
Abstract
Background
Spreading depression (SD) is the electrophysiological substrate of migraine aura and a potential trigger for headache. Since its discovery by Leão in 1944, SD has transformed from being viewed as an epiphenomenon into a therapeutic target relevant in the pathophysiology of migraine and brain injury.
Aim
Despite decades of research, the underpinnings of SD are still poorly understood, hampering our efforts to selectively block its initiation and spread. Experimental models have nevertheless been useful to measure the likelihood of SD occurrence (i.e. SD susceptibility) and characterize genetic, physiological and pharmacological modulation of SD in search of potential therapies, such as in migraine prophylaxis and stroke. Here, I review experimental SD susceptibility endpoints and surrogates, and minimum essential model requirements to improve their utility in drug screening.
Conclusion
A critical reappraisal of strengths and caveats of experimental models of SD susceptibility is needed to set standards and improve data quality, interpretation and reconciliation.
Introduction
Spreading depression (SD) is an intense neuronal and glial depolarization wave that slowly propagates in brain tissue (∼3 mm/min) by way of gray matter contiguity. The near-complete loss of membrane potential is a result of massive transmembrane ionic and water shifts, including a K+ and glutamate efflux, and Na+, Ca2+ and water influx, which last up to a minute and does not lead to injury in otherwise normal brain tissue. It is believed that the rise in extracellular K+ and glutamate act as chemical signals diffusing to and depolarizing adjacent cells, and in this way the depolarization slowly spreads regardless of functional or vascular divisions. For the released K+ and glutamate to reach the critical depolarization threshold of adjacent cells, high neuronal and synaptic density and low extracellular space volume are required; therefore, white matter is characteristically resistant to SD. All these ionic and water shifts create a signature extracellular negative slow potential shift (aka DC shift) accompanied by suppression of action potentials and synaptic activity (Figure 1). As a result, electrocorticogram (ECoG) is depressed, hence the historical term “spreading depression” originally coined by Leão (1**).
Cortical spreading depression (SD) triggered by a suprathreshold stimulus delivered on to the frontal cortex is detected by a glass micropipette a few mm away in the parietal cortex after a latency accounting for its slow propagation speed. Note the large negative slow (DC) potential shift that lasts approximately 30 sec (upper tracing) and the concurrent electrocorticogram (ECoG) suppression (lower tracing) that outlasts the DC shift by several minutes. The DC shift accompanied by ECoG suppression is the most reliable electrophysiological signature of SD. Tracings are from a rat under isoflurane and nitrous oxide anesthesia. Vertical scale 20 mV DC, 0.4 mV ECoG; horizontal scale 1 min.
Since the discovery of SD decades ago, similarities between the electrophysiological properties of SD and the neurological signs and symptoms during migraine aura suggested a causative link between the two (2–4). A large body of evidence now supports the SD theory of migraine (5). Moreover, experimental evidence also suggests that SD can trigger headache (6–10**,11). Although whether an asymptomatic SD triggers migraine headache without a perceived aura is still debated, suppression of SD susceptibility by migraine prophylactic drugs as a class effect (12–14**,15–18) supported this notion since these drugs have been equally efficacious in migraine with or without a perceived aura. Therefore, SD is now considered a potential therapeutic target in migraine, and experimental models of SD susceptibility are increasingly being used to screen for migraine drug candidates. A range of experimental models of SD has been developed, although the sensitivity and specificity and the predictive value of each model have not been systematically investigated, and a consensus is still lacking.
In vitro vs. in vivo models
In vitro models, such as isolated retina and brain slices, have been used in the past to study the effects of various pharmacological agents on SD properties. These models bypass the blood-brain barrier and eliminate hemodynamic, pharmacokinetic, anesthesia-related and systemic physiological factors. However, tissue oxygenation and metabolism differ from the in vivo state because of the absence of blood flow (19,20), exposure to hypoxia and trauma during experimental preparation may confound the results, and it is difficult to judge the clinically relevant drug concentrations when directly superfused in vitro. Therefore, convenience of the relatively high-throughput in vitro drug screening is offset by difficulties in extrapolating the in vitro results to in vivo efficacy.
In vivo models have been for the most part limited to cerebral cortex because of ease of access. Although most studies have been in rodents, a wide variety of species from pigeons to cats and monkeys have been used (21,22). There is a fair degree of interspecies variability in SD susceptibility. In general, the larger the brain is, the lower the susceptibility (e.g. mouse vs. rat). In addition, gyrencephalic species (e.g. cats) are less susceptible than lissencephalic species (e.g. rodents). Species differences in SD susceptibility, and their determinants, have not been systematically studied. Although increasing astrocyte/neuron ratios in higher species have frequently been cited as the cause (23,24), it is unlikely to be the only determinant.
Methods to trigger SD
SD is triggered when a sufficiently intense depolarizing stimulus raises extracellular [K+] above a threshold in a minimum critical volume of tissue, estimated to be approximately 12 mM and 1 mm3, respectively, in rodent brain (25–27). The depolarizing stimulus is most commonly electrical, chemical or mechanical.
Electrical stimulation is one of the most direct methods to assess SD susceptibility (14**,28,29). A single square pulse of stepwise escalating cathodal charge is applied at regular intervals (e.g. every 4–5 minutes) until an SD is triggered (Figure 2). The product of stimulus current (ampere) and duration (seconds) that triggers an SD is then taken as the charge intensity threshold (coulomb). High-frequency train stimulation of increasing intensity or duration is an alternative approach. The absolute threshold values in any experimental setting critically depend on methodological details. Electrode properties and electrode-tissue contact determine the stimulus geometry, which heavily influences the charge intensity required to trigger an SD. Stimulus geometry is best controlled by a cortical cup electrode (26); however, this is not always available or practical. Instead, both unipolar and concentric or parallel bipolar electrodes can be used. We favor the latter since stimulus polarity can be alternated to minimize oxidation. In all cases, slight differences in tip separation or insulation between two electrodes, or changes in the same electrode due to wear and tear over time, can result in different absolute threshold values. Therefore, it is important not to rely on historical controls when studying modulation of electrical threshold. Electrical threshold yields categorical (i.e. non-parametric) data, and often shows higher variability compared to chemical depolarizing agents because of irregularities at the electrode-tissue contact (e.g. bleeding or drying).
Direct electrical stimulation of cortex with stepwise escalating charge intensities (single square pulses of alternating polarity using a bipolar electrode in this example) triggers a spreading depression (SD) when the threshold is reached. The stimulus intensity steps depend on the electrode and the species, and can be adjusted according to the hypothesis being tested; if testing a drug or mutation that significantly lowers the threshold, it would be important to start at a low stimulus intensity with small step increases. Tracing is from a representative rat under isoflurane and nitrous oxide anesthesia. Vertical scale bar 10 mV; horizontal scale bar 2 min.
The most common chemical depolarizing agent used to determine SD susceptibility is concentrated KCl solution (often 50 mM or higher), although glutamate receptor agonists (e.g. N-methyl- Topical application of stepwise increasing concentrations of KCl solution triggers a spreading depression (SD) at a threshold concentration of 40 mM. As with electrical stimulation, the KCl concentration steps can be tailored to the hypothesis; for example, when studying a drug or mutation that is anticipated to significantly lower the threshold, testing should start at a lower concentration with smaller step increases. Tracing is from a representative mouse under isoflurane and nitrous oxide anesthesia. Vertical scale bar 10 mV; horizontal scale bar 1 min.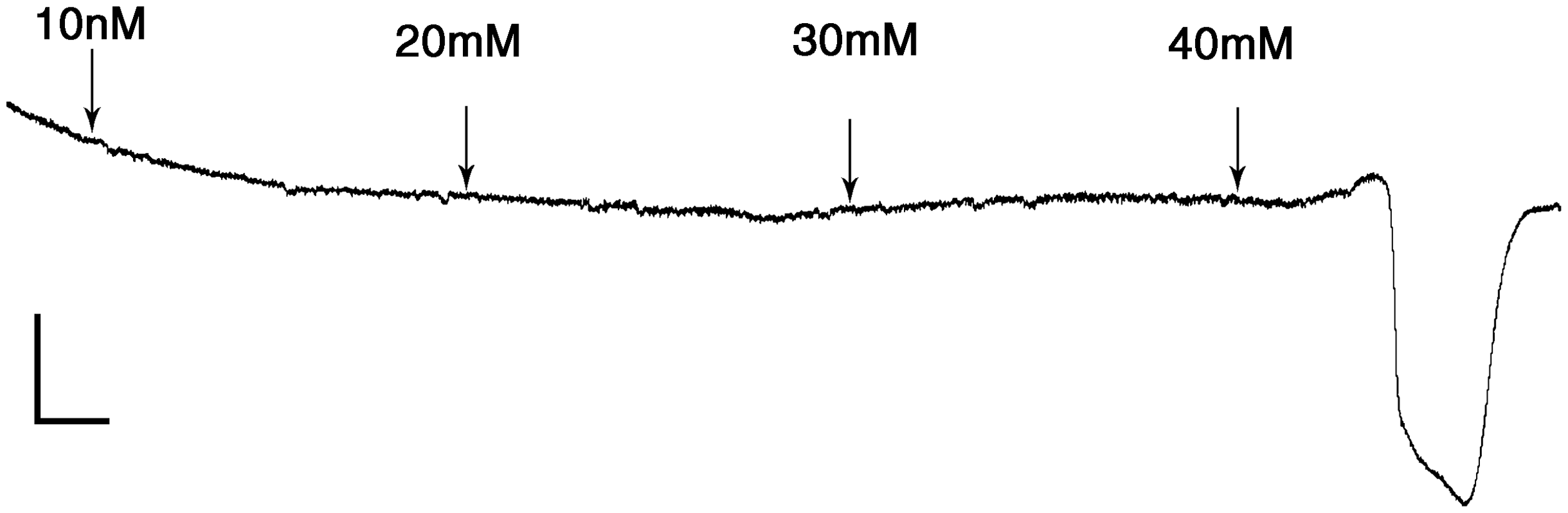
Alternatively, a constant suprathreshold concentration (e.g. 1 M KCl) can be applied continuously to trigger repetitive SDs (Figure 4) and determine their frequency (14**,30,35). An important potential caveat of SD frequency endpoint is the absolute and relative refractory periods during which a subsequent SD cannot be triggered (36). Importantly, longer SD durations are associated with prolonged refractory periods and lower SD frequencies, which may be misinterpreted as lower susceptibility. In such cases, the total cumulative SD duration (e.g. per hour) may be an alternative endpoint. Because SD frequency yields continuous data and is less prone to cortical surface irregularities, it tends to be statistically more powerful compared to electrical threshold. Both KCl concentration threshold and KCl-induced SD frequency critically depend on the cranial window diameter and the presence of dura. Therefore, these parameters must be identical across animals and experimental groups, different experimenters, and if possible, different labs.
Continuous topical KCl application on occipital cortex (horizontal line on top) triggers repetitive spreading depression (SD) waves recorded from the parietal cortex. Occasional small-amplitude SDs are included in the count if they are larger than 5 mV (arrowhead). Note the suppression and recovery of the electrocorticogram (ECoG) after each SD (arrows), which often helps differentiate electrical noise from true SDs when the latter is very small (dot). Tracing is from a representative rat under isoflurane and nitrous oxide anesthesia. Vertical scale bar 10 mV; horizontal scale bar 5 min.
Mechanical stimulation has also been used to assess SD susceptibility. However, it is difficult to titrate to determine a threshold. Therefore, SD susceptibility is measured by how often a single mechanical stimulus triggers SD, i.e. an all or none response (37). Repetitive assessment can be problematic because of poor reproducibility of the stimulus and cumulative traumatic injury. Furthermore, mechanically induced SD appears to have a different pharmacological profile compared to chemical or electrical SD induction (38,39). Other methods to trigger SD include high-frequency afferent pathway stimulation and focal cerebral ischemia (40), but these are not practical for high-throughput drug screening.
Methods to detect SD
Electrophysiologically, the characteristic extracellular negative slow potential shift is the gold standard to detect SD. This is often briefly preceded by a burst of neuronal firing that can be detected by single unit recordings, followed by a longer lasting suppression of neuronal firing and ECoG activity. Although this pattern of single unit activity can be used as a surrogate for the slow potential shift (37–39), it is neither more sensitive nor more specific than the latter, does not directly inform about the depolarization duration or amplitude, and cannot be used if the test drug has a direct inhibitory effect on neuronal firing (e.g. tetrodotoxin).
An alternative method to detect SD in vivo is diffusion-weighted magnetic resonance imaging (MRI) (41). The advantages of being non-invasive and three dimensional are offset by relatively low spatial and temporal resolution. Therefore, MRI may be superior to traditional electrophysiological methods only in larger gyrencephalic species and low SD repetition rates. SD is also associated with characteristic optical intrinsic signal transients (42). These are caused by changes in light absorption and scattering properties of the tissue due to transmembrane ionic and water shifts, as well as changes in tissue hemoglobin concentration. Alternatively, one can also monitor the large amplitude blood flow transients to detect SD (38,43). These are complementary surrogates, but should not replace the slow potential shift as the standard method to detect SD.
Relevant SD attributes
SD susceptibility is defined as the likelihood of the brain tissue to develop and sustain SD. There are various experimental models and approaches that have been used by different labs to assess SD susceptibility. Physiological processes interrogated by each model, and the translational value of their readouts, have not been established. Of course, each model has strengths and pitfalls, and agreement of results among models remains to be established.
Arguably the most relevant measure of SD susceptibility is the threshold intensity of electrical charge or threshold concentration or volume of the depolarizing agent (e.g. KCl) that triggers an SD, as described above. The threshold topical electrical stimulation intensity and the threshold KCl concentration for SD induction show excellent concordance (34). Interestingly, continuous topical KCl-induced SD frequency has also showed complete concordance with electrical SD threshold when studied in the same animal in our hands (14**,16,17,28). The relationship is inverse, such that lower SD thresholds yield higher SD frequencies and vice versa. Importantly, SD frequency has a theoretical ceiling determined by the SD duration and the absolute refractory period during which a subsequent SD cannot be initiated, which is estimated to be two to three minutes. Therefore, one cannot trigger more than 20–30 SDs in one hour regardless of the intensity of stimulation, a ceiling that we have observed in highly susceptible mutant mouse strains (30).
The relationship between SD susceptibility and other SD attributes.

SD: spreading depression.
Numbers indicate percentage distribution of susceptibility outcome (threshold and/or frequency) vs. speed, duration and amplitude outcomes when measured in the same cohort, categorized based on statistically significant increase or decrease, or no change. Sensitivity, specificity, and positive and negative predictive values of SD speed to predict SD susceptibility were calculated from true positives (attribute changes in the same direction as susceptibility), true negatives (neither susceptibility nor attribute shows a change), false negatives (attribute fails to show a change when susceptibility is changed), and false positives (attribute shows a change when susceptibility is unchanged, or attribute shows a change in the direction opposite to the change in susceptibility). Kappa (κ)statistic was calculated with quadratic weighting. The corrected κ value as proportion of maximum possible (given the observed marginal frequencies) is also provided in parentheses. κ = 1 is the theoretical maximum, while κ > 0.75 is regarded as excellent agreement.
Although based on these data, propagation speed may be an acceptable surrogate to detect a change in SD susceptibility, its clinical relevance is not clear. Moreover, there are potential caveats associated with these surrogates. For example, the propagation speed as well as the duration decrease with each successive SD to reach a plateau as much as 25% lower than the initial values after a few SDs. Therefore, care should be taken to not trigger SDs inadvertently during cranial surgery as these will make subsequent SD speeds and durations appear lower than normal. We recommend monitoring accidental SD occurrence, for example, by non-invasive laser Doppler flowmetry, during experimental preparation, particularly in rodents where SD susceptibility is inherently high.
When monitoring SD occurrence for threshold determination, one should keep in mind that SDs can fail to propagate, especially when SD susceptibility is dramatically reduced (34). Because of this, the rate of SD detection can vary inversely with the distance between the recording and stimulation sites. To minimize this confounder, we adopted the use of two recording sites, one sufficiently close to the stimulation site (but not too close to be directly depolarized during stimulation, typically a couple of mm), and one farther away. This arrangement allows the detection of most if not all SDs, as well as the calculation of propagation failure rate between the two recording sites.
Recommended quality measures
In vivo vs. in vitro testing
In general, in vitro models (e.g. isolated retina) may provide a high-throughput system for initial SD susceptibility testing (46,47). However, especially in drug screening, promising hits must then be fully characterized in vivo upon systemic administration, preferably in more than one species and both sexes. Although testing in a gyrencephalic species may be intuitive, there is yet no evidence to suggest that efficacy in gyrencephalic species better predicts efficacy in human compared to rodent models. Migraine is a genetic disease, and the recent development of transgenic mouse models expressing human migraine mutations (28,30,45,48–50) provides an excellent opportunity to test drug efficacy in a genetically susceptible population.
Anesthesia
Anesthesia modulates SD susceptibility (35,51–57). Inhalational anesthetics such as isoflurane and nitrous oxide, and particularly the NMDA receptor blocker and dissociative anesthetic ketamine, suppress SD induction and hinder propagation. Propofol was recently reported to suppress SD as well (58), although this remains to be confirmed. In our experience, barbiturates, urethane and α-chloralose do not significantly inhibit SD, and should be the first choice when feasible. However, an additional consideration is the ease with which anesthesia can be induced and maintained over time while preserving systemic physiology (e.g. arterial blood gas and pressure). Deep anesthesia often results in hypotension, which is a potential confounder when determining SD frequency (see below). Significant respiratory depression necessitates mechanical ventilation, a particularly challenging task in mice. These side effects are common with barbiturates, urethane and α-chloralose. In general, an anesthetic that acts on the same target as the drug to be tested should be avoided (e.g. barbiturate anesthesia when testing a drug that acts on γ-amino butyric acid (GABA)A receptors, or ketamine when testing the efficacy of an NMDA receptor blocker). Anesthesia must be carefully tailored for each study in order not to confound the results and diminish the sensitivity of the employed assays. The convenience of anesthesia induction and maintenance by a given anesthetic should be weighed against its direct impact on SD susceptibility, and possible interactions between the anesthetic and the modulator (e.g. mutation or drug) to be tested.
Systemic physiology
It is imperative that systemic physiology be monitored, and when necessary, controlled (e.g. mechanical ventilation) in experimental models of SD susceptibility. Arterial blood pressure is inversely related to SD duration (59,60). Hypotension, even if mild, prolongs SD duration, which in turn prolongs the absolute refractory period. As a result, SD frequency during continuous topical KCl stimulation will be reduced. Hence, a drug that consistently reduces blood pressures may yield lower SD frequencies, and vice versa (6,61). Whether blood pressure also directly alters SD threshold has not been tested. Arterial blood gas and pH are also expected to influence SD susceptibility, but the relationship is less straightforward and has not been well investigated. Nevertheless, maintaining these values within normal physiological ranges is good practice. We perform endotracheal intubation and mechanical ventilation routinely in rats and when necessary in mice (e.g. barbiturate anesthesia), as the latter species is more fragile and tends to get hypotensive with prolonged anesthesia and mechanical ventilation. Of note, certain mouse strains are more resilient and can maintain their blood pressure for longer periods of anesthesia and ventilation. Blood glucose is also an important factor. Hyperglycemia inhibits SD susceptibility (62–65), whereas hypoglycemia prolongs SD durations (64). Therefore, blood glucose measurements further improve the consistency, particularly when genetic, physiological or pharmacological interventions are anticipated to alter this parameter. Overnight fasting generally stabilizes the systemic physiology and ameliorates hyperglycemia during surgery and subsequent interventions, but also tends to enhance SD susceptibility directly. Lastly, body temperature must also be regulated under anesthesia.
Surgical preparation and maintenance
Surgical preparation and maintenance of the craniotomy are also critical for reliable and consistent results and intra- and interobserver reproducibility. Of course, cortical injury due to trauma or heat during craniotomy, and bleeding or drying can all affect SD susceptibility. The cortex must be kept moist by artificial cerebrospinal fluid (CSF) or physiologic saline to maintain isotonicity during the experiments. Dura must be removed prior to electrical and chemical stimulation in rats and larger animals, but can be left intact in mice, as it is sufficiently thin and difficult to remove in an atraumatic fashion.
SD susceptibility
The most direct and relevant SD attribute is susceptibility, monitored using intraparenchymal extracellular microelectrode recordings of slow potential shift combined with ECoG activity. We prefer to determine the electrical threshold on one hemisphere, and KCl-induced SD frequency on the other hemisphere of the same animal; these two methods are non-overlapping but complementary measures of SD susceptibility and correlate well. We also routinely measure SD speed, duration and amplitude, and use them to obtain additional physiological insight.
Pharmacokinetic factors
Careful consideration of pharmacokinetic factors (e.g. absorption and elimination rates and routes, plasma and brain levels), seeking a dose-response relationship, testing the efficacy of repeated or chronic dosing paradigms in addition to a single dose, and including a positive control (e.g. a drug known to produce the sought effect) among the cohorts, all minimize the type II error.
Conduct of research
Last but not least, responsible conduct of research must be ensured by single or double blinding, and independent confirmation of data in a second laboratory. Inclusion of a negative control (e.g. a drug known to be ineffective) helps minimize type I error. In this context, Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines for reporting animal research are worth adopting universally (66).
Future directions and unmet needs
As SD is increasingly recognized as a potential therapeutic target not only in migraine but also in stroke and other brain injury states, research into the determinants and modulators of SD susceptibility (Figure 5) has been gaining momentum (67). As more experimental data on SD susceptibility become available, its predictive and translational therapeutic value will be better defined. In order to ensure efficient progress in the field, there is a need for an in-depth and timely discussion of relevant models and attributes toward a consensus standardization of the experimental approaches used in different labs.
There are numerous modulators of spreading depression (SD) susceptibility, intrinsic or environmental, some of which are modifiable. Checkmarks indicate existing supportive evidence experimentally or clinically. A detailed discussion of pharmacological targets can be found in Ayata (13).
Footnotes
Funding
This work was funded in part by the United States National Institutes of Health (NIH) (NS061505) and the Andrew David Heitman Neuroendovascular Research Fund from the Andrew David Heitman Foundation.
Conflict of interest
None declared.