN1A gene, 1,999 sequence variants were identified, encompassing 796 missense variants and 18 protein-truncating variants (see supplemental material, Table S4C). The majority of the variants were predicted to be either neutral or GoF whereas only three variants were LoF (Table 2). The carrier frequencies of the variants in SCN1A were common, with 203,793 individuals (43.4% of UKB cohort) being heterozygous and 43,296 individuals (9.2%) being homozygous carriers of at least one SCN1A missense or protein-truncating variant. The majority of homozygous SCN1A variants were predicted to be neutral, whereas six homozygous carriers of GoF variants were detected (Table 2; see also supplemental material, Dataset S2). No homozygous carriers of LoF variants were found. Although commonly found in UKB, the carriers of heterozygous variants showed an increased risk of self-reported migraine (Figure 1A). Homozygous carriers of SCN1A missense and protein-truncating variants showed an increased risk of diagnosed unspecified migraine, whereas the association with self-reported migraine did not achieve statistical significance after adjusting for multiple testing. Unexpectedly, neutral variants drove the associations with migraine in heterozygous and homozygous carriers (Figure 1D), whereas carriers of GoF variants did not show an increased disease risk of migraine (Figure 1B). The homozygous carriers of SCN1A variants showed a lower disease risk of diagnosed unspecified epilepsy (Figure 2A), an association that was also driven by carriers of neutral variants (Figure 2D). This association was not driven by generalized idiopathic epilepsy, which showed a similar prevalence among the homozygous carriers (Figure 2A,D). Thirty-one of the SCN1A variants were reported in ClinVar (Table 2). When exclusively analyzing these variants, heterozygous carriers showed an increased disease risk of self-reported migraine (see supplemental material, Dataset S1). Homozygous carriers of ClinVar-reported variants were associated with increased risk of diagnosed unspecified migraine but were negatively correlated with the diagnosis of unspecified epilepsy. Only one SCN1A variant were classified as pathogenic or likely pathogenic, and none of the carriers of this variant had self-reported or diagnosed migraine or epilepsy.
Discussion
This study adopted a genotype-first strategy to investigate the potential link between the three FHM-associated genes with migraine and epilepsy. The results unveiled a link between migraine and the genes CACNA1A, ATP1A2 and SCN1A. The association with migraine distinctly varied based on the specific gene under investigation. In the SCN1A gene, the link with migraine was observed when heterozygous and homozygous carriers of all detected missense and protein-truncating variants were included in the analysis. This is particularly interesting considering that 43 and 9.2% of the UKB participants were heterozygous and homozygous carriers of SCN1A missense or protein-truncating variants, respectively. The association between migraine and ATP1A2 became apparent when examining LoF variants. Conversely, the link between migraine and CACNA1A was driven by GoF variants. None of the FHM-associated genes was linked to an increased risk of epilepsy. By contrast, homozygous carriers of SCN1A missense and protein-truncating variants demonstrated a lower risk of epilepsy, while exhibiting an increased risk of migraine. These results provide important new insights into the genetic compound of migraine and the previously suggested link between FHM-associated genes and epilepsy.
Although genomic research often focuses on characterizing specific gene profiles in individuals with heritable diseases, the decreasing cost of sequencing has enabled reverse phenotyping based on the gene variants. Given the complex interplay of genes in pathology, this genotype-first approach should not be viewed as definitive diagnoses but rather as indicators of potential risk for developing certain conditions. This is, however, crucial for uncovering novel genotype-disease associations, broadening the phenotypic spectrum, and facilitating further exploration of the functional significance of genetic variants (46).
This is not the first study that used a genotype-first approach to investigate the link between FHM and migraine in the UKB. Markel and Curtis (47) investigated the association with self-reported and diagnosed migraine in UKB participants carrying rare variants with minor allele frequency ≤0.01 in migraine-associated variants, including the three FHM-associated genes. It was concluded that none of the FHM-associated genes were significantly associated with migraine in the UKB (47). Notably, at the time of the study by Markel and Curtis (47), the sample size of the exome sequence data was on approximately 200,000 individuals. In contrast, the current dataset comprises 454,706 individuals (32), more than doubling the available whole-exome sequences and significantly increasing the statistical power to identify associations. The difference in sizes of the UKB cohorts may therefore account for the discrepancy of the results of these studies employing similar burden analysis. Another notable difference from previous studies is the comprehensive approach employed in the present study. This approach includes distinction between predicted GoF and LoF variants, while also considering if variants were previously associated with migraine and their pathogenicity. This multi-faceted strategy provides a more detailed and nuanced understanding of the genetic underpinnings of FHM and its relationship with epilepsy and migraine. Indeed, this approach revealed intriguing differences between the associations in the three genes. For example, an increased risk of self-reported migraine was observed for the FHM3-associated SCN1A when all detected missense and protein-truncating variants were included for analysis. This association did not reach statistical significance for ATP1A2 where a link with migraine was found only when analyzing LoF variants and pathogenic variants. For CACNA1A, the association with migraine only reached statistical significance when including GoF and pathogenic variants. These results are in line with previous literature suggesting that LoF ATP1A2 variants and GoF variants in CACNA1A and SCN1A are associated with the migraine phenotype (14,48–51). A recent study by Hautakangas et al. (52), which also adopted a genotype-first strategy, suggested that the risk loci in CACNA1A are associated with >95% probability with migraine with aura but not with migraine without aura (52). In the present study, pathogenic/likely pathogenic CACNA1A variants were linked to both migraine with aura and unspecified migraine, although these results should be interpreted with caution due to the small number of cases and carriers. While the similarities between the study by Hautakangas et al. (52) and the present study are noteworthy, it is important to recognize their differing objectives. The present study aimed to test whether missense and protein-truncating variants within FHM-associated genes were linked to migraine, whereas Hautakangas et al. (52) sought to identify new specific variants associated with migraine across the whole genome.
All three migraine-associated genes addressed in the present study encode membrane ion transporters important for ion homeostasis in the brain and thus its neuronal excitability, as shown in cell culture and animal models (53). Missense GoF variants of CACNA1A gene encoding the neuronal P/Q-type Ca2+ channel are linked to excessive Ca2+ influx into the presynaptic terminal resulting in abnormal neurotransmission (54). Missense LoF ATP1A2 mutations suppress the astrocytic α2 Na,K-ATPase transport activity leading to neuronal hyperexcitability because of extracellular glutamate accumulation and membrane depolarization due to elevation of synaptic potassium (55). However, the α2 Na,K-ATPase plays also an important role in the wall of cerebrovascular microvessels (10), suggesting possible neurovascular implications of these mutations in migraine pathology, as suggested previously (52). The effect of disturbed function of the SCN1A gene-coded voltage-gated Na+ channel is rather complex (23,56) and has been proposed to be associated with disbalance in inhibitory neuronal activities (14,48). Hence, the genetic underpinnings of FHM appear to involve disruptions in ion transporters that increase the excitatory state of neuronal networks, highlighting a complex interplay among various ion transporters in its pathophysiology. The disturbances in ion transport may not only be associated with migraine but also predispose to other neurological disorders. Migraine and epilepsy, despite being distinct neurological disorders, share some pathophysiological features, including abnormal brain electrical activity, suggesting potential comorbidity as evidenced by shared clinical manifestations and treatment responses (57,58). This hypothesis is supported by reports linking FHM-associated gene mutations with ataxia and epilepsy (15–23,48,59). Gene testing of epilepsy patients has revealed heterozygous variants in the FHM3-associated SCN1A gene (60). Accordingly, Dravet syndrome characterized by early-onset epileptic encephalopathy are caused by LoF variants in SCN1A (30). Knockout and LoF variants in SCN1A results in hypoexcitability of GABAergic neurons associated with epilepsy, whereas GoF variants have been linked to migraine (48,49). In this context, the finding in the present study of a reduced risk of epilepsy among homozygous carriers of neutral SCN1A variants is compelling. Remarkably, the same group of homozygous neutral SCN1A variants exhibited an elevated disease risk of migraine, resembling the phenotype previously associated with GoF variants (48,49). This suggests that not all the predicted neutral SCN1A variants appear to be entirely neutral, at least not when the variant is present on both alleles. Another possibility is that some variants classified by the machine-learning model as neutral were misclassified. To the best of our knowledge, the present study is the first to report the association of homozygous SCN1A variants with a decreased risk of epilepsy. The study is also the first to identify an association with an increased risk of migraine among the homozygous carriers of SCN1A variants. Further investigations are required to clarify how disturbance in neuronal function associated with homozygous SCN1A variants can be associated with epilepsy and migraine in contradictory directions.
The identification of only a few predicted LoF variants in the SCN1A gene in the present study is noteworthy. This may reflect evolutionary pressures, as these variants are linked to a severe epileptic phenotype, potentially associated with reduced survival and reproductive fitness (61). By contrast, SCN1A GoF variants, associated with migraine (48,49), were more common and likely result in milder phenotypes. Interestingly, a reverse pattern was seen with ATP1A2, where LoF variants were more prevalent and GoF extremely rare, suggesting gene-specific selective pressures. While ATP1A2 GoF variants may have strong pathogenic effects, their role in disease remains largely unexplored. These observations suggests the potential impact of evolutionary pressures on the prevalence of LoF and GoF variants in FHM-associated genes, which may influence the results of association analyses of these variant types with migraine and epilepsy.
One of the limitations of the present study lies in the absence of a specific diagnosis code for FHM and the lack of FHM as a distinct self-reported disease in the UKB database. This study design carefully considered the balance between statistical robustness and the inclusiveness of disease categories to ensure the reliability of findings. Therefore, the study was restricted to exploring associations with unspecified migraine and migraine with aura. It was not possible to independently reassess whether the standardized diagnostic criteria for migraine and epilepsy were fully met in subjects assigned an ICD-10 diagnosis and in cases of self-reported disease. The study found lower prevalences of self-reported and diagnosed migraine in the UKB cohort compared to previous global estimates (62), potentially limiting the power of analysis. There may be several reasons for the lower prevalence of migraine in UKB. First, the healthy-volunteer bias, which is inherent to the UKB resource, does call for caution when interpreting the results (63). Second, migraine studies in biobanks generally have some assured delicacy (47), as not all patients with migraine have been assigned the migraine diagnosis code unless migraine was related to the contact with the hospital system. This may explain the low number of cases of diagnosed migraine without aura in the UK Biobank. Another possible explanation may be that the general trend in UK is to register migraine by the diagnostic code for Migraine, Unspecified G43.9, unless aura is present. The low prevalence of diagnosed migraine in the UKB may lead to misclassification, with undiagnosed cases in the control group, potentially underestimating the associations with migraine in this study. Due to the low number of diagnosed cases of migraine without aura, this diagnosis was excluded from our analysis. This exclusion may have biased our findings by limiting the study to certain migraine subtypes, potentially underestimating associations that could be present in broader migraine phenotypes. Another limitation of the study is the absence of stratification by biological sex due to the limited number of carriers and cases. Instead, adjustment for sex was made within the statistical analyses. Finally, in addition to clinically diagnosed migraine and epilepsy, the study included self-reported conditions, which is a limitation because self-reported conditions were not necessarily diagnosed by a healthcare professional and may lack clinical validation. The self-reported diagnoses are also potentially limited by recall bias, influencing the accuracy of participants’ recollections of past events or experiences. A previous study demonstrated a good agreement between self-reported migraine and diagnosed migraine based on the International Classification of Headache Disorders (ICHD-II) criteria (64) suggesting a value of including self-reported migraine in the analysis. Thus, migraine without aura or probable migraine without aura is confirmed in 87% of self-reported cases in females (64). By contrast, another study showed that 23.8% of migraine patients are not identified in self-reported assessment (65), potentially leading to underestimation. This may lead to bias in the analysis, but its specific direction is difficult to predict.
In silico prediction was used to classify variants as neutral, GoF or LoF, which is associated with a risk of misclassification. While LoGoFunc demonstrates superior performance compared to other in silico prediction algorithms, it remains a predictive tool (42). As such, it cannot fully capture the biological complexity of genetic variants. To gain more precise and reliable insights into the effects of different types of genetic variants, laboratory-based functional assays would be valuable, although they are beyond the scope of the present study. These assays would allow for direct measurement of the impact of genetic changes on the structure and function of the encoded proteins, providing a better understanding of how these alterations may contribute to disease development and progression. We cannot be certain whether some genetic variants have been misclassified as GoF, LoF or neutral, but, as the statistics rely on a burden test (i.e. the joint analysis of multiple genetic variants), single cases of misclassification will doubtfully have strong consequences on overall conclusions in this study.
Common polygenic variation significantly contributes to the familial aggregation of migraine (66) as observed with CACNA1A variants (52). This suggests the potential for multiple genes to synergistically influence the phenotype of migraine with aura, limiting the effectiveness of a single gene analysis. This may highlight the limited predictive power of monogenetic variants for general migraine diagnoses, emphasizing that clinical characterization should serve as the foundation. Moreover, incorporating genetic analyses that consider the complex interplay of multiple genes may refine diagnostic accuracy.
Despite these limitations, the findings of the present study hold significant promise for clinical practice. The findings provide valuable insights into the genetic component of migraine, highlighting the potential role of FHM-related genes in broader migraine phenotypes. By identifying associations between specific genetic variants and migraine, this research enhances our understanding of the genetic underpinnings of the disorder. These insights have the potential to inform future diagnostic tools and treatment strategies. Understanding mutation-related alterations in protein function can catalyze research into novel treatment paradigms, driving innovation in migraine medicine. This knowledge may pave the way for personalized treatment, allowing for targeted therapies that address the functional disturbances caused by specific genetic abnormalities. This is highly relevant in a therapeutic landscape, where, for example, choices of primary prophylactic treatment of sporadic migraine today are based on trial and error. The choice between an antihypertensive, anticonvulsant or antidepressant drug as a first and second choice is purely empirical rather than based on pathophysiological considerations.
The future development of this research will require a more detailed categorization of diagnoses that may be linked to the FHM-related mutations. It is crucial to establish clear connections between specific genetic changes and their functional impacts on the pathogenicity of migraine and related disorders. Although these initial mechanistic insights may be derived from experimental models, it is essential to consider these findings in the context of multicellular organisms and validate them in future studies within a polygenic environment similar to that of our current study.
Conclusions
This study offers a unique genotype-first analysis in the UKB, utilizing almost half a million exome-sequenced participants to investigate the link between variants in the FHM-associated genes CACNA1A, ATP1A2 and SCN1A with migraine and epilepsy. An association was identified between migraine and all three FHM genes, yet the nature of these associations varied across genes based on pathogenicity and functional impact of the variants. The study revealed a novel link between homozygous SCN1A variants and migraine. No association was observed between epilepsy and the three FHM-associated genes, except for a reduced prevalence of epilepsy among carriers of homozygous SCN1A missense and protein-truncating variants. These findings offer significant advancements in our understanding of the genetics of migraine and epilepsy, and enhance future ability to provide an effective, targeted and personalized care for migraine patients.
Clinical implications
The study highlights the role of FHM-related genes in migraine, offering insights that could inform future diagnostic tools and treatment strategies. In today's therapeutic landscape, primary prophylactic treatment of migraine is often based on trial and error. Understanding the genetic associations of CACNA1A, ATP1A2 and SCN1A can shift this toward treatments grounded in underlying pathophysiological mechanisms. This understanding of genetic abnormalities paves the way for personalized, targeted therapies that address the functional disturbances caused by these genetic variants, leading to more effective and individualized treatment for migraine patients. Homozygous carriers of SCN1A variants were associated with an increased risk of migraine and a lower risk of epilepsy. These variants were commonly observed in the UK Biobank cohort, suggesting a potential role for SCN1A in more common types of migraine.
Supplemental Material
sj-docx-1-cep-10.1177_03331024241306103 - Supplemental material for Exploring the association between familial hemiplegic migraine genes (CACNA1A, ATP1A2 and SCN1A) with migraine and epilepsy: A UK Biobank exome-wide association study
Supplemental material, sj-docx-1-cep-10.1177_03331024241306103 for Exploring the association between familial hemiplegic migraine genes (CACNA1A, ATP1A2 and SCN1A) with migraine and epilepsy: A UK Biobank exome-wide association study by Christian Staehr, Mette Nyegaard, Flemming W. Bach, Palle Duun Rohde and Vladimir V. Matchkov in Cephalalgia
Supplemental Material
sj-xlsx-2-cep-10.1177_03331024241306103 - Supplemental material for Exploring the association between familial hemiplegic migraine genes (CACNA1A, ATP1A2 and SCN1A) with migraine and epilepsy: A UK Biobank exome-wide association study
Supplemental material, sj-xlsx-2-cep-10.1177_03331024241306103 for Exploring the association between familial hemiplegic migraine genes (CACNA1A, ATP1A2 and SCN1A) with migraine and epilepsy: A UK Biobank exome-wide association study by Christian Staehr, Mette Nyegaard, Flemming W. Bach, Palle Duun Rohde and Vladimir V. Matchkov in Cephalalgia
Supplemental Material
sj-xlsx-3-cep-10.1177_03331024241306103 - Supplemental material for Exploring the association between familial hemiplegic migraine genes (CACNA1A, ATP1A2 and SCN1A) with migraine and epilepsy: A UK Biobank exome-wide association study
Supplemental material, sj-xlsx-3-cep-10.1177_03331024241306103 for Exploring the association between familial hemiplegic migraine genes (CACNA1A, ATP1A2 and SCN1A) with migraine and epilepsy: A UK Biobank exome-wide association study by Christian Staehr, Mette Nyegaard, Flemming W. Bach, Palle Duun Rohde and Vladimir V. Matchkov in Cephalalgia
Footnotes
Acknowledgements
The data used in the presented study were obtained from the UKB Resource (project ID 60032).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The research was supported by the Danish Cardiovascular Academy and Riisfort Fonden.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.