
Abstract
Background
Persistent headache attributed to traumatic injury to the head is divided into two subtypes, one attributed to moderate or severe traumatic injury and another attributed to mild traumatic injury (i.e., concussion). The latter is much more prevalent, in part because more than 90% of cases with traumatic brain injury are classified as mild. The pathophysiology of persistent post-traumatic headache is poorly understood and the underlying mechanisms are likely multifactorial. There is currently no approved treatment specifically for persistent post-traumatic headache, and management strategies rely on medications used for migraine or tension-type headache. Therefore, high-quality trials are urgently needed to support clinical decision-making and optimize management strategies. International guidelines can facilitate appropriate trial design and ensure the acquisition of high-quality data evaluating the efficacy, tolerability, and safety of available and novel pharmacological therapies for the preventive treatment of persistent post-traumatic headache.
Methods
The development of this guideline was based on a literature review of available studies in MEDLINE, Embase, and the Cochrane Central Register of Controlled Trials, along with a review of previously published guidelines for controlled trials of preventive treatment for episodic and chronic migraine. The identified literature was critically appraised, and due to the scarcity of scientific evidence, recommendations were primarily based on the consensus of experts in the field.
Objective
To provide guidelines for designing state-of-the-art controlled clinical trials aimed at evaluating the effectiveness of preventive treatments for persistent post-traumatic headache attributed to mild traumatic brain injury.
Introduction
The Clinical Trials Committee of the International Headache Society (IHS) has actively developed and published guidelines for controlled clinical trials aimed at treating headache disorders since 1991 (1). Over the years, the IHS has been continuously updating and releasing new guidelines. These include guidelines on acute and preventive treatment of migraine in adults, preventive treatment for children and adolescents with episodic migraine, and trials using neuromodulation devices for the treatment of migraine (2–6). Until now, with the exception of medication-overuse headache (MOH) in the setting of chronic migraine, guidelines for controlled clinical trials of secondary headaches have not been available (4).
In the context of the treatment of persistent post-traumatic headache (PPTH), often involving the utilization of medications used for migraine or tension-type headache due to the absence of approved PPTH treatments, this guideline presents recommendations for trials exploring pharmacological preventive interventions for PPTH linked to mild traumatic brain injury (TBI), commonly referred to as concussion. Of note, our guidelines pertain exclusively to the preventive pharmacological treatment of individuals with manifest PPTH after mild TBI (i.e., once PPTH has developed). Separate guidelines will need to be developed for clinical trials that aim to study the effects of early therapeutic intervention in preventing the transition from acute post-traumatic headache to PPTH.
It is imperative to highlight that this guideline does not specifically address the issue of MOH or any other secondary headache disorder. Such matters lie beyond the scope of this guideline, which is primarily centered on controlled trials for PPTH, but inclusion or exclusion of comorbid secondary headache disorders should always be taken into account when performing PPTH trials, as with other trials on any primary or secondary headache.
As trial designs have evolved, so have data collection methods, incorporating advancements like the integration of electronic diaries. There is a growing emphasis on patient-reported outcomes and the cost-effectiveness of novel treatments. Stakeholders utilize these factors to inform decisions regarding reimbursement for emerging indications and therapies. Building upon innovative strategies and drawing from the insights gained from guidelines for primary headaches, this guideline strives to enhance the quality of forthcoming preventive treatment trials for individuals with PPTH attributed to mild TBI, effectively addressing unmet therapeutic needs.
A point that merits emphasis is the current scarcity of data from controlled trials. To our knowledge, the available data is limited to two small, controlled trials of preventive treatment for PPTH attributed to mild TBI (7,8). This underscores the substantial unmet needs faced by these patients. In response, our approach leverages the positive experiences gained from crafting guidelines for migraine management. Furthermore, in instances where empirical evidence is absent, we adopt a consensus-based methodology, utilizing an iterative process that involves circulating subsequent versions for comment and refinement. This approach aims to judiciously integrate the absence of evidence and ensure a comprehensive framework for guiding clinical trial design decisions.
1. Selection of participants
1.1 Definition of PPTH
Recommendation
Persistent headache attributed to traumatic injury to the head (i.e., PPTH) can be divided into two groups: persistent headache attributed to moderate or severe traumatic injury to the head and persistent headache attributed to mild traumatic injury to the head. The latter is much more prevalent, with over 90% of TBI cases classified as mild (9). To diagnose PPTH attributed to mild TBI, clinicians should adhere to the latest edition of the International Classification of Headache Disorders (ICHD) and fulfill the criteria for persistent headache attributed to mild traumatic injury to the head as specified in the ICHD (10). Inclusion of subjects with more than one episode of mild TBI should be decided on a case-by-case basis. If there is an episode of mild TBI not followed by headache and then a subsequent TBI followed by PPTH such an individual can be included. If an episode of mild TBI is followed by PPTH and then a second head injury occurs with exacerbation of PPTH before or during the trial, we recommend exclusion to reduce heterogeneity. Conversely, we recommend permitting inclusion of individuals who report no exacerbation of PPTH after a second head injury before or during the trial.
Comments
Individuals with PPTH attributed to mild TBI are eligible for a preventive treatment trial if they report at least four monthly headache days with moderate-to-severe pain intensity during the month prior to enrollment. It is important to mention that controlled trials can adopt specific frequency intervals as an inclusion criterion to ensure a more homogenous study population in order to facilitate accurate outcome assessment. For example, eligible individuals with PPTH attributed to mild TBI might be required to report eight to 14 monthly headache days with moderate-to-severe pain intensity in one trial and 20–28 days in another. Participants diagnosed with moderate to severe TBI, whiplash injury, or craniotomy must be excluded from clinical trials investigating the treatment of PPTH attributed to mild TBI. The inclusion of individuals with more than one episode of mild TBI should be determined on a case-by-case basis. If a patient has experienced an episode of mild TBI without subsequent headache, followed by a subsequent TBI with PPTH, they can be eligible for inclusion in the trial. However, if an episode of mild TBI is followed by PPTH and then a second TBI occurs, exacerbating the PPTH, we recommend exclusion to reduce heterogeneity. In the context of PPTH, the ICHD-3 criteria do not differentiate between specific headache phenotypes, such as migraine-like or tension-type headache-like. This might nonetheless be relevant from both clinical and therapeutic standpoints. Investigators can therefore opt to focus on specific phenotypes in their clinical trials. However, it is important to consider the complexities in defining a migraine-like phenotype, which appears to be the most frequent one in clinical samples. The presence of sensory hypersensitivities, such as photophobia and phonophobia, can occur independently of headache after mild TBI and might confound phenotype-based subject stratification. This observation necessitates a nuanced approach in assigning specific headache phenotypes, particularly in clinical trials, to ensure the validity and applicability of the findings.
1.2 Other headache types
Recommendations
Individuals with pre-existing primary headache disorders are eligible for enrollment if their headache has significantly worsened in close temporal relation to the TBI, such as a two-fold or greater increase in headache frequency and/or intensity, or if they develop headaches of a new phenotype following the mild TBI. Individuals with pre-existing chronic migraine, chronic tension-type headache, hemiplegic migraine, migraine with brainstem aura, trigeminal autonomic cephalalgias, and cranial neuralgias should not be included in the trial. Individuals with a current diagnosis of a secondary headache disorder other than PPTH should not be included in the trial, except for those with MOH, which should be diagnosed upon enrollment.
1.3 Duration of disease
Recommendations
To be considered eligible for study enrollment, an individual should have experienced post-traumatic headache for at least three months. This recommendation is based on the ICHD-3 criteria where PPTH is diagnosed when a headache persists for more than three months after the onset of acute post-traumatic headache. The onset of acute PTH and the date of mild TBI should be documented and included in the study. The duration of PPTH should be established using appropriate data sources, such as medical records or subject self-report collected during a clinical interview by the site investigator.
1.4 Duration of screening and baseline phase
Recommendations
Controlled trials investigating the efficacy of preventive treatments for PPTH must include a four-week baseline phase, which can begin as early as 13 to 16 weeks after the onset of acute PTH. The baseline phase is used for determining continued eligibility and prospective recording of baseline variables, providing a basis for measuring change subsequent to treatment. Participants should ideally record outcome data in an electronic diary.
Comments
To avoid confusion caused by variations in the length of a calendar month, we recommend using the term “four weeks” instead of “one month”. This corresponds to a precise duration of 28 days. A prospective baseline phase of four weeks is necessary to establish the baseline frequency of headache days and to classify each headache day based on pain intensity (mild/moderate/severe). This is to ensure that the threshold number of four headache days per 28 days of moderate to severe pain intensity is met. Using a diary during the baseline phase is also important for assessing headache characteristics such as pain quality, intensity, and relationship with routine physical activity. In addition, the presence of aura symptoms, use of acute headache medication, and subject compliance of at least 80% per four-week period with the diary should also be recorded.
1.5 Age at entry
Recommendation
The minimum age for participation in clinical trials should be six years of age, and appropriate age strata must be defined for trials in children, adolescents, and adults (≥18 years), respectively.
Comments
Regulatory agencies have specific requirements for conducting trials in children and adolescents to ensure the safety, tolerability, and efficacy of the investigational product. In some countries, data from participants over 65 years of age must be collected and analyzed separately. Elderly subjects should only be excluded if there is a safety concern. Children should be defined as participants aged six to 11 years, and adolescents as participants aged 12 to 17 years. Regulatory agencies might require separate trials for these age groups (5,11). It is essential to consider the appropriate age strata to obtain meaningful results from clinical trials.
1.6 Sex
Recommendation
Controlled trials must aim to include both male and female participants to improve the generalizability of the study findings. However, in some cases, trials can be restricted to a single sex, such as in studies conducted within a specific population, like military service members. In such cases, it is important to acknowledge that the generalizability of the findings might be limited due to the restricted sample.
Comments
The sex ratio of PPTH is not well-established and can vary between study populations, such as civilians versus military populations. If assessing treatment benefits in military populations is of interest, it is important to ensure that the enrolled sample reflects the sex ratio among military service members. In female adults, it is important to collect information on their reproductive life stages such as fertile, premenopausal, and postmenopausal stages. Furthermore, it is important to take appropriate precautions to exclude women of childbearing potential unless they are using adequate contraception, especially when the treatment has known or potential obstetrical risks. Pregnant and lactating women must be excluded from trials of treatments with potential toxicity to the infant or unknown potential for toxicity. All nonsterile women must agree to use appropriate contraceptive measures throughout the trial. Results of studies should provide separate data on females and males, which can be provided in the supplementary appendix. If the power is adequate, planned post-hoc analyses should be conducted to identify differences in treatment effects by biological sex.
1.7 Enrollment
Recommendation
Participants must fulfill all the predefined inclusion criteria and should not meet any of the predefined exclusion criteria. This information must be documented at the time of enrollment and again at randomization to ensure that the participants are eligible to participate in the study. Before enrollment, participants must receive a clear explanation of the purpose of the trial, their role in the study, and the potential risks and benefits of participation. The information provided must be formulated in a way that does not exaggerate placebo or nocebo responses (12). According to the Good Clinical Practice Guideline (13), the participant’s informed consent must include an explanation of how the data will be used, as well as their rights concerning data privacy and exiting the study. Participants with a known allergy or hypersensitivity to compounds similar to the trial drug, including excipients, must be excluded from the study.
Comment
Recommended data collection elements.

1.8 Concomitant disorders
Recommendations
Prior to enrollment, participants must undergo a comprehensive medical evaluation to identify any concomitant medical conditions, including severe psychiatric disorders, that could affect participation in the trial or the interpretation of its results. Depending on the research question and the nature of the medical condition, its presence may justify exclusion due to the potential for exacerbation or confounding of the results or preventing adherence to the trial obligations (14). Participants with comorbid TBI-related sequelae such as major depressive disorder or post-traumatic stress disorder can be included in PPTH trials if they have been prospectively identified and are on a stable treatment regimen (or no treatment regimen) for at least three months, with no anticipated changes during the study. It is important to specify and preferably quantify these comorbid conditions using specific and validated questionnaires. If these conditions are expected to influence treatment outcomes, exclusion of rare disorders and stratified randomization for more common disorders should be considered. Alternatively, planned post-hoc analysis of effect modification by treatment is an option. The effect of treatment on common comorbid conditions can be studied as secondary outcomes, moderators, or mediators of treatment effects. However, individuals at high risk for suicide and those with alcohol or illicit drug use disorders should be excluded from participation.
Comments
TBI-related sequelae are common in people with PPTH and include anxiety, depression, cognitive impairment, PTSD, and sleep disturbances (15). To ensure the accuracy of the trial’s results, it is necessary to assess the presence, characteristics, and treatment of TBI-related sequelae before including participants reporting them in the trial. In cases where the treatment of comorbid or concomitant illnesses can interfere with the preventive treatment of PPTH, exclusion of these participants might be necessary.
1.9 Concomitant drug use
Recommendations
Monotherapy studies are essential to establish the safety, tolerability, and efficacy of novel preventive therapies in Phase 2 clinical trials. In Phase 3 trials, subjects may be permitted to take one concomitant headache preventive medication during treatment with the study drug if the other conditions specified below are met. Concurrent administration of preventive medication with a drug from the same class as the study drug should be avoided. The dose of the non-study medication should remain stable for at least two months before randomization and should not be altered during the trial (16). Investigators should note that medications well-documented as preventive headache treatments, but prescribed for other indications, must also be considered as preventive headache medications if they are used in therapeutic doses for the treatment of headache. If participants taking concomitant preventive medication are enrolled, stratified randomization should be considered to ensure that treatment groups are balanced in terms of concomitant medication use (see Section 2.5). In Phase 4 trials, concurrent medications for the same or other indications can be permitted as long as participants are on a stable regimen during the study.
Comments
The trial protocol should clearly specify any concomitant medications that are allowed or prohibited at the time of enrollment and/or during the trial. Any concomitant treatments used during the trial must be documented, including the indication for use, dosing, and duration of treatment.
1.10 Subjects from previous headache trials
Recommendations
Participants are not permitted to participate in more than one clinical trial simultaneously. However, if a trial extension, such as an open-label phase to assess long-term safety, is offered as part of the original trial protocol, it should be considered as part of the same trial and not a separate trial. Participants should not enroll in more than one trial that evaluates the same preventive treatment. However, it is possible for participants to concurrently participate in non-interventional studies and prospective registries.
1.11 Data collection and monitoring
Recommendations
To ensure the prospective collection of trial data, it is strongly recommended to use an electronic diary that is capable of time stamps, remote monitoring, and alerts. While paper diaries are less desirable, they can be used if electronic diaries are not available. Participants must be instructed to record specific headache characteristics, such as pain quality, intensity, accompanying symptoms, and relationship with routine physical activity. Additionally, they must record the presence of aura, use of acute headache medication, and compliance with treatment. Adverse events (AEs) should be recorded in real-time in the diary by the patient according to regulatory guidance. The characteristics of the AEs and their relationship with the treatment under investigation should be ascertained during follow-up visits or phone calls. The data collected on AEs should include a list of specific side effects and open-ended questions. Any serious AEs must be reported within 24 hours of their occurrence. Participants who have incomplete diaries during the baseline or treatment phases (e.g., <23 days of diary data within 28 days) should be excluded from the trial. To handle the issue of missing data in clinical trials, investigators must develop a pre-planned missing data strategy. This should be an integral part of the trial design and detailed in the study protocol.
Comments
To ensure accurate patient data collection, it is essential to balance the need for information with the response burden placed on patients when entering data into their diary. When conducting international trials, standardized diary designs should be used, with translations adapted to the linguistic and sociodemographic characteristics of the targeted populations.
1.12 Response to previous treatments
Individuals who have previously failed preventive headache treatments can be included in clinical trials of PPTH.
Treatment failure is defined as any of the following: 1) No meaningful reduction in headache frequency, duration, and/or severity after an adequate trial of medication, usually for two-three months for oral drugs and depending on injection intervals for subcutaneously- or intravenously-administered drugs used for headache prevention; 2) Intolerable adverse events; or 3) Contraindications precluding use.
It is important to document the type(s) of treatment(s) that previously failed or were not well tolerated.
Comments
To document previous failures of preventive medication, it is generally recommended to use the participant’s medical record, which should include the medication name, treatment duration, dose level, and reason for discontinuation. This information should be verified for completeness and accuracy with the participant. However, if the medical record is unavailable, the investigator can obtain this information from the participant as an alternative method, although this is less desirable.
2. Trial design
2.1 Blinding
Recommendation
Double-blind controlled trials are essential to establish the efficacy, tolerability, and safety of a preventive treatment for PPTH.
Comments
Blinding of both participants and investigators/site personnel is essential to minimize bias and the effects of placebo in clinical trials evaluating preventive treatments for PPTH. Unblinding due to adverse events can be a significant factor in placebo-controlled trials (see Section 2.2). At the end of the trial, participants and investigators can be asked to guess whether the participant was assigned to active treatment or placebo, and these data should be recorded to confirm the success of blinding. The success of blinding can be evaluated using the Bang Index (17).
2.2 Placebo control and comparative studies
Recommendations
Placebo control is recommended for comparing treatments used for the prevention of PPTH. When comparing two presumably active drugs, a placebo control is strongly recommended to provide a measure of assay sensitivity, if appropriate. At present, there is no standard-of-care drug for PPTH with proven efficacy. As standard treatments become available, comparative effectiveness studies are recommended.
Comments
The extent of the placebo effect associated with preventive medications for PPTH is not well-established. However, based on insights from clinical trials in patients with migraine, higher placebo response rates are likely to be observed when the treatment is administered parenterally rather than orally, and when there is unbalanced randomization with more participants allocated to active treatment than to placebo (16,18). To establish the efficacy of an active treatment for PPTH, it must demonstrate superiority to placebo. A trial that shows no significant difference between two active treatments does not prove the efficacy of either treatment. It is essential to include a placebo control to demonstrate that the active treatment is superior to no treatment at all.
2.3 Parallel-group and crossover design
Recommendations
Parallel-group designs are preferred over crossover designs due to their ability to avoid carryover effects. In crossover studies, it is important to ensure that an adequate assessment of the effects in each treatment phase is achieved. Ideally, headache frequency should return to baseline levels before beginning a new treatment phase. However, this can be challenging to achieve within reasonable timeframes, especially when treating secondary headaches such as PPTH. To offset this challenge, counterbalancing and analysis of differences from baseline can be used to obtain more reliable results.
Comments
Crossover designs have significant disadvantages that should be considered when designing a study. These include fluctuations in treatment effects over time, carry-over effects, which cannot be controlled with certainty even with washout periods, and the need for a longer study duration. The longer duration increases the likelihood of withdrawals and protocol deviations, as well as spontaneous changes in headache frequency, which can impact the results. Therefore, parallel-group designs are recommended over crossover designs to minimize these potential sources of bias.
2.4 Run-in
Recommendations
To ensure that participants meet the diagnostic criteria for PPTH and collect other essential information, including headache frequency and duration, headache characteristics and associated symptoms, impact on functional ability and work, and use of acute (pain) medication(s), a prospective four-week baseline phase is recommended. Electronic diaries that include time stamps to reduce recall bias and allow remote monitoring of data entered by subjects are the optimal way to capture data. To effectively reduce recall bias, it is advisable to set a 24-hour window for diary entry, balancing the need for timely data capture with practical considerations of subject adherence. If electronic diaries are not available, paper diaries can be used (see Section 1.12).
Comments
The baseline phase serves multiple purposes, including confirming subjects’ eligibility for the study, assessing adherence to data collection procedures, and collecting pre-treatment data to calculate outcome measures (19,20). The primary efficacy endpoint in trials of preventive treatments for PPTH is typically the change from baseline in moderate-to-severe headache days; therefore, the accuracy of measurements during the baseline phase directly impacts study results. A minimum of four weeks is recommended for the baseline phase, although eight weeks can be used. Since headache frequency can vary weekly and monthly in individuals with PPTH (21,22), longer baseline periods might provide more accurate assessments of baseline status. However, a long baseline phase can complicate enrollment, increase pre-randomization dropout rates, and delay treatment for patients with unmet treatment needs. Highly variable baseline frequency estimates can reduce the statistical power of the primary efficacy analysis, but this can be minimized by carefully considering inclusion and exclusion criteria during screening. One approach is to exclude participants with atypical or highly variable headache patterns that might introduce inappropriate variance. However, such criteria should not be so restrictive as to exclude a considerable proportion of the PPTH population, thereby limiting the generalizability of the results. Setting reasonable and clinically relevant threshold for headache frequency and severity might help achieve this balance.
2.5 Randomization
Recommendations
To ensure unbiased assignment of participants to treatment groups, controlled trials necessitate randomization of participants, preferably in relatively small blocks, after the run-in period (23). The process for randomization should be well-defined and appropriately documented to reduce the potential for errors or bias. This might include the use of centralized randomization systems or computer-generated randomization lists. In parallel-group trials, stratified randomization is recommended to ensure that important confounding variables, such as monthly headache day frequency category, comorbidities, or current use of concomitant preventive medications, are balanced across treatment groups. Stratification variables should be clearly defined and chosen based on clinical and statistical relevance.
Comments
Participants in trials of preventive treatment for PPTH are typically recruited over extended periods. To ensure balanced randomization across treatment groups, it is preferable to randomize subjects in relatively small blocks of varying sizes, such as four-eight or four-ten (23). This approach reduces the risk of imbalanced group allocation due to chance, helps to ensure that the study groups are well-matched, and reduces the potential for bias in the trial results. Randomization alone is not sufficient to ensure that treatment groups are balanced for factors that may impact treatment response, especially for uncommon confounders or modest sample sizes. As the sample size increases, randomization becomes increasingly effective in balancing the treatment groups for potential confounders. There are two methods to address the issue of imbalance in randomization: statistical adjustments in analysis and stratified randomization. Statistical adjustments in analysis are commonly used and involve incorporating potential confounders into planned statistical analyses. On the other hand, stratified randomization is used to assign participants to treatment groups based on the confounder and ensures group balance. Stratified randomization may be appropriate for known confounders that are easily measurable at baseline, such as the number of prior preventive medications. However, the number of stratification factors should be limited by the sample size. Candidate variables for stratification may include important comorbidities (e.g., PTSD), time since the inciting TBI (to control for spontaneous remissions), among others.
2.6 Duration of treatment phases
Recommendations
The duration of the treatment phase is determined, in part, by the time required for full treatment effects to become apparent. It is generally recommended to have a double-blind treatment phase lasting at least 12 weeks to allow for the evaluation of treatment efficacy. A 24-week double-blind treatment phase may be considered to assess the cumulative benefit and persistence of efficacy, as well as to further analyze the safety and tolerability of the treatment. In Phase 3 trials, a long-term open-label phase of at least three months following the double-blind treatment phase is useful for collecting additional information on the persistence of treatment effects and the delayed emergence of treatment effects, as well as assessing long-term tolerability and safety.
Comments
Prolonging the duration of the double-blind treatment phase can enhance the trial’s statistical power by allowing sufficient time for delayed treatment benefits to fully emerge. Certain preventive medications require gradual dose titration and adjustment before an optimal dosage is attained. Subsequently, treatment effects may accrue gradually, especially for orally administered medications, and some drugs may require several weeks or longer at an optimal dosage before reaching their full preventive potential. A longer double-blind phase may result in higher drop-out rates, especially in the placebo arm, and delay access to effective treatment. After completing the placebo-controlled phase, a long-term observation phase could be useful to identify additional adverse events (AEs), assess the cumulative benefits of treatment, and evaluate the maintenance of treatment effects. For trials evaluating preventive treatments that have not been approved, the IHS recommends an open-label, long-term extension study to provide participants who received placebo in the controlled trial with access to the investigational therapy while collecting useful information about tolerability, safety, and adherence to treatment. In the open-label phase, re-randomization of participants to one of two active dose regimens is a viable approach to assess dose-dependent AEs (24).
2.7 Post-treatment phase
Recommendations
Following the completion of a randomized treatment or open-label extension phase, it is important to prospectively monitor subjects for safety evaluations. The duration of follow-up should be determined based on the treatment being investigated and any potential long-term effects. To ensure accurate monitoring during this period, it is recommended that subjects continue to maintain a daily diary, including any usage of other preventive medications.
2.8 Dosage
Recommendations
In Phase 2 trials, it is important to explore a broad range of dosages, including both the minimally effective and maximally tolerated doses, to identify the optimal dose range for further study. Phase 3 trials typically involve testing one or two doses, as these have been identified as the most promising doses from the Phase 2 trials. Investigating more than two doses in Phase 3 trials can increase the complexity of the study design, the number of treatment arms, and the potential for placebo effects. Therefore, careful consideration should be given to the number of doses tested and the balance between scientific rigor and practical feasibility.
Comments
The selection of dosage and/or intensity of intervention for a preventive treatment depends on available preliminary evidence on the tolerability and pharmacological characteristics of the compound, particularly when the basis of efficacy is unknown. When the mechanism of action is known, dose-response curves from in vitro or in vivo models can provide information on the range of doses to be tested.
2.9 Use of acute and preventive treatments
Recommendations
During the trial, acute treatment of PPTH should be permitted, provided that the agent and dosage remain consistent throughout the baseline phase and for the entire duration of the trial, and intake is recorded in the diary. Concomitant preventive headache treatments that could affect the trial outcomes should not be initiated or discontinued during the double-blind study period, except for safety reasons or intolerable side effects. If concomitant preventive medication is permitted, the preventive medication and its dosage should be stable for at least two months before the start of the baseline run-in phase. Examples of these medications include drugs approved for the preventive treatment of headache disorders, such as migraine and tension-type headache. Moreover, it is worth noting that dual-purpose medications, such as rimegepant, present a unique challenge, since they can be used as both acute and preventive headache treatments. It is therefore recommended that rimegepant be considered only as an acute treatment option when its use is limited to no more than eight days per month. This arbitrary restriction is to reduce the likelihood of rimegepant serving as a concomitant preventive treatment.
Comments
It is important to allow participants to use acute headache medication during the trial. However, during the baseline phase, it is important to instruct participants not to change the type, dosage, or formulation of their acute medication, or the manner in which it is taken (such as during mild pain versus moderate-to-severe pain). Standardization of instruction on acute medication usage across treatment centers is important to avoid confounding the interpretation of study results.
2.10 Study visits
Recommendations
Regular follow-up of participants is essential throughout the trial. Study visits should be scheduled at screening, at the beginning and end of the baseline phase, and after randomization/initiation of treatment to monitor safety, efficacy, and treatment compliance. During the treatment phase, frequent site visits or remote follow-up visits are necessary, typically every four to eight weeks. The frequency of these visits may be adjusted depending on the nature of the treatment being tested, the expected AEs, and the duration of the trial. To improve adherence and reduce the burden of in-person visits, remote monitoring methods should be encouraged. Telephone, messaging (email or text), and video conferencing can be used for this purpose.
Comments
Regular communication with participants in clinical trials is essential to determine their eligibility, ensure adherence to the study protocol, and monitor for AEs. This contact includes both in-person visits and remote monitoring methods.
3. Evaluation of endpoints
Recommendations
All primary and secondary endpoints must be prospectively and operationally defined in a clinical trial. This includes identifying specific comparison groups and time points for each endpoint. These definitions ensure that the trial’s objectives are clear and that the collected data are relevant and reliable for analysis. The selection of endpoints should be based on the study’s objectives. Prior to the initiation of a clinical trial, power and sample size calculations must be performed for the primary and key secondary endpoints.
Comments
Multiple comparison issues can arise when there are multiple primary and key secondary endpoints or when there are three or more treatment groups in a trial. Hierarchical testing procedures can be used to avoid these multiplicity issues in the case of multiple primary endpoints (25). However, if investigators or sponsors choose to use a multiple-comparison adjustment, it is important to consider this adjustment in the sample size calculation and statistical power calculation. This will ensure that the trial is appropriately powered to detect significant differences in all endpoints while controlling for the increased risk of false positive findings due to multiple comparisons.
3.1 Primary endpoints
Recommendations
The primary endpoint for controlled trials of preventive treatment for PPTH should be one of two options. The first option is the change from baseline in the number of moderate-to-severe headache days per unit time, which is typically a four-week period. The second option is the 50% responder rate for the reduction from baseline in the number of moderate-to-severe headache days per unit time, which is also typically measured over a four-week period.
Comments
The time-period selected for assessing the primary endpoint depends on the expected time to achieve maximal treatment effect. For oral agents that require dose titration, the primary endpoint is defined by the change from baseline to weeks 9–12, as this period allows sufficient time for dose escalation and for the treatment to reach its maximum effect. For treatments with rapid onset, the four-week average of moderate-to-severe headache days (MHD) frequency over weeks 1–12 can be selected, as the longer follow-up time provides more stable estimates. In 24-week trials, the recommended period for analysis is the last 12 weeks (i.e., weeks 13–24), which provides a sufficient time frame to assess the long-term effects of the treatment. Alternatively, the results over the entire period can be considered either as a primary or key secondary endpoint. However, evaluations of efficacy should be based on information obtained from electronic diaries, as these provide a more accurate and reliable assessment of headache frequency and severity compared to retrospective paper-based diaries.
Definition of moderate-to-severe headache day
A moderate-to-severe headache day is defined as a day with headache pain of moderate or severe intensity that lasts at least 30 minutes without medication, or a day with headache pain of at least moderate intensity that responds to acute treatment with medication. For migraine, the definition of 30 minutes seems accurate as most patients are instructed not to wait for a full-blown attack before they start treating their attacks (26). However, it is important to note that in some cases, the headache duration required to classify a day as moderate-to-severe might be longer than 30 minutes. For example, if a participant goes to bed with headache and wakes up without symptoms, the headache should be considered to have ended at the onset of sleep. Conversely, if a participant wakes up with a headache that is already in progress, the onset of the headache should be considered to be the time of awakening. These conventions ensure consistent recording of headache days across participants and trials.
Definition of responder rate
The responder rate is a commonly used measure in clinical trials to evaluate the efficacy of a treatment in reducing the frequency of moderate-to-severe headache days. It is defined as the percentage change from baseline in the number of such headache days during each dosing interval. The responder rate can also be defined for all headache days or for days meeting other prespecified criteria, such as mild headache days or moderate-to-severe headache days. In PPTH trials, participants who achieve at least a 50% reduction from baseline in moderate-to-severe headache days are considered responders. Other percent changes from baseline (such as 30%, 75%, or 100%) are not recommended as primary endpoints, but can be considered for secondary endpoints. It is important to prospectively define the responder rate used in controlled trials to ensure consistency and comparability across studies. By using a standardized definition, the results of different trials can be more easily compared and combined in meta-analyses.
Comments
The definitions of moderate-to-severe headache days and responder rates are considered more clinically relevant and easier to explain to patients than reductions in monthly headache days. If 75% of patients achieve at least a 50% reduction in moderate-to-severe headache days, patients can be informed that three of four patients who receive the treatment will reduce their moderate-to-severe headache days by at least 50%. In contrast, change in monthly headache days averages together people who respond well and people who do not respond at all, resulting in numbers that are difficult to interpret. Responder rates reduce this heterogeneity and provide a more straightforward measure of treatment effectiveness. Moreover, responder rates can be determined with a relatively simple headache diary.
3.2 Secondary endpoints
The secondary outcomes listed below are organized by the domains they assess and are not prioritized.
3.2.1 Headache-related
3.2.1.1 Moderate-to-severe headache days
Should be considered as an end point if not selected as the primary endpoint.
3.2.1.2 50% responder rate
Should be considered as an endpoint if not selected as the primary endpoint.
3.2.1.3 Headache days
Defined as the change from baseline in the number of headache days per unit time, which is typically a 4-week period. A headache day is typically defined as a day with headache pain lasting at least 30 minutes without the use of an acute headache medication or not relieved by a previously taken acute headache medication. In cases where investigators wish to include subjects with mild headache, the change in headache days of any intensity should be used as the primary endpoint.
3.2.1.4 Cumulative hours per 28 days of moderate-to-severe pain
Reductions in this endpoint might be clinically meaningful and should be considered. Cumulative hours per 28 days of moderate-to-severe pain is an outcome measure that can be calculated using data from electronic diaries, provided participants log hourly entries. To facilitate this process, the use of electronic diaries or mobile applications is recommended. These tools can simplify the process for participants, encouraging regular and accurate data entry. Additionally, clear instructions and support should be provided to participants to ensure understanding and adherence to the hourly logging requirement. This approach aims to balance the need for detailed and meaningful data with the practical considerations of participant engagement and compliance.
3.2.1.5 Onset of effect
Assessing the onset of action of a preventive treatment is helpful in refining management strategies. In a clinical trial, the onset of effect can be determined by analyzing data from each four-week treatment period. Typically, onset is defined as the time period when statistically significant separation from placebo is achieved.
3.2.2 Acute headache treatments
3.2.2.1 Acute treatment utilization
To ensure accurate evaluation of headache severity and treatment efficacy, it is important to record the use of acute medication, including the specific drug used, number of days, and dose of drugs. It is essential that participants do not receive any special counsel to alter the frequency of acute headache medication use during the treatment phase. This ensures that any changes in medication use, whether an increase or decrease, can be attributed to changes in headache frequency and severity and accurately evaluated.
3.3 Patient-reported outcome measures (PROMs)
Patient-reported outcome measures (PROMs) play a crucial role in evaluating the burden of illness and the benefits of treatment for PPTH. While several PROMs have been included in the label for migraine treatments, they have not yet been deemed fit for purpose for PPTH by regulatory bodies. However, it is recommended to measure PROMs at baseline and at the end of the double-blind treatment period, or more frequently depending on the recall interval for the PROM and study objectives.
Depending on the investigators’ priorities, the following PROMs may be considered:
3.3.1 Patient Global Impression of Change (PGIC)
The Patient Global Impression of Change scale (PGI-C) is a subjective measure that can be used to evaluate subjects’ impression of their clinical status (27). This scale asks participants how they are doing overall at specified post-baseline time points (e.g., four, eight, or 12 weeks) relative to their pre-treatment baseline.
3.3.2 Functional Impairment Scale (FIS)
The Functional Impairment Scale (FIS) is a four-point scale that assesses functional status and the intensity of impairment during daily activities (28). It can be used in conjunction with the four-point pain intensity scale and is usually completed daily and summarized over four-week intervals.
3.3.3 Headache Impact Test (HIT-6)
Headache Impact Test (HIT-6) is a recommended tool for evaluating headache-related disability with a one-month recall period (29). Note that HIT-6 needs to be licensed, and it has been deemed fit for purpose by the European Medicines Agency.
3.3.4 EuroQoL-5 Dimension Questionnaire (EQ-5D)
EuroQoL-5 Dimension Questionnaire (EQ-5D) is a standardized measure of health status that requires registration for use (30,31). Since this measure is obtained on a single day, it is subject to temporal sampling error. If EQ-5D is used, multiple days of administration are recommended, along with recording whether headache was present on the day of administration.
3.3.5 Short Form 36-Item Health Survey (SF-36)
Short Form 36-Item Health Survey (SF-36) is a well-known generic instrument for evaluating quality of life (32). It is useful for determining the magnitude of the quality-of-life burden and the benefits of treatment across diseases.
3.3.6 Pittsburgh Sleep Quality Index (PSQ-I)
Pittsburgh Sleep Quality Index (PSQ-I) is a 19-item self-report instrument that comprises 7 components used to assess the quality of sleep (33).
3.3.7 Rivermead Post-Concussion Symptoms Questionnaire (RPQ)
Rivermead Post-Concussion Symptoms Questionnaire (RPQ) is a 16-item self-report instrument used to screen for post-concussion symptoms, including headache (34).
3.3.8 Quality of Life After Brain Injury-Overall Scale (QoLIBRI-OS)
Quality of Life After Brain Injury-Overall Scale (QoLIBRI-OS) is a 6-item instrument used to assess health-related quality of life in domains that are often affected following brain injury (35).
3.4 Outcomes associated with comorbidities or complications
3.4.1 Depression and anxiety
Depression and anxiety are common comorbidities that should be assessed in clinical trials.
3.4.1.1 Validated scales for depression
Validated scales should be used to measure symptoms suggestive of depression. Examples of validated scales include the Patient Health Questionnaire-9 (PHQ-9), the Hospital Anxiety and Depression Scale (HADS), and the Beck Depression Inventory – Second Edition (BDI-II) (36–38).
3.4.1.2 Validated scales for anxiety
Validated scales should be used to assess symptoms suggestive of anxiety. Examples of validated scales include the HADS, the Generalized Anxiety Disorder (GAD-7), and the State-Trait Anxiety Inventory (STAI) (38,39).
3.4.2 PTSD Checklist for DSM-5
The PTSD Checklist for DSM-5 (PCL-5) is a widely used measure of PTSD symptoms that should be included in clinical trials (40).
3.4.3 Insomnia Severity Index (ISI)
The Insomnia Severity Index (ISI) is a 7-item self-report instrument that should be used to screen for insomnia in clinical trials (41).
3.5 Pharmacoeconomic endpoints
Studies evaluating the economic value of preventive treatment for PPTH should consider both the direct costs of medical treatment and the indirect costs of lost productivity. The reduction in work productivity and activity due to PPTH should be considered as important components of disability and headache-associated costs. The Work Productivity and Activity Impairment (WPAI) instrument can be used to measure the mean change from baseline in work productivity and activity (42).
3.6 Adverse events
Documentation of AEs and serious adverse events (SAEs) during treatment should follow the requirements of local institutional review boards, regulatory authorities, and Good Clinical Practice guidelines.
Acceptable methods for documenting AEs include compiling lists of events, collecting spontaneous reports from study participants, recording observations made during physical exams, using open-ended questions (if the event is not covered by the AE listing), and conducting direct questioning.
AEs should be reported separately for the active treatment and placebo groups to allow for appropriate comparisons.
Comments
Adverse events are a common occurrence during preventive treatment, and they can occur before maximum efficacy is reached. Therefore, the incidence of AEs, particularly those that lead to subject discontinuation from a trial, should be considered a critical measure of the tolerability of preventive treatments for PPTH. Adverse events may not always be related to the treatment. However, during the development of a new treatment, it is essential to record AEs to detect any unexpected and unwanted effects. Investigators need to assess the likelihood of AEs being treatment related. It should be noted that regulatory authorities mandate detailed reporting of AEs using the Medical Dictionary for Regulatory Activities (MedDRA) system.
4. Statistics
Recommendation
The analysis of data for PPTH trials should be preplanned, and issues that need to be prospectively defined should be considered. Table 2 provides a list of recommended issues that should be addressed during preplanning of the data analysis for PPTH trials.
Analytic issues to be prospectively defined during preplanning.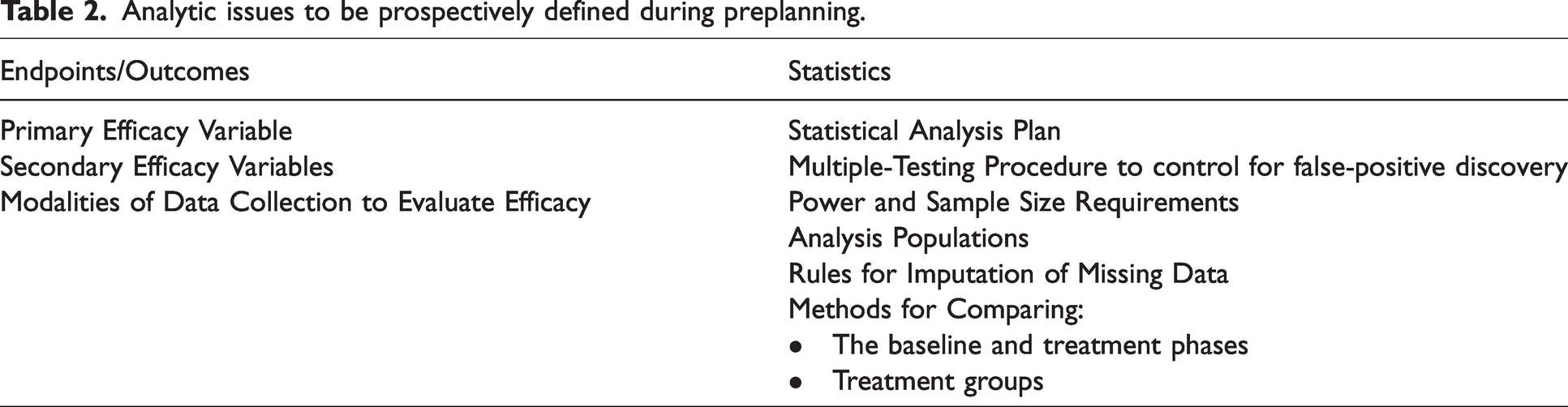
Comments
Statistical analyses are dependent on certain assumptions, and statistical analysis plans should include methods and tests that are designed to evaluate these assumptions. Investigators should be ready to propose an alternative analysis plan if assumptions are not met during the study. To ensure the validity and reliability of clinical trial data, it is recommended that efficacy endpoints be analyzed based on the randomization assignment of subjects, regardless of the actual treatment they received. This approach should be carried out using the intent-to-treat principle, with a clearly defined full analysis set analyzed as randomized (13). For safety variables, analyzing participants based on the treatment actually received may be appropriate, regardless of the assigned treatment. To include data for all participants in the full analysis set, missing data can be imputed for at least the primary variable of interest, either as a primary analysis or a sensitivity analysis. It is also possible to use alternative statistical methods, but these must be verified by a statistician. Summary tables for each treatment and measurement timepoint should present the number of participants analyzed, as well as descriptive statistics such as mean, standard deviation, median, minimum, and maximum values, and/or response frequencies. Randomization is a critical step in clinical trials to reduce the risk of selection bias and ensure that treatment groups are comparable at baseline. However, it does not guarantee complete balance on all baseline characteristics. If significant imbalances are found in key variables of interest between treatment groups, regression methods should be used for analysis. To improve evaluations of the efficacy of different interventions, the effect size for the primary outcome measure(s) should be calculated with available statistical methods. This approach will also facilitate comparisons of findings from different studies (42).
5. Trial registration
Prior to the initiation of a trial, it is required that the trial be registered with a clinical trial registry such as clinicaltrials.gov, euclinicaltrials.eu/home, anzctr.org.au, or a similar regional or national official database. Registration should occur prior to the enrolment of the first participant. The registry must include information such as the trial design, primary and secondary endpoints, eligibility criteria, interventions, and outcomes. Any changes made to the trial after registration should be updated promptly in the registry.
6. Publication of results
Recommendations
A publication plan should be developed prior to the start of the trial, including a timeline for publication. Investigators and sponsors (if applicable) should agree upon publication timelines before the trial is initiated, ideally as part of the protocol. All research results, including primary and secondary endpoints and safety data, whether positive or negative, must be published in manuscript form. A design paper with baseline data may be published at the time of trial initiation or at the end of recruitment. Authorship should follow the recommendations of the International Committee of Medical Journal Editors. To ensure transparency, all authors must declare their conflicts of interest. Investigators should avoid entering into agreements with sponsors, both for-profit and non-profit, that restrict access to study data, limit its analysis and interpretation, or interfere with the independent preparation and publication of manuscripts.
Comments
In cases where applicable, controlled trials should be published on behalf of a study group consisting of investigators who have enrolled participants in the trial. Conflicts of interest may arise when a professional’s judgment concerning a primary interest, such as participants’ welfare or research validity, may be influenced by a secondary interest, such as a financial relationship with the sponsor. Potential conflicts of interest arise from financial ties such as employment, consultancies, grants, fees and honoraria, patents, royalties, stock or share ownership, and paid expert testimony. Therefore, it is essential to disclose all potential conflicts of interest to ensure transparency and integrity in reporting clinical trial results.
7. Independent data safety monitoring board
Recommendations
To ensure participant safety, it is recommended that phase 3 trials have an independent data safety monitoring board and predefined rules for futility or safety. The independent data safety monitoring board must be composed of individuals with relevant expertise who are independent of the sponsor and investigators.
8. Steering committee
Recommendations
In industry-sponsored trials, it is strongly recommended to establish a steering committee that includes independent academic researchers, statisticians, and company representatives, where appropriate. For investigator-initiated trials, the establishment of a steering committee is not mandatory but may still be beneficial.
Comments
Regardless of the establishment of a steering committee, investigators and sponsors share the responsibility for study conception, design, operational execution, investigator training, data handling, data analysis and interpretation, subsequent reporting and publication, and compliance with local laws and regulations.
9. Post-approval registries
Recommendations
To evaluate newly approved drugs and biologics in clinical practice, prospective post-approval registries, open-label or observational studies should be utilized. Registries and studies should consider including participants excluded from randomized trials, such as individuals with comorbid and concomitant conditions (e.g., cardiovascular disease) and those using concomitant treatments, as well as participants following the double-blind treatment phase of randomized trials. This would allow for a more comprehensive evaluation of the drugs and biologics in real-world settings.
Comments
Registries play a key role in generating long-term data on the efficacy, tolerability, and safety of treatments. They can provide valuable insights into compliance and adherence and may also shed light on withdrawal symptoms. In addition, pregnancy registries are highly recommended.
10. Health Technology Assessment
In certain countries, Health Technology Assessment (HTA) bodies mandate dedicated studies to establish the cost-effectiveness of a treatment and calculate the cost-benefit ratio as a precondition to granting reimbursement. Such studies might require a comparison with an approved drug treatment. To conduct these studies, healthcare costs associated with office and emergency department visits, diagnostic tests, hospital admission, and medication must be carefully documented, while working days lost (i.e., the total number of days missed from work due to illness or injury) may also be measured (43).
11. Methodology used for guideline development
The IHS Clinical Trials Standing Committee developed the present guideline independently and without bias. The process of developing this guideline involved two phases. In the first phase, the initial draft of the guideline was reviewed by the Clinical Guidelines Committee of the IHS, who proposed multiple changes. This version of the guideline was then shared with representatives of the European Medicines Agency, the US Food and Drug Administration, pharmaceutical manufacturers, and patient associations. Their suggestions were discussed in a series of meetings with members of the Committee. After incorporating the views of these stakeholders, the Committee posted a pre-final version on the IHS website in May 2023, inviting comments from IHS members. The Committee incorporated member comments to finalize this edition, ensuring that it represents a consensus of stakeholders in the field of PPTH research.
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the official policy of the Uniformed Services University of the Health Sciences or the United States Department of Defense.
Conflict of interest
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: HA reports personal fees from Lundbeck and Teva, outside of the submitted work. H-CD received honoraria for participation in clinical trials, contribution to advisory boards or oral presentations from: AbbVie, Lilly, Lundbeck, Novartis, Pfizer, Teva, Weber & Weber and WebMD. The German Research Council (DFG) and the German Ministry of Education and Research (BMBF) support headache research by H-CD. H-CD serves on the editorial boards of Cephalalgia, Lancet Neurology and Drugs. H-CD is a member of the Clinical Guidelines Committee of the German Society of Neurology and of the Clinical Trials Committee of the IHS. CT has received personal fees for the participation in advisory boards and/or scientific symposia from Abbvie, Dompé, Eli Lilly, Medscape, Novartis, Lundbeck and Teva. Her institution has received payments for sponsored clinical trials or investigator-initiated trials from Abbvie, Eli Lilly, Novartis, Lundbeck and Teva. AIS reports receiving research support from Eli Lilly. RBL reports support from the NIH, FDA as well as the National Headache Foundation and the Marx Foundation. He also receives research support from Allergan/Abbvie, Amgen, Eli Lilly and Satsuma. He receives personal fees as a consultant or advisor from Allergan/Abbvie, Amgen, Biohaven Holdings, GlaxoSmithKline, Grifols, Impel NeuroPharma, Pfizer, Eli Lilly, Lundbeck, Satsuma and Teva Pharmaceuticals. He holds stock or options in Biohaven and Manistee. PP-R reports that within the prior 36 months, having received honoraria as a consultant and speaker for: AbbVie, Amgen, Biohaven, Chiesi, Eli Lilly, Lundbeck, Medscape, Novartis, Pfizer and Teva. Her research group has received research grants from Novartis, Teva, AbbVie, EraNET Neuron, RIS3CAT FEDER, AGAUR, ISCIII, International Headache Society; has received funding for clinical trials from Alder, Amgen, Biohaven, ElectroCore, Eli Lilly, Lundbeck, Novartis, Teva. She is the Honorary Secretary of the International Headache Society. She is a member of the Clinical Trials Guideline Committee of the International Headache Society. She serves as an associate editor for Cephalalgia, Headache, The Journal of Headache and Pain, Neurologia, and Revista de Neurologia. She is the founder of www.midolordecabeza.org. PP-R does not own stocks from any pharmaceutical company. AJS reports honoraria for speaker events from Teva. Consulting fees from Invex therapeutics; salary and stockholding with Invex therapeutics. Received research funding from the Medical Research Council, National Institute for Health Research, Sir Jules Thorn Charitable Trust, UK Space Agency, Ministry of Defence UK and the Department of Defense US. CDC reports receiving research support from the American Heart Association, Amgen, National Institutes of Health, and United States Department of Defense. She is on the Editorial Board of Neurology. AGF reports no potential conflicts of interest. MA reports receiving personal fees from AbbVie, Amgen, Eli Lilly, GlaxoSmithKline, Lundbeck, Novartis, Pfizer, and Teva Pharmaceuticals outside of the submitted work. MA also serves as an associate editor of Brain, Cephalalgia, and The Journal of Headache and Pain. TJS reports that within the prior 24 months he has received personal compensation for consulting from Abbvie, Allergan, Amgen, Axsome, Biodelivery Science, Biohaven, Collegium, Eli Lilly, Ipsen, Linpharma, Lundbeck, Novartis, Satsuma, and Theranica. He has received royalties from Up To Date. He has stock options in Aural Analytics and Nocira. He has received research support from American Heart Association, Amgen, Henry Jackson Foundation, National Institutes of Health, Patient Centered Outcomes Research Institute, Spark Neuro, and United States Department of Defense. He is on the Board of Directors of the American Headache Society and American Migraine Foundation and on the Editorial Boards of Cephalalgia and Journal of Headache and Pain. DWD reports Consulting: Amgen, Atria, CapiThera Ltd., Cerecin, Ceruvia Lifesciences LLC, CoolTech, Ctrl M, Allergan, AbbVie, Biohaven, GlaxoSmithKline, Lundbeck, Eli Lilly, Novartis, Impel, Satsuma, Theranica, WL Gore, Genentech, Nocira, Perfood, Praxis, AYYA Biosciences, Revance, Pfizer. Honoraria: American Academy of Neurology, Headache Cooperative of the Pacific, Canadian Headache Society, MF Med Ed Research, Biopharm Communications, CEA Group Holding Company (Clinical Education Alliance LLC), Teva (speaking), Amgen (speaking), Eli Lilly (speaking), Lundbeck (speaking), Pfizer (speaking), Vector Psychometric Group, Clinical Care Solutions, CME Outfitters, Curry Rockefeller Group, DeepBench, Global Access Meetings, KLJ Associates, Academy for Continued Healthcare Learning, Majallin LLC, Medlogix Communications, Medica Communications LLC, MJH Lifesciences, Miller Medical Communications, WebMD Health/Medscape, Wolters Kluwer, Oxford University Press, Cambridge University Press. Non-profit board membership: American Brain Foundation, American Migraine Foundation, ONE Neurology, Precon Health Foundation, International Headache Society Global Patient Advocacy Coalition, Atria Health Collaborative, Arizona Brain Injury Alliance, Domestic Violence HOPE Foundation/Panfila. Research support: Department of Defense, National Institutes of Health, Henry Jackson Foundation, Sperling Foundation, American Migraine Foundation, Henry Jackson Foundation, Patient Centered Outcomes Research Institute (PCORI). Stock options/shareholder/patents/board of directors: Aural analytics (options), Axon Therapeutics (shares/board), ExSano (options), Palion (options), Man and Science, Healint (options), Theranica (options), Second Opinion/Mobile Health (options), Epien (options), Nocira (options), Matterhorn (shares), Ontologics (shares), King-Devick Technologies (options/board), EigenLyfe (shares), AYYA Biosciences (options), Cephalgia Group (shares/board), Atria Health (options/employee). Patent 17189376.1-1466:vTitle: Onabotulinum Toxin Dosage Regimen for Chronic Migraine Prophylaxis (Non-royalty bearing). Patent application submitted: Synaquell® (Precon Health). GMT reports consultancy or industry support from Abbvie/Allergan, Lilly, Lundbeck, Novartis, Pfizer, Teva, and Interactive Studios BV, and independent support from the European Community, Dutch Heart and Brain Foundations, Dutch Research Council, and Dioraphte.