
Abstract
Background
OnabotulinumtoxinA (onabotA), is assumed to achieve its therapeutic effect in migraine through blocking activation of unmyelinated meningeal nociceptors and their downstream communications with central dura-sensitive trigeminovascular neurons in the spinal trigeminal nucleus (SPV). The present study investigated the mechanism of action of onabotA by assessing its effect on activation and sensitization of dura-sensitive neurons in the SPV by cortical spreading depression (CSD). It is a follow up to our recent study on onabotA effects on activation and sensitization of peripheral trigeminovascular neurons.
Methods
In anesthetized male and female rats, single-unit recordings were used to assess effects of extracranial injections of onabotA (five injections, one unit each, diluted in 5 μl of saline were made along the lambdoid (two injection sites) and sagittal (two injection sites) suture) vs. vehicle on CSD-induced activation and sensitization of high-threshold (HT) and wide-dynamic range (WDR) dura-sensitive neurons in the SPV.
Results
Single cell analysis of onabotA pretreatment effects on CSD-induced activation and sensitization of central trigeminovascular neurons in the SPV revealed the ability of this neurotoxin to prevent activation and sensitization of WDR neurons (13/20 (65%) vs. 4/16 (25%) activated neurons in the control vs. treated groups, p = 0.022, Fisher's exact). By contrast, onabotA pretreatment effects on CSD-induced activation and sensitization of HT neurons had no effect on their activation (12/18 (67%) vs. 4/7 (36%) activated neurons in the control vs. treated groups, p = 0.14, Fisher's exact). Regarding sensitization, we found that onabotA pretreatment prevented the enhanced responses to mechanical stimulation of the skin (i.e. responses reflecting central sensitization) in both WDR and HT neurons. In control but not treated WDR neurons, responses to brush (p = 0.004 vs. p = 0.007), pressure (p = 0.002 vs. p = 0.79) and pinch (p = 0.007 vs. 0.79) increased significantly two hours after CSD. Similarly, in control but not treated HT neurons, responses to brush (p = 0.002 vs. p = 0.79), pressure (p = 0.002 vs. p = 0.72) and pinch (p = 0.0006 vs. p = 0.28) increased significantly two hours after CSD. Unexpectedly, onabotA pretreatment prevented the enhanced responses of both WDR and HT neurons to mechanical stimulation of the dura (commonly reflecting peripheral sensitization). In control vs. treated WDR and HT neurons, responses to dural stimulation were enhanced in 70 vs. 25% (p = 0.017) and 78 vs. 27% (p = 0.017), respectively.
Conclusions
The ability of onabotA to prevent activation and sensitization of WDR neurons is attributed to its preferential inhibitory effects on unmyelinated C-fibers. The inability of onabotA to prevent activation of HT neurons is attributed to its less extensive inhibitory effects on the thinly myelinated Aδ-fibers. These findings provide further pre-clinical evidence about differences and potentially complementary mechanisms of action of onabotA and calcitonin gene-related peptide-signaling neutralizing drugs.
Introduction
OnabotulinumtoxinA (onabotA) is a US Food and Drug Administration-approved prophylactic drug for chronic migraine (1,2). When used for migraine prevention, onabotA is injected into discrete pericranial, neck and shoulder muscles (3). Until recently, it was considered to be a neuromuscular blocker that acts through the inhibition of transmitter release, and is mainly used in the treatment of muscle hyperactivity such as dystonia and spasticity (4). However, recent findings have provided convincing evidence for the ability of onabotA to interfere with sensory transmission in peripheral meningeal nociceptors that are at the origin of the trigeminovascular pathway (4). In the context of migraine, it was shown that local administration of onabotA to pericranial muscles and along calvarial suture lines can preferentially inhibit activation of unmyelinated (C-fiber) but not myelinated (A-delta-fiber) meningeal nociceptors by capsaicin (TRPV1 agonist), mustard oil (TRPA1 agonist) and cortical spreading depression (CSD) (5–7), comprising stimuli used commonly in animal models of migraine. Outside the migraine field and trigeminovascular pathway, animal as well as human studies have provided evidence for the selective onabotA effects on the unmyelinated C-fibers. Scientifically, administration of onabotA was shown to decrease allodynia induced by sciatic nerve damage in rats (8), an effect attributable to its inhibitory effects on neurogenic inflammation, C-fiber sensitization and, consequently, the development of central sensitization (9–12). Clinically, onabotA mitigated capsaicin-induced pain and neurogenic vasodilation without altering thermal and pain perception, a set of findings attributed to the selective inhibition of C-type nociceptors and their TRPV1 receptors (9–12). Therapeutically, it appears that the selective inhibition of C-fibers by onabotA (6,7,12) may be sufficient to produce a sustained analgesic effect on peripheral neuropathic pain (13–15).
To date, no prior in vivo electrophysiological study exists that describes the impact of inhibition of the unmyelinated meningeal nociceptors by onabotA on the activation, sensitization and response magnitude of the different classes of central trigeminovascular neurons in the spinal trigeminal nucleus (SPV). Because the reduction in the perception of headache by onabotA cannot be fully explained without complete understanding of its effects on activation and sensitization of the second- and third-order neurons along this migraine-specific nociceptive pathway, the present study attempted to determine the effects of onabotA on trigeminovascular wide-dynamic range (WDR) and high-threshold (HT) neurons in the SPV.
CSD, a slowly propagating wave of neuronal depolarization assumed to underlie the aura phase of migraine, is one of the most commonly used and best characterized animal models of migraine (16–18). In the present study, which is a follow up to our recent study on the effects of onabotA on meningeal nociceptors (5), we tested onabotA effects on responsiveness of HT and WDR neurons in the SPV.
Methods
Experiments were approved by the Beth Israel Deaconess Medical Center and Harvard Medical School standing committees on animal care and were in accordance with the US National Institutes of Health's Guide for the Care and Use of Laboratory Animals. In total, 36 female and 29 male Sprague–Dawley rats (240–300 g) were used.
Overview of experimental protocol
An experimental protocol was designed to test the effect of onabotA on the activity of central trigeminovascular neurons in the dorsal horn (spontaneous and induced activity in response to peripheral stimulation and CSD) (Figure 1). Based on the time course of action, the onabotA was administered seven days prior to the day of neuronal recording. Two groups of rats were studied with this protocol: (i) onabotA and (ii) vehicle (saline). In this protocol, single-unit recordings of activity of central trigeminovascular neurons were obtained in anesthetized rats. After characterization of responses to dural and facial stimulation, their spontaneous activity (SA) was recorded for one hour. CSD was then induced and recording of neuronal activity continued for an additional two hours. Characterization of responses to dural and facial stimulation was repeated two hours after CSD induction (Figure 2). Ongoing discharge was recorded continuously throughout the experiment. Methodological details are described below.

Experimental design. Treatment group received onabotA injections seven days prior to the experiment. Control group received saline (vehicle) injections seven days prior to the experiment. Stars depict times at which responses to mechanical stimulation of the dura and skin were determined. Inset depicts locations of the five injection sites of onabotA. onabotA = onabotulinumtoxinA; CSD = cortical spreading depression.

Recording sites (A, B), dural (C, D) and facial (E, F) receptive fields of all studied trigeminovascular neurons in the spinal trigeminal nucleus. Black and gray circles depict locations of lesions of high threshold ( HT) and wide-dynamic range (WDR) neurons in the different laminae of the upper cervical spinal cord segment. Red indicates locations and sizes of most sensitive areas of dural and cutaneous receptive fields. The inset in (C) depicts dural receptive fields.
Surgical preparation
Rats were anesthetized with intraperitoneal urethane (0.9–1.2 g/kg) and surgically prepared for recording of neuronal activity in the SPV as described in detail previously (5,19–21). The rats were intubated to allow artificial ventilation, and the femoral vein was cannulated for drug infusion. Core temperature and end-tidal CO2 were monitored and kept within physiological range. Rats were paralyzed with rocuronium and ventilated. A craniotomy was made to expose the left transverse sinus, and a second craniotomy was made in the left parietal bone to allow recording of electrocorticogram activity and induction of CSD by pinprick. A segment of the spinal cord between the obex and C2 was exposed for recording of activity from central neurons in the left SPV (C1–2 dorsal horn).
Identification and characterization of central trigeminovascular neurons
To record neuronal activity, a tungsten microelectrode (impedance 1–4 MΩ; FHC Co., Bowdoin, ME, USA) was lowered repeatedly into the SPV (C1–2 dorsal horn) in search of central trigeminovascular neurons receiving input from the dura. Trigeminovascular neurons were first identified based on their responses to electrical stimulation of the dura. They were selected for the study if they exhibited discrete firing bouts in response to ipsilateral electrical (0.1–3.0 mA, 0.5 ms, 0.5 Hz pulses) and mechanical (with calibrated von Frey monofilaments) stimulation of the exposed cranial dura.
Dural receptive fields were mapped by indenting the dura (4.19 g von Frey hair (VF) monofilament). Cutaneous receptive fields were mapped by applying innocuous and noxious mechanical stimulation to all facial skin areas as described previously (5,19–21). Responses to mechanical stimulation of the skin were determined by applying brief (10 s) innocuous and noxious stimuli to the most sensitive portion of the cutaneous receptive field. Innocuous stimuli consisted of slowly passing a soft bristled brush across the cutaneous receptive field and pressure applied with a loose arterial clip. Noxious stimuli consisted of pinch with a strong arterial clip. Two classes of neurons were thus identified: WDR neurons (incrementally responsive to brush, pressure and pinch) and HT neurons (unresponsive to brush). A real-time waveform discriminator was used to create and store a template for the action potential evoked in the neuron under study by electrical pulses on the dura; spikes of activity matching the template waveform were acquired and analyzed online and offline using Spike 2 software (Cambridge Electronic Design Limited, Cambridge, UK). Data were used only in cases in which the physiological condition of the rats (heart rate, blood pressure, respiration, end-tidal CO2) and the neuronal isolation signal (signal-to-noise ratio ∼1:3) were stable throughout the experimental period.
At the conclusion of all experiments, a small electrolytic lesion was made at the recording site, and its localization in the dorsal horn was determined postmortem using histological analysis as described previously (5,19–21). Only one neuron was studied in each animal.
Treatment with onabotA
Seven days prior to surgical preparation, male and female Sprague–Dawley rats (250–300 g) were briefly anesthetized (2% isoflurane) and injected with onabotA (final dose = 5 U) or vehicle (normal saline). Five injections of onabotA (each containing 1 U diluted in 1 μL of saline, total dose 5 U/5 μL) or saline (5 μL) were made along the lambdoid sutures (two injection sites on each side), and at the site where the sagittal and lambdoid sutures meet (on the midline) (Figure 1, inset). To reach the suture lines, a small needle (30 guage) was lowered through the layers of the scalp until it hit the bones of the calvaria (i.e. between galea aponeurotica and periosteum). The targeting of the suture lines for the injections is based on findings from anatomical studies of intra- and extracranial sensory innervations that pass through the sutures (22,23) (see Discussion).
CSD induction and electrocorticogram recording
Seven days after onabotA injections, CSD was induced in the occipital cortex by pinprick (inserting a glass micropipette, 25 μm in diameter, approximately 1 mm into the cortex for 10 s) (5). For verification of CSD, electrocorticogram activity was recorded with a glass micropipette (0.9% saline, ∼1 MΩ, 7 μm tip) placed just below the surface of the parietal cortex (∼100 μm). We opted to use a single wave of CSD because patients rarely describe/experience more than one wave of aura in a single migraine attack; thus, this paradigm adheres better to the clinical experience (24).
Statistical analysis
Analyses of neuronal firing before and after induction of CSD and in responses to mechanical stimulation of the dura and skin, as well as their classification, was carried out by an experimenter who was blinded to the treatment each rat received (i.e. onabotA or vehicle). Randomization was applied to recording order of neuronal class (WDR and HT) and treatment (control, onabotA). To determine neuronal responses to CSD, the mean firing frequency occurring before the onset of CSD (calculated from measuring the SA during the one-hour period before CSD onset) was compared to the mean firing frequency recorded for one hour (0–60 min after CSD) and two hours (recorded from 60–120 min after CSD) after CSD induction. A neuron was considered activated if its mean firing rate after CSD exceeded its mean baseline activity by 1 SD for a period >10 min, which translated to an approximately >33% increase in activity. To calculate the response to mechanical stimulation of the dura (VFH) and skin (brush, pressure, pinch), the mean firing frequency occurring before the onset of the first stimulus (mean spontaneous activity for 30 min) was subtracted from the mean firing frequency that occurred throughout the duration of each stimulus. Mean firing rates of respective values were compared using a non-parametric repeated measures test (Friedman test) and post-hoc analysis (Tukey’s honestly significant difference). p < 0.05 was considered statistically significant. For comparisons on paired groups, a Wilcoxon signed-rank test was used.
Results
Recordings were made from 36 WDR (20 control and 16 treatment) and 29 HT (18 control and 11 treatment) neurons in male and female rats. Locations of their recording sites in the different laminae of the medullary dorsal horn and their dural and facial receptive fields are shown in Figure 2.
Male vs. female
Control group (saline)
Data collected pre- and post-CSD revealed no differences between neurons recorded in male and neurons recorded in female rats, regardless of whether they were HT or WDR. CSD activated 75% (15/20) of the neurons in males and 55% (10/18) in females (p = 0.301 Fisher’s exact). Pre-CSD, there were no significant differences between male and female in spontaneous firing (female: 3.3 spikes/second (0–8.8); male: 2.3 spikes/second (0–6.5), p = 0.613), responses to dural indentation with VFH (female: 9.7 spikes/second (6.6–21.1); male: 8.5 spikes/second (5.1–17.4), p = 0.491) or skin brush (female: 7.4 spikes/second (0–27.6); male: 0.65 spikes/second (0–11.5), p = 0.286), pressure (female: 15.3 spikes/second (8.3–25.9); male: 8.4 spikes/second (4.2–21.2), p = 0.092) and pinch (female: 26.5 spikes/second (17.1–40.2); male: 22.1 spikes/second (9.3–29.7), p = 0.266). Post-CSD, spontaneous firing increased by 0.7 spikes/second (−2.8–10.1) in females vs. 2.3 spikes/second (0.4–6.1) in males (p = 0.298); responses to dural indentation with VFH increased by 8.2 spikes/second (0.4–19) in females vs. 11.5 spikes/second (6.5–18.4) in males (p = 0.254), and skin brush increased by 17.1 spikes/second (4.3–27.7) in females vs. 14.1 spikes/second (0–29.7) in males (p = 0.988), pressure increased by 11.2 spikes/second (0.3–29.6) in females vs. 15.2 spikes/second (1.2–24.1) in males (p = 0.989) and pinch increased by 11.3 spikes/second (2.5–29.7) in females vs. 18.2 spikes/second (1.1–36.6) in males (p = 0.988). All values are reported as the median (interquartile range (IQR)) and the statistical test used for the comparisons was the Mann–Whitney U-test.
Treatment group (onabotA)
Although pre-CSD spontaneous firing and responsiveness to dural and skin stimuli appeared higher in males than females, and their post-CSD values seemed to be further affected in males vs. females, none of the differences reached statistical significance (regardless of whether they were HT or WDR). CSD activated 22% (2/9) of neurons in males and 33% (6/18) in females (p = 0.675, Fisher’s exact). Pre-CSD, no male-female differences were found in spontaneous firing (female: 2.8 spikes/second (0–5.6); male: 4.4 spikes/second (0.5–8.6), p = 0.623), responses to dural indentation with VFH (female: 9 spikes/second (3.4–15.2); male: 11.6 spikes/second (4.4–15.7), p = 0.553) or skin brush (female: 4.9 spikes/second (0–13.7); male: 8.7 spikes/second (0–20.1), p = 0.451), pressure (female: 11.9 spikes/second (5.1–20.3); male: 21.5 spikes/second (12.3–35.3), p = 0.075) and pinch (female: 18.8 spikes/second (12.5–31.3); male: 31.9 spikes/second (23.6–45.1), p = 0.064). Post-CSD, spontaneous firing increased by 0.1 spikes/second (−1.7–5.1) in females vs. −0.5 spikes/second (−4.7–0.4) in males (p = 0.207), responses to dural indentation with VFH decreased by −0.7 spikes/second (−6.7–8.8) in females vs. −2.3 spikes/second (−9.1–3.1) in males (p = 0.425), and skin brush increased by 5.3 spikes/second (−0.2–22.8) in females vs. 0 spikes/second (−4.4–9.3) in males (p = 0.142), pressure decreased by −0.05 spikes/second (−1.8–18.3) in females vs. −4 spikes/second (−19.2–4.9) in males (p = 0.081) and pinch increased by 4.4 spikes/second (−5.9–21.3) in females vs. −5.4 spikes/second (−21.9–14.5) in males (p = 0.207). All values are reported as the median (IQR) and the statistical test used for the comparisons was the Mann–Whitney U-test.
We therefore combined all data collected in males and females in the overall analyses below.
Baseline activity
The median (IQR) firing rates at baseline, prior to CSD induction, were 3.3spikes/second (0–9.42) for HT neurons in the control group vs. 5.6 spikes/second (2.3–9.3) in the treatment group (p = 0.41 (Z = 0.81, n1 = 18, n2 = 11), Mann–Whitney U-test) and 2.15 spikes/second (0–6.62) for WDR neurons in the control group vs. 0.95 spikes/second (0.12–5.4) in the treatment group (p = 0.83 (Z = 0.20, n1 = 20, n2 = 16), Mann–Whitney U-test). Analysis of all neurons (HT plus WDR) also revealed no significant differences in baseline spontaneous firing between control (2.6 spikes/second (0–7.05)) vs. treatment (3.3 spikes/second (0.5–6.8)) groups (p = 0.82 (Z = 0.22, n138, n2 = 27), Mann–Whitney U-test).
Activation by CSD
HT neurons
Rate of activation
Examples of individual neurons responses to CSD are shown in Figure 3. In the control group, CSD triggered distinct and prolonged activation in 12/18 (66%) neurons (Figures 3Aii and 4Ai) and in the treatment group, it activated four of 11 (36%) neurons (p = 0.142, χ2=2.53, d.f. = 1, Fisher’s exact) (Figures 3Bii and 4Aii).

Examples of cortical spreading depression (CSD) effects on activation and sensitization of individual wide-dynamic range (WDR) (Ai, Bi) and HT (Aii, Bii) neurons in untreated (vehicle) and treated (onabotulinumtoxinA) animals. (Ai, Aii) Plots of firing rate before (blue) and one and two hours after (red) CSD induction in an animal treated with saline (vehicle). Note that spontaneous firing and responses to mechanical stimulation of the dura and skin increased after the CSD. (Bi, Bii) Plots of firing rate before (blue) and one and two hours after (green) CSD induction in an animal treated with onabotulinumtoxinA. Note that spontaneous activity and responses to stimulation of the dura and skin did not increase after the CSD. Recording sites and locations of dural and cutaneous receptive fields of each of the four neurons are shown. VFH = von Frey hair.

Percentage of high threshold ( HT) neurons, wide-dynamic range (WDR) neurons and all neurons activated by cortical spreading depression (CSD) in the 38 animals treated with saline (control, Ai, Bi, Ci) and 27 animals treated with onabotulinumtoxinA (treatment, Aii, Bii, Cii). Fisher’s exact test was used to calculate the level of significance of the percentage differences between the groups. Plots of changes in spontaneous firing rate recorded in individual HT (Aiii–iv), WDR (Biii–iv) and all (Ciii–iv) neurons before and two hours after the occurrence of CSD. Red and blue lines depict neurons classified as activated and not activated, respectively.
Magnitude of activation
Actual changes in spontaneous firing rate recorded in each HT neuron before and two hours after the occurrence of CSD are shown in Figure 5Ai (control) and Figure 5Aii (treatment). Firing rate analyses of all HT neurons (activated and non-activated) showed that, in the control group, baseline spontaneous activity (3.3 spikes/second (0–9.42) (median (IQR))) increased significantly at one hour (by 2.5 spikes/second (0–7.26)) and two hours (by 4.3 spikes/second (0.37–18.02)) after CSD (χ2=10.75, d.f. = 2, p = 0.0032, Friedman test). Post-hoc (Tukey’s honestly significant difference) comparisons between baseline and one and two hours post-CSD yielded p values of 0.046 and 0.0029, respectively (Figure 5Ai). By contrast, in the treatment group, spontaneous activity remained unchanged after CSD. The neurons’ baseline firing rate (5.6 spikes/second (2.3–9.3) (median (IQR))) did not change significantly at one hour (decrease of 0.18 spikes/second (−2.3–4.7)) or two hours (−0.33 spikes/second (−4.33–4.9)) after CSD (χ2=0.72, d.f. = 2, p = 0.70, Friedman test) (Figure 5Aii).

Response magnitude. Spontaneous activity changes (from baseline) recorded in high threshold ( HT) (Ai, Aii), wide-dynamic range (WDR) (Bi, Bii) and all (Ci, Cii) neurons at one and two hours after CSD induction. Box-and-whisker plots depict the median and interquartile range (Ai-Aii, n = 18 in control group, n = 11 in treatment group; Bi, Bii, n = 20 in control group, n = 16 in treatment group; Ci, Cii, n = 38 in control group, n = 27 in treatment group). Scatterplots depict changes from baseline of individual neurons. *p < 0.05, statistical difference compared to baseline, Friedman test; post-hoc/Tukey’s honestly significant difference.
WDR neurons
Rate of activation
Examples of individual neurons responses to CSD are shown in Figure 3. In the control group, CSD triggered distinct and prolonged activation in 13/20 (65%) neurons (Figures 3Ai and 4Bi), whereas, in the treatment group, it activated four of 16 (25%) neurons (p = 0.022, χ2=5.70, d.f. = 1, Fisher’s exact) (Figures 3Bi and 4Bii).
Magnitude of activation
Actual changes in spontaneous firing rate recorded in each WDR neuron before and two hours after the occurrence of CSD are shown in Figure 5Bi (control) and Figure 5Bii (treatment). Firing rate analyses of all WDR neurons (activated and non-activated) showed that, in the control group, baseline spontaneous activity (2.15 spikes/second (0–6.62)) increased significantly at one hour (by 0.8 spikes/second (0–5.27)) and two hours (by 0.85spikes/second (0–3.4)) after CSD (χ2=4.9, d.f. = 2, p = 0.044, Friedman test). Post-hoc (Tukey’s honestly significant difference) comparisons between baseline and one and two hours post-CSD onset yielded p values of 0.025 and 0.032, respectively (Figure 5Bi). By contrast, in the treatment group, spontaneous activity remained unchanged after CSD. The neurons’ baseline firing rate (0.95 spikes/second (0.25–5.4) (median (IQR))) did not change significantly at one hour (decrease of 0.15 spikes/second (−0.73–1.1)) or two hours (0.15 spikes/second (−1.02–1.67)) after CSD (χ2=0.21, d.f. = 2, p = 0.91, Friedman test) (Figure 5Bii).
All neurons
Rate of activation
In the control group, CSD triggered distinct and prolonged activation in 25/38 (65%) neurons (Figure 4Ci), whereas, in the treatment group, it activated eight of 27 (29%) neurons (p = 0.005, χ2 = 8.25, d.f. = 1, Fisher’s exact) (Figure 4Cii).
Magnitude of activation
Actual changes in spontaneous firing rate recorded in all neurons before and two hours after the occurrence of CSD are shown in Figure 5Ci (control) and Figure 5Cii (treatment). Firing rate analyses of all neurons (activated and non-activated) showed that, in the control group, baseline spontaneous activity (2.6 spikes/second (0–7.05)) increased significantly at one hour (1.6 spikes/second (0–6.04)) and two hours (by 1.25 spikes/second (0–6.65)) after CSD (χ2=11.7, d.f. = 2, p = 0.0002, Friedman test). Post-hoc (Tukey’s honestly significant difference) comparisons between baseline and one and two hours post-CSD onset yielded p values of 0.003 and 0.001, respectively (Figure 5Ci). By contrast, in the treatment group, spontaneous activity remained unchanged after CSD. The baseline firing rate of neurons (3.3 spikes/second (0.5–6.8) (median (IQR))) did not change significantly at one hour (0 spikes/second (−1.4–3)) or two hours (0.1 spikes/second) (−2.2–2.6)) after CSD (χ2=0.12, d.f. = 2, p = 0.94, Friedman test) (Figure 5Cii).
Responses to mechanical stimulation of the dura before and after CSD
HT neurons
In the control group, baseline responses to dural indentation with calibrated VFH monofilament (4.1 g) (8.5 spikes/second (5.5–21.12)) increased by 10.5 spikes/second (4.12–24.12) 2 h after CSD (p = 0.0005, Z = 3.43, n1 = 18, n2 = 18, Wilcoxon signed-rank test) (Figure 6A). By contrast, in the treatment group, baseline responses (13 spikes/second (9–25.5)) to dural indentation with the same VFH monofilament decreased (rather than increased), although not significantly, by 6.5 spikes/second (−7.3–4.6) two hours after CSD (p = 0.44, Z = 0.80, n1 = 11, n2 = 11, Wilcoxon signed-rank test) (Figure 6B). At the individual level, 14/18 (77%) neurons in the control group exhibited enhanced responses to dural stimulation (>50% increase) after the CSD, whereas, in the treatment group, only 4/11 (36%) neurons exhibited such enhanced responses (p = 0.017, χ2=7.17, d.f. = 1, Fisher’s exact).
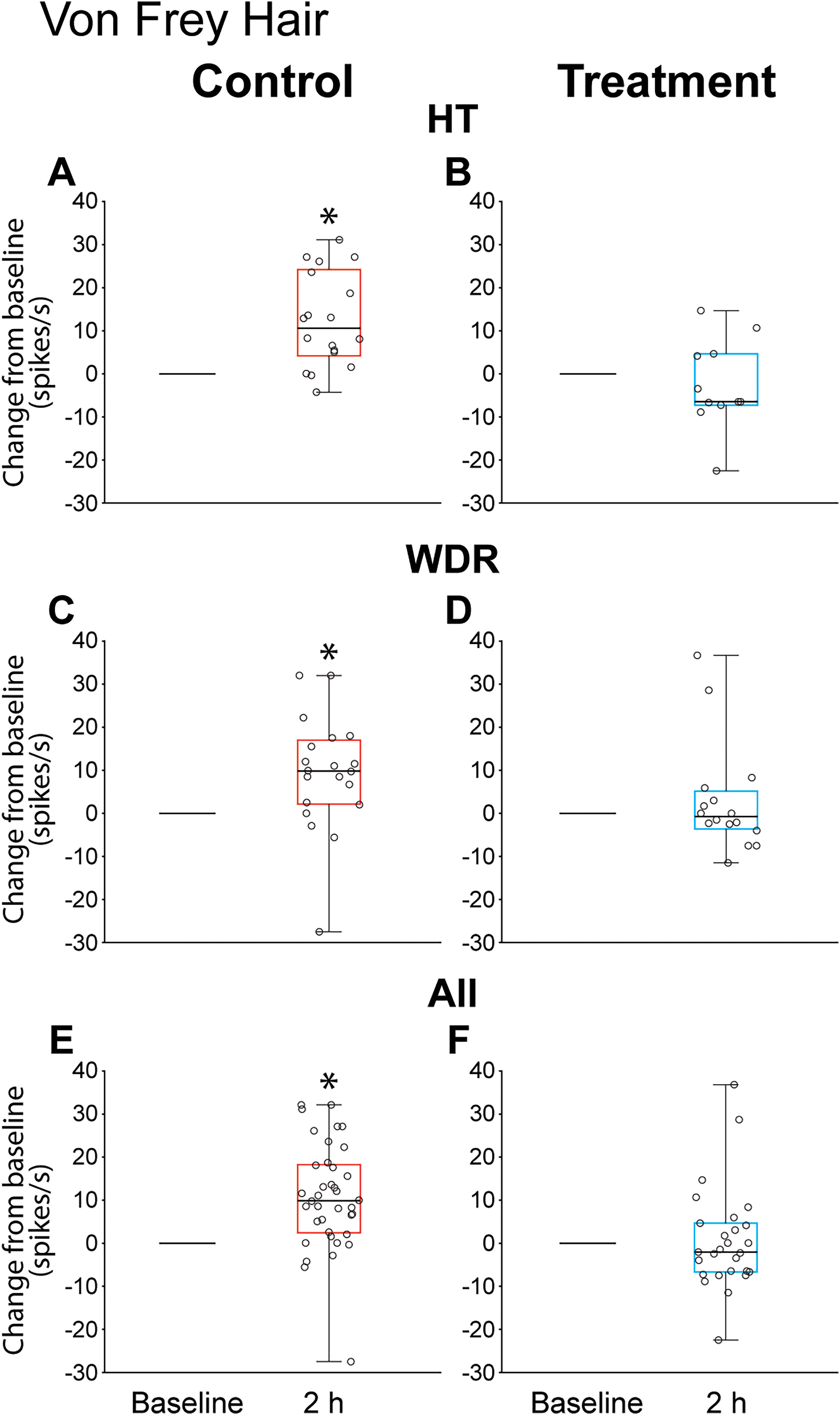
Changes from baseline in response to dural indentation two hours after cortical spreading depression (CSD) induction. (A, C, E) animals treated with saline. (B, D, F) animals treated with onabotulinumtoxinA. Box-and-whisker plots depict the median and interquartile range (A, B, n = 18 in control group, n = 11 in treatment group; C, D, n = 20 in control group, n = 16 in treatment group; E, F, n = 38 in control group, n = 27 in treatment group). Scatterplots depict changes from baseline of individual neurons. *p < 0.05, statistical difference compared to baseline, Wilcoxon signed-rank test.
WDR neurons
In the control group, baseline responses (8.8 spikes/second (5.5–11.82)) to dural indentation with calibrated VFH monofilament increased by 9.8 spikes/second (2.12–17)) two hours after CSD (p = 0.004, Z = 2.85, n1 = 20, n2 = 20, Wilcoxon signed-rank test) (Figure 6C). By contrast, in the treatment group, baseline responses (5.5 spikes/second (2.32–11.27)) to dural indentation with the same VFH monofilament remained unchanged (−0.75 spikes/second (−3.62–5.17) two hours after CSD (p = 0.92, Z = 0.09, n1 = 16, n2 = 16, Wilcoxon signed-rank test) (Figure 6D). At the individual level, 14/20 (70%) neurons in the control group exhibited enhanced responses to dural stimulation after the CSD, whereas, in the treatment group, only four of 16 (25%) exhibited such enhanced responses (p = 0.017, χ2=7.2, d.f. = 1, Fisher’s exact).
All neurons
In the control group, baseline responses (8.55 spikes/second (5.57–19.5)) to dural indentation with calibrated VFH monofilament increased by 9.8 spikes/second (2.37–18.15) two hours after CSD (p = 0.000001, Z = 4.38, n1 = 38, n2 = 38, Wilcoxon signed rank-test) (Figure 6E). In the treatment group, baseline responses (9 spikes/second (4–13.5)) to dural indentation with the same VFH monofilament remained unchanged (−2.1 spikes/second (−6.7–4.6)) two hours after CSD (p = 0.67, Z = 0.41, n1 = 27, n2 = 27, Wilcoxon signed-rank test) (Figure 6F). At the individual level, 28/38 (73%) neurons in the control group exhibited enhanced responses to dural stimulation after the CSD, whereas, in the treatment group, only 7/27 (25%) exhibited such enhanced responses (p = 0.0001, χ2=14.4, d.f. = 1, Fisher’s exact).
Responses to mechanical stimulation of the skin before and after CSD
HT neurons
In the control group, baseline responses to skin stimulation with brush, pressure and pinch were 0 spikes/second (0–0), 11.7 spikes/second (5.47–22.45) and 21.25 spikes/second (13.5–35.55), respectively. Two hours after CSD, responses to brush (increased by 22.05 spikes/second (0–38.8), p = 0.002, Z = 3.05, n1 = 18, n2 = 18), pressure (increased by 14.35 spikes/second (−1.05–26.97), p = 0.002, Z = 3.02, n1 = 18, n2 = 18) and pinch (increased by 24.65 spikes/second (1.4–37.32), p = 0.0006, Z = 3.41, n1 = 18, n2 = 18) increased significantly compared to baseline (Figure 7Ai, Bi, Ci). In the treatment group, baseline responses to skin stimulation with brush, pressure and pinch were 0 spikes/second (0–0), 13.4 spikes/second (5–34.2) and 20.16 spikes/second (11.75–51), respectively. Two hours after CSD, responses to brush increased significantly (increased by 7.5 spikes/second (0–24.5), p = 0.017, Z = 2.36, n1 = 11, n2 = 11) compared to baseline, whereas responses to pressure (−0.1 spikes/second (−4.3–10.7), p = 0.72, Z = 0.35, n1 = 11, n2 = 11) and pinch (5.7 spikes/second (−6.1–13.5), p = 0.28, Z = 1.06, n1 = 11, n2 = 11) remained unchanged compared to baseline (Figure 7Aii, Bii, Cii).

Changes from baseline in responses to mechanical stimulation of the skin (A, Brush; B. Pressure; C, Pinch) two hours after cortical spreading depression (CSD) induction in animals treated with saline (control) and onabotulinumtoxinA (treatment). Box-and-whisker plots depict the median and interquartile range (HT, n = 18 in control group, n = 11 in treatment group; WDR, n = 20 in control group, n = 16 in treatment group; All, n = 38 in control group, n = 27 in treatment group). Scatterplots depict changes from baseline of individual neurons. *p < 0.05, statistical difference compared to baseline, Wilcoxon signed-rank test. HT = high threshold; WDR = wide-dynamic range.
WDR neurons
In the control group, baseline responses to skin stimulation with brush, pressure and pinch were 13.15 spikes/second (6.32–24.8), 15.7 spikes/second (6.17–22.77) and 26.65 spikes/second (9.35–35.1), respectively. Two hours after CSD, responses to brush (increased by 14.12 spikes/second (6.25–26), p = 0.004, Z = 2.85, n1 = 20, n2 = 20), pressure (increased by 15.25 spikes/second (1.27–30.97), p = 0.002, Z = 3.17, n1 = 20, n2 = 20) and pinch (increased by 14.05 spikes/second (2.12–30.3), p = 0.007, Z = 2.68, n1 = 20, n2 = 20) increased significantly compared to baseline (Figure 7Aiii, Biii, Ciii). In the treatment group, baseline responses to skin stimulation with brush, pressure and pinch were 12 spikes/second (6.27–20.42), 15.65 spikes/second (7.45–23) and 26.15 spikes/second (13.92–34.67). Two hours after CSD, responses to brush (0.3 spikes/second (−5.02–7.45), p = 0.79, Z = 0.25, n1 = 16, n2 = 16), pressure (1.15 spikes/second (−6.17–21.65), p = 0.79, Z = 0.25, n1 = 16, n2 = 16) and pinch (0.9 spikes/second (−19.75–18.82), p = 0.95, Z = 0.051, n1 = 16, n2 = 16) remained unchanged compared to baseline (Figure 7Aiv, Biv, Civ).
Activation and central sensitization
For this analysis, a neuron was considered activated if its mean firing rate after CSD exceeded its mean baseline activity by 1 SD for a period >10 min, and sensitized if its responses to skin stimulation with brush, pressure and/or pinch increased by >50% at two hours post-CSD. We did not consider the central neurons as sensitized if their response magnitude increased to dural but not skin stimulation. Enhanced responses of the central neurons to dural stimulation could result from peripheral sensitization alone (CSD-induced sensitization of the meningeal nociceptors (25), which provide synaptic input to the central neurons). By contrast, enhanced responses of the central neurons to facial stimulation following CSD has been established as a sign of central rather than peripheral sensitization (sensitization of the central neurons themselves, making them more responsive to primary afferent inputs) (26) because CSD is an intracranial event and cannot directly affect extracranial sensory nerve endings. Accordingly, of the 38 neurons studied in the control group, CSD triggered activation and central sensitization in 57.9%, activation without central sensitization in 7.9% and central sensitization without activation in 15.9% (Table 1). By contrast, of the 27 neurons studied in the treatment group, CSD triggered activation and central sensitization in 29.6%, activation without central sensitization in 0% and central sensitization without activation in 18.5% (Table 2). Table 3 provides a summary of onabotA effects on activation and sensitization of WDR and HT neurons.
Activation (SA), peripheral sensitization (VFH) and central sensitization (brush, pressure, pinch) in untreated wide-dynamic range (WDR) and high threshold (HT) neurons.

Activation (SA), peripheral sensitization (VFH) and central sensitization (brush, pressure, pinch) in botulinum neurotoxin type A treated wide-dynamic range (WDR) and high threshold (HT) neurons.
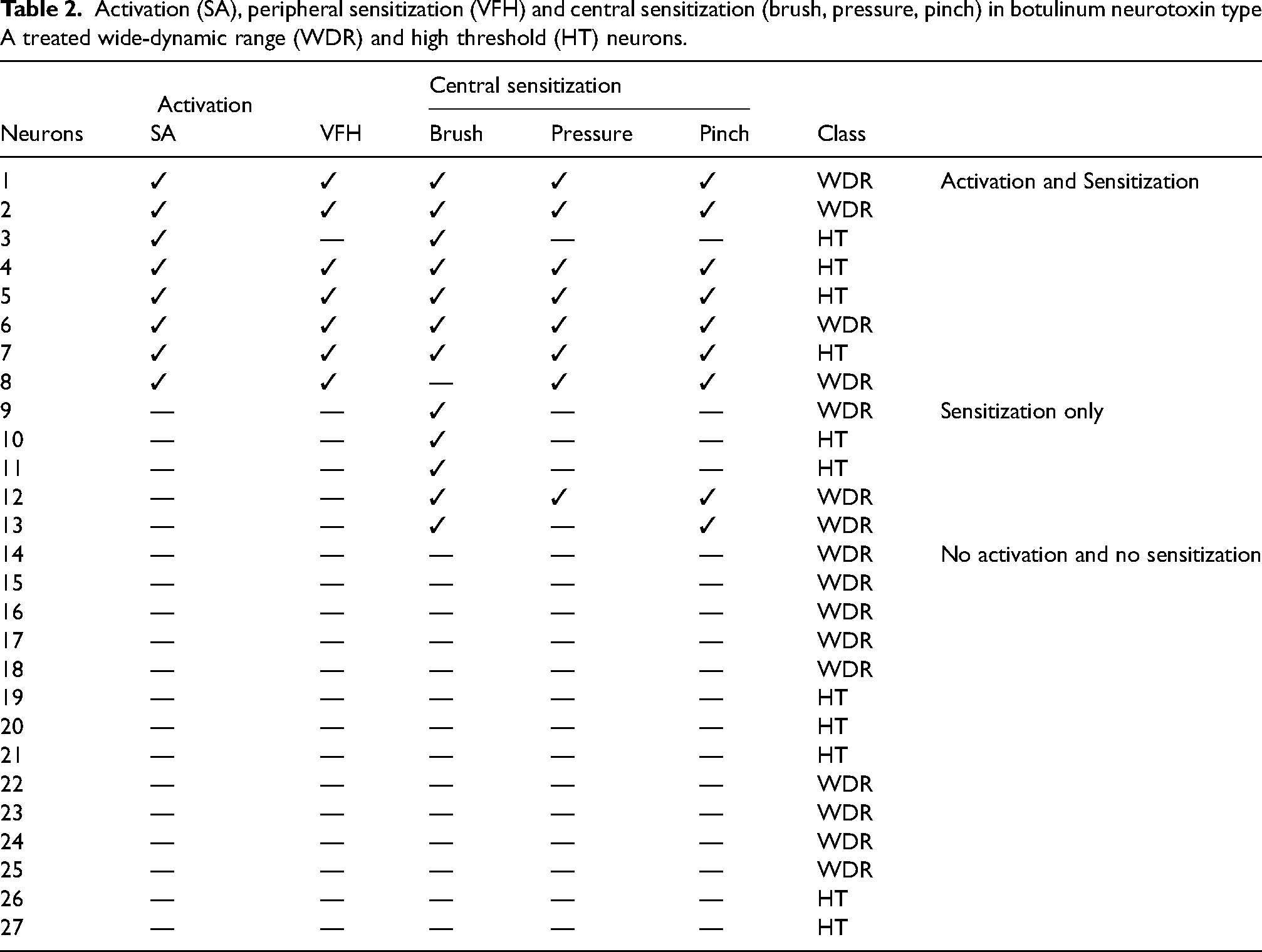
OnabotA effects on activation and sensitization of WDR and HT trigeminovascular neurons.

CSD = cortical spreading depression; HT = high threshold neurons; VFH = von Frey hair; WDR = wide-dynamic range neurons.
Discussion
Using in vivo single unit recording, we demonstrate the ability of onabotA to prevent the development of (central) sensitization in both WDR and HT neurons, and noted that the prevention of their sensitization occurred despite onabotA preventing the activation of WDR but not HT neurons in the SPV. The prevention of activation and sensitization of WDR neurons could be explained by the selective inhibitory effects of onabotA on the unmyelined C-fiber meningeal nociceptors (5,7,27) and their preferential termination on WDR neurons (28,29). By contrast, the ability of onabotA to prevent the development of central sensitization and the consequential attenuation of the response magnitude of both activated and non-activated WDR and HT neurons, may be explained by the possibility that secondary to the inhibition of meningeal C-fibers is a potential decrease in the amount of CGRP release in the SPV during the headache phase of a migraine attack. This explanation is based on C-fibers ability to release CGRP in the SPV (30) and the known role of CGRP in facilitating neuronal sensitization (31,32). Mechanistically, these findings suggest that even partial reduction in the nociceptive signals that originate in the meninges and reach the SPV through the unmyelinated fibers may be sufficient to reduce the overall activity of central dura-sensitive neurons. Clinically, these finding can explain cases in which onabotA reduces the intensity of the remaining headaches (33–35) (commonly described by both responders and non-responders). It is also worth noting that all onabotA non-responsive neurons exhibited central sensitization (Table 2) and that their sensitization was not necessarily related to their activation. This observation raises the possibility that onabotA non-responders are more likely to be those exhibiting cutaneous allodynia at the ictal as well as interictal phase. However, the translatability of these observations to clinical practice cannot be predicted at this time because of the absence of quantitative controlled data that measure changes in skin sensitivity in responders and non-responders during ictal and interictal phases.
WDR vs. HT
The present study demonstrates the ability of onabotA to prevent the development of (central) sensitization in both WDR and HT neurons, and that the prevention of their sensitization occurred despite onabotA preventing the activation of WDR but not HT neurons in the SPV. The preferential inhibition of WDR neurons by onabotA may be explained by three pre-clinical and four clinical studies showing inhibitory effects of this neurotoxin on the unmyelinated C-type meningeal nociceptors, but not the thinly myelinated Ad fibers. The first study showed that direct administration of onabotA to the dura inhibited naïve/non-sensitized C-fiber (but not Aδ) responses to mechanical indentation of the dura, reversed inflammatory soup (IS)-induced mechanical hypersensitivity and prevented the development of IS-induced peripheral sensitization if given hours before establishment of peripheral sensitization (6). The second study showed that, seven days after onabotA injections along suture lines and in pericranial muscles, unmyelinated C-type meningeal nociceptors (but not Aδ) lose their ability to respond to stimulation of their dural receptive fields with TRPV1 and the TRPA1 agonists (7). The third study showed that one to three hours after induction of a single wave of CSD in animals treated with onabotA seven days earlier, the level of activation of C-type meningeal nociceptors was significantly lower than in the untreated rats (5). The four corroborating clinical studies showed that onabotA mitigated capsaicin-induced pain and neurogenic vasodilation without altering thermal and pain perception, comprising a set of findings attributed to the selective inhibition of C-type nociceptors and their TRPV1 receptors (9–12). Functional and anatomical evidence for preferential innervation of WDR neurons by C-fibers and HT neurons by Aδ-fibers (28,29) defines the structural-functional ballpark used in the development of the presented reasoning.
Central to the concept that extracranial injections of onabotA attenuate activation of intracranial meningeal nociceptors are three anatomical studies demonstrating the existence of C2 DRG axons that branch from the extracranial occipital nerve and reach the intracranial meninges after crossing the bones of the calvaria through five different pathways (36) and intracranial meningeal nociceptors that issue collateral branches that cross the calvarial sutures and reach the extracranial periosteum and pericranial muscles (4,6,22,23).
Response magnitude
Analysis of response magnitude (comparing firing rate before and after CSD) revealed that the overall firing frequency of activated and non-activated WDR and HT neurons was reduced significantly in the onabotA treated animals. This finding has scientific and clinical implications. Scientifically, these data provide an independent verification for our assertion that onabotA inhibits WDR neurons to a far greater extent than it inhibits HT neurons. Mechanistically, it is likely that the greater inhibition of the WDR neurons is sufficient to reduce the firing frequency to an extent that eliminated their activation, whereas the less extensive inhibition of the HT neurons is sufficient for reduction in overall firing but insufficient for complete elimination of activation. Clinically, it is reasonable to suggest that the reduction in response magnitude in both WDR and HT neurons can explain the well-described effect of onabotA on reducing headache intensity in both responders (patients achieving at least a 50% or greater reduction in monthly migraine days (MMDs) as well as non-responders (patients achieving less than a 50% reduction in MMDs) (30–32). Often, this reduction leads patients to report that the treatment is effective for them even if their MMDs remain unchanged.
Activation vs. sensitization
Our analysis of the relationship between activation and sensitization revealed that, in the absence of treatment, 58% of the neurons got activated and sensitized, whereas, in the treatment group, only 30% of neurons were activated and sensitized. As proposed previously, in animal models of migraine, activation and sensitization state of central trigeminovascular neurons represents a state of ictal cephalic allodynia (37–39). Along this line, it is also worth noting that, in the treatment group, there was complete elimination of activation in non-sensitized neurons. Although these findings raise the possibility that onabotA therapy may be more effective in patients for whom migraine attacks are not accompanied by cephalic allodynia, in the absence of well-controlled studies (i.e. studies using quantitative sensory testing rather than the ASC-12), this is a speculation that lacks clinical correlates.
Although central sensitization can be expressed as allodynia only, hyperalgesia only, or both, the mechanistic distinction between them is based on the principle that, in the hyperalgesic state, sensitized neurons respond more robustly to stimuli that were painful even before they were sensitized, whereas, in the allodynic state, these sensitized neurons begin to respond to innocuous stimuli to which they did not respond in their non-sensitized state. The finding that the HT neurons exhibited signs of sensitization to brush but not pressure or pinch further supports the scientific rationale that underlies interictal allodynia (rather than hyperalgesia).
Male vs. female
In the present study, we sampled neurons from male and female rats and found that onabotA treatment was similarly effective in preventing activation and sensitization of WDR neurons and in attenuating the response magnitude in both activated and non-activated WDR and HT neurons. Although the onabotA treatment was effective in both males and females, we noted a higher variability in the female group that suggests potential sex-related differences warranting further investigation (because this variability was not statistically significant, we combined data from both sexes in our overall analysis). Our pre-clinical results may be used to explain a recent clinical study showing that the response to onabotA is significant in both men and women (50% responders’ rates are 27.7, 29.2 and 35.6 in cycles 1, 2 and 3 in men vs. 26.6, 33.5 and 41.0 in cycles 1, 2 and 3 in women), and that, although these response rates are not statistically different, women may have a slightly favorable outcome (40).
Caveat
An important consideration in this study, which applies broadly to research involving the meningeal sensory pathway, is the potential confounding effect of surgical intervention. The necessary surgical exposure of the dural receptive fields of neurons can cause some inevitable disturbance to the dura and damage to the overlying extracranial tissues. Such interventions may result in a certain degree of sensitization of the nociceptive innervation. Although this is a common issue across electrophysiological studies of nociceptive sensory pathways, it remains a limitation that must be acknowledged. To mitigate the degree of sensitization in our studies, care was taken to minimize the degree of sensitization, including the subcutaneous injection of lidocaine at the incision site. One indication of the degree of sensitization is the level of baseline spontaneous activity, which generally is fairly low in our studies (and in many cases is 0), suggesting no sensitization at all. In considering possible implications of our data to migraine patients, one must keep in mind the fundamental difference between preclinical studies in anesthetized animals and treatment effects in awake patients.
Conclusions
The ability of onabotA to prevent activation of WDR (but not HT neurons) and sensitization of both WDR and HT neurons may explain its ability to prevent the increase in migraine frequency over treatment duration (i.e. prevent disease progression). The ability of onabotA to attenuate response magnitude in all neurons may explain its ability to reduce the overall intensity of the remaining attacks.
Clinical implications
OnabotA prevents activation of WDR but not HT neurons, and sensitization of both WDR and HT neurons. The ability of onabotA to prevent the development of central sensitization calls attention to the possibility that its most significant role in migraine treatment may be the prevention of a further increase in monthly migraine days. OnabotA attenuates the response magnitude in all neurons. This finding may explain its ability to reduce the overall intensity of the remaining attacks.
Footnotes
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RB is the John Hedley-Whyte Professor of Anesthesia and Neuroscience at the Beth Israel Deaconess Medical Center and Harvard Medical School. He has received research support from the NIH: R01 NS094198-01A1, R37 NS079678, R01NS095655, R01 NS104296, R21 NS106345, Allergan, Teva, Dr Ready, Eli Lilly, Trigemina and the Migraine Research Foundation. He is a reviewer for NINDS, holds stock options in AllayLamp and Percept; serves as consultant, advisory board member, or has received honoraria from: Alder, Allergan, Biohaven, Dr Reddy's Laboratory, Eli Lilly, GlaxoSmithKline, Merck, Teva and Trigemina. CME fees from Healthlogix, Medlogix, WebMD/Medscape, and Patents 9061025, 11732265.1, 10806890, US2021-0015908, WO21007165, US2021-0128724, WO210054
RBr, BD, AMA and MFB are employees of Allergan, an AbbVie Company.
The other authors declare that they have no conflicts of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: this work was supported by the AbbVie, National Institute of Neurological Disorders and Stroke, (grant number R37-NS079678, RO1 NS069847, RO1 NS094198 ).