
Abstract
Background
Migraine and epilepsy are highly co-morbid neurological disorders associated with episodic dysfunction of both cortical and subcortical networks. The study examined the interrelation between cortical spreading depression, the electrophysiological correlate of migraine aura and seizures triggered at cortical and brainstem levels by repeated sound stimulation in rats with acoustic hypersensitivity (reflex audiogenic epilepsy).
Method
In awake, freely moving rats with innate audiogenic epilepsy, 25 episodes of running seizure (brainstem seizures) were induced by repeated sound stimulation. Spreading depression and seizures were recorded using implanted cortical electrodes.
Results
The first sound-induced brainstem seizures evoked neither spreading depression nor seizures in the cortex. With repetition, brainstem seizures began to be followed by a single cortical spreading depression wave and an epileptiform discharge. Spreading depression was more frequent an early cortical event than seizures: spreading depression appeared after 8.4 ± 1.0 repeated stimulations in 100% rats (n = 24) while cortical seizures were recorded after 12.9 ± 1.2 tests in 46% rats. Brainstem seizure triggered unilateral long-latency spreading depression. Bilateral short-latency cortical spreading depression was recorded only after intense cortical seizures.
Conclusion
These data show that episodic brainstem activation is a potent trigger of unilateral cortical spreading depression. Development of intense seizures in the cortex leads to initiation of spreading depression in multiple cortical sites of both hemispheres.
Introduction
Migraine and epilepsy are highly co-morbid neurological disorders (1,2). Incidence of migraine in epilepsy patients is 2.4 times higher than in the general population (3) and post-ictal migraine-like headache is the most frequent type of headache associated with seizures (4,5).
Genetic and environmental risk factors for migraine and epilepsy often overlap (6). Hyper-reactivity to sensory stimuli, especially repetitive, is typical for patients with migraine (7) and some epileptic syndromes (8). Visual stimulation is a commonly reported trigger of migraine attacks (9) and epileptic seizures in photosensitive epilepsy (8,10).
Cortical hyper-excitability is believed to play a crucial role in pathogenesis of both disorders (11) but neuroimaging studies have also shown abnormal function of subcortical structures in patients with migraine (12) and photosensitive epilepsy (13). Alterations in the same cortico-subcortical network involved in sensory processing (the thalamus and superior colliculus) have been shown in migraine and photo-induced seizures.
Whereas abnormal sensitivity to light stimuli (photosensitivity) is the most common mode of sensory seizure precipitation in humans (8,10), sound-induced (audiogenic) seizures are the most frequent type of epileptic activity triggered by sensory stimulation in rodents (14,15). The genetic predisposition to audiogenic seizures is typical of many strains of rats and mice (16). Audiogenic seizures depend on neural substrates in the brainstem and do not require the integrity of the forebrain for their expression (15,16).
Triggering seizures by sound is determined by innate or acquired hyper-excitability of the inferior colliculus, the major auditory processing centre of the brainstem (15,16). Localized epileptic activation of the brainstem region behaviourally manifests as paroxysmal running (16). Subsequent involvement of other brainstem structures such as the periaqueductal grey and reticular formation leads to transition from running (focal brainstem seizure) to generalized tonic-clonic convulsions (15,16). The cortex is recruited into the seizure network only after repeated brainstem seizures when epileptic activity spreads along ascending pathways and leads to a gradual increase in cortical excitability and development of seizure activity in the cortex (15–17). The process of secondary brainstem-to-cortex seizure generalization is known as audiogenic kindling (17).
Our study in Wistar rats with innate acoustic hypersensitivity (reflex audiogenic epilepsy) has shown that repeated focal brainstem audiogenic seizures (episodes of running) reliably trigger unilateral cortical spreading depression early in audiogenic kindling when seizure activity is still absent in the cortex (18). Thus, repeated sensory stimulation of rats with audiogenic epilepsy can provoke both seizures (primary brainstem seizure and secondary cortical seizure) and cortical spreading depression. Since cortical spreading depression represents the electrophysiological correlate of migraine aura (19,20), the model offers a valuable opportunity to study neurophysiological mechanisms of migraine-epilepsy co-morbidity. Tonic-clonic convulsions induced by sound in rodents closely mimic tonic-clonic seizures in humans (16). Brain-imaging studies in patients with idiopathic generalized epilepsy have demonstrated abnormally increased activity of the brainstem in tonic-clonic seizures (13,21,22). Since post-ictal headache is observed significantly more often in patients with generalized tonic-clonic seizures than in those with other seizure types (4,5), investigation of the triggering role of brainstem audiogenic seizures in cortical spreading depression may be useful for understanding the migraine/epilepsy co-morbidity.
On the other hand, migraine and epilepsy obviously overlap at the cortical level and the present electrographic study examined the temporal and spatial association between spreading depression and seizures synaptically triggered in the cortex by repeated brainstem audiogenic seizures. Additionally, involvement of the brainstem in triggering cortical spreading depression in the audiogenic seizure model makes it possible to compare the roles of cortical and brainstem mechanisms in synaptic initiation of migraine aura. This is of particular interest because imaging data have shown profound activation of the brainstem during migraine attacks (23) and chronic dysfunction of subcortical areas in migraineurs (12).
Method
Male Wistar rats weighing 350–380 g at the beginning of the experiment (Stolbovaya breeding nursery, Russia) were used. After preliminary screening for audiogenic susceptibility, 30 rats with acoustic hypersensitivity (displaying an episode of paroxysmal running in response to sound stimulation) were selected for the study. Rats were housed in individual cages under controlled environmental conditions (a 12-h light–dark cycle, lights on at 7:00 a.m., 20–23℃) with free access to food and water. Experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and our protocol was approved by the Institutional Animal Care Committee. All efforts were made to minimize animal suffering.
Electrodes for recording spreading depression and seizure activity were implanted bilaterally under chloral hydrate (360 mg/kg, i.p.) anaesthesia. Glass electrodes with inner carbon fibre (a tip diameter of 50–100 µm) were implanted at the following coordinates (mm from bregma): anterior 2, lateral 2.5 for the motor cortex; posterior 3, lateral 3 for the somatosensory cortex; posterior 6, lateral 3 for the occipital (visual) cortex; posterior 5, lateral 6,5, ventral 4 for the temporal (auditory) cortex (24). A stainless steel screw positioned on the scull bone over the cerebellum was used as a reference electrode. All electrodes were soldered to a pin connector and secured with acrylic cement. The experiment began 2 weeks after surgery.
Experiments were performed in awake, freely moving rats with simultaneous video-monitoring of behaviour and recording full-band electroencephalography (EEG). Each rat was individually placed in a shielded experimental chamber (60 × 40 × 40 cm) and the implanted connector was attached to the recording cable. Electrical activity of the cortex (in a frequency band from 0 to 500 Hz) was recorded with a six-channel, high-input impedance (1 gΩ) DC amplifier and A/D converter (E14–440, L-Card, Russia). The data were digitized at a 2 kHz sampling rate, stored on a PC and analysed off-line. In some rats, slow potential shifts were recorded using a paper recording device.
After a 5-min period of habituation in the chamber and a 15-min period of baseline recording electrical activity of the cortex, a rat was submitted to standard sound stimulation (‘key ringing’, a frequency range of 13–85 kHz with a spectral maximum of 20–40 kHz and an intensity of 50–60 dB) produced by an electromechanical vibro-device. The sound lasted until the onset of running or for 60 s. Each rat was subjected to 25 repeated sound stimulations once per day at 3–4-day intervals between stimulations. Cortical seizure was defined as post-running spike/spike-wave discharge in the EEG with an amplitude of at least twice the background activity and duration of at least 5 s. Latency of cortical spreading depression was defined as a time lag between the end of running and appearance of spreading depression in a recording site.
Data were expressed as mean ± SEM. Numbers of repeated stimulations needed to trigger the first cortical seizure and spreading depression within and between groups were compared using Wiscoxon and Mann–Whitney tests, respectively. Regional differences in the spreading depression latencies and effect of cortical seizures on the latency of spreading depression were estimated using two-way analysis of variance followed by Tukey’s post hoc test. Fisher’s exact test was used to compare number of rats expressing cortical spreading depression and seizure discharge. The level of significance was set to p < 0.05.
Results
The first sound stimulation induced a brief (5.2 ± 0.3 s), self-sustained episode of running (focal brainstem seizure) in all rats with acoustic hypersensitivity (n = 30). In the first tests, neither spreading depression nor epileptic discharge was recorded in the cortex.
Repeated sound stimulation reliably induced a brainstem seizure (an episode of running) in 24 rats. In repeated tests, a proportion of these rats (46%, 11/24) developed secondary seizure activity in the cortex and the rest (54%, 13/24) did not show cortical seizures (Figures 1 and 2). The cortical seizure represented by an epileptiform spike/spike-wave discharge appeared immediately after the end of running (a brainstem seizure) and behaviourally expressed as a clonic convulsion. The first post-running cortical seizures were short (7.1 ± 0.9 s) and mild. But in repeated tests these seizures progressively increased in duration and amplitude, reflecting a gradual enhancement of cortical excitability during kindling. At the late kindling stages, brainstem seizures were followed by a prolonged (22.9 ± 3.1 s) high-voltage cortical spike-wave discharge in all areas of the cortex. The generalized cortical seizure behaviourally manifested as a severe clonic or tonic-clonic convulsion.
Incidence of spreading depression and seizure discharge in the cortex after repeated sound-induced brainstem seizures in rats showing (a, n = 11) and not showing (b, n = 13) seizure activity in the cortex. Percentages of rats, in which spreading depression and seizure discharge were recorded in the cortex, are marked by grey and black bars, respectively. Abscissa: number of sound stimulation inducing brainstem seizure (an episode of running). Incidence of cortical spreading depression progressively increases with repetition of audiogenic brainstem seizure, irrespectively of seizure development in the cortex. Representative electrographic tracings recorded in the cortex after the end of 1st (a), 10th (b), 15th (c) and 25th (d) sound-induced episodes of brainstem seizures in two representative rats not showing (I) and showing (II) secondary development of cortical seizures. Simultaneous recordings of both EEG and slow potential shifts (DC) in the somatosensory cortex of the right (R) and left (L) hemispheres are presented. The beginning of each trace coincides with the end of running. Calibration bars: 30 s, 1 mV (EEG) and 4 mV (DC). In fragments C and D (II), horizontal bars and extended fragments of recording above tracings indicate a post-running cortical seizure discharge. Note that unless intense seizure activity developed in the cortex, unilateral long-latency spreading depression was triggered. Bilateral short-latency cortical spreading depression developed only after intense cortical seizure discharge.

A single spreading depression wave was recorded after repeated brainstem running seizures in all rats. Fisher’s exact test has shown that spreading depression was a significantly more frequent cortical event than seizure discharge (expressed by 100% (24/24) and 46% (11/24) rats, respectively; p < 0.0001, Figures 1 and 2). In rats that developed cortical seizures, spreading depression began to appear significantly earlier than seizures (after 8.4 ± 1.0 versus 12.9 ± 1.2 repeated stimulations, respectively, n = 11; Wilcoxon test, p < 0.05). There was no significant correlation between the number of stimuli required for the first appearance of cortical spreading depression and that for triggering the first cortical seizure (Spearman’s correlation coefficient, r = 0.529, p = 0.094). In rats showing no cortical seizure, spreading depression was registered after 11.9 ± 1.2 repeated stimulations, which was significantly later than in rats exhibiting cortical seizures (n = 13, Mann–Whitney test, p < 0.05). Once appeared in the cortex, spreading depression and seizure discharge became reliable consequences of repeated sound-induced brainstem seizures.
Repeated audiogenic brainstem seizures regularly triggered a spreading depression wave in the cortex of one hemisphere unless prolonged and intense seizure activity developed in the cortex (Figure 2). A brainstem seizure alone and a brainstem seizure followed by a brief and mild cortical seizure elicited a unilateral spreading depression in the cortex. Only after intense cortical seizures spreading depression was triggered bilaterally (Figure 2(d) II).
The unilateral spreading depression wave appeared in the motor, somatosensory, temporal (auditory) and occipital (visual) areas of the cortex with a long 1–2-min delay after the end of sound-induced brainstem seizure (Figures 2 and 3). The mean latency of the unilateral spreading depression waves was minimal in the auditory and somatosensory cortices and maximal in the motor cortex. But even the shortest spreading depression latency exceeded 1 min, which suggests initiation of the spreading depression in a remote cortical site(s) and its non-synaptic propagation to the recording electrodes. Latencies of unilateral spreading depression waves triggered by brainstem seizures alone and brainstem seizures with mild cortical seizures did not differ (Figures 2 and 3). Audiogenic seizures with intense epileptic activation of the cortex were followed by a short-latency bilateral spreading depression wave, indicating multi-focal triggering of cortical spreading depression (Figures 2(d) and 3). Despite identical seizure pattern in all cortical regions, there was a significant regional difference in the latency of spreading depression triggered by the seizures. Spreading depression appeared immediately after the end of high-voltage cortical seizure discharge in sensory (somatosensory, auditory and visual) cortical areas and after a delay of 40 ± 12.6 s in the motor cortex (Figure 3).
Latencies of spreading depression SD appearance in the motor (M Cx), somatosensory (SS Cx), auditory (Aud Cx) and occipital (Occ Cx) areas of the cortex after the end of sound-induced seizures of three types – a brainstem seizure alone, a brainstem seizure followed by a mild cortical seizure and a brainstem seizure followed by a severe cortical seizure. * p < 0.05, ** p < 0.005 and *** p < 0.0005 significant regional difference as compared to the frontal cortex within each group with a particular seizure type; # p< 0.0005 significant between-group difference for a particular area of the cortex. Data are expressed as mean ± SEM.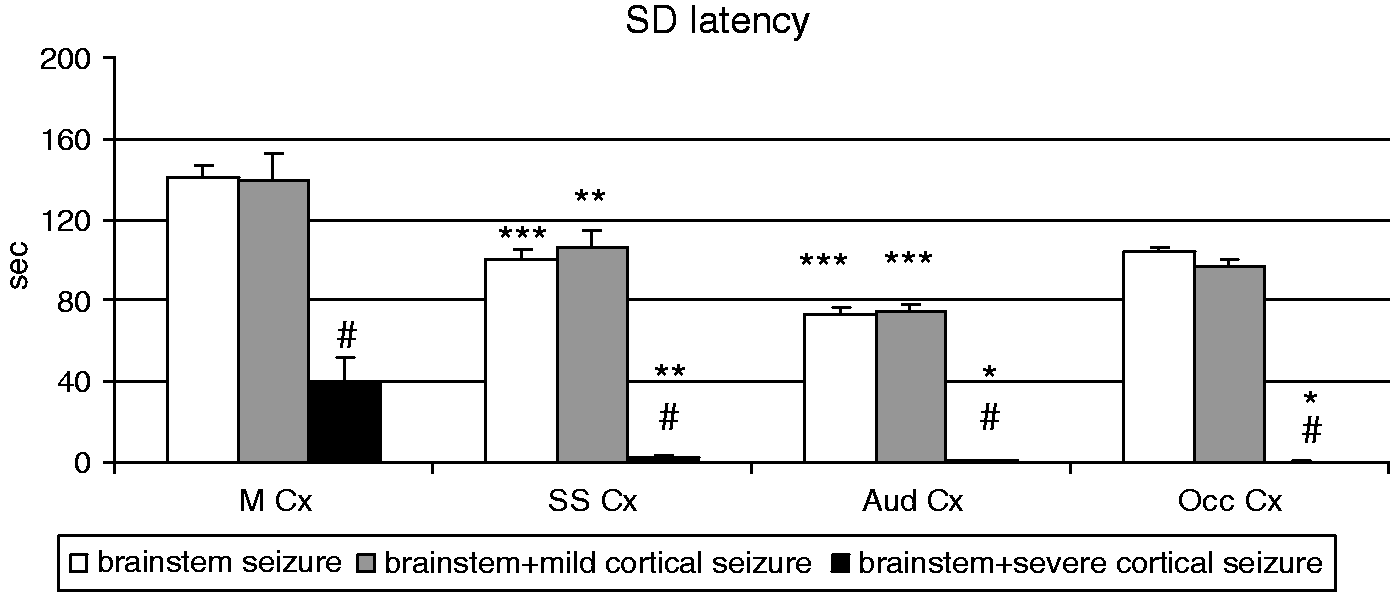
Discussion
The present experimental study in awake rats with innate hypersensitivity to acoustic stimulation (reflex audiogenic epilepsy) investigated spreading depression and epileptic activity, which are triggered in the cerebral cortex by repeated brainstem audiogenic seizures and reflect projected activity of the brainstem. It has been found that in the cortex spreading depression occurs significantly more often (in 100% rats) than seizures (in 46% rats). This result is in line with reports about more frequent occurrence of spreading depression as compared to seizures in the acutely injured human cortex (25).
The present study has also confirmed the tight association of cortical spreading depression with brainstem audiogenic seizures. Spreading depression was recorded in the cortex both after a brainstem seizure propagating to the cortex and after a local brainstem seizure. Consistent with the previous report that examined early cortical responses to repeated running seizures (18), unilateral long-latency cortical spreading depression began to be triggered by the brainstem seizures when epileptic activity was still absent in the cortex. Furthermore, cortical spreading depression was induced later in kindling along with development of cortical seizures. It is accepted that cortical spreading depression, a slowly propagating wave of transient neuronal depolarization (26), is responsible for generation of neurological symptoms of migraine aura (19,20,27) and probably for triggering headache (20,28–31). The close association between spreading depression and sound-induced seizures found in the present study may be a neurophysiological basis of the well-known co-morbidity between migraine and epilepsy.
Cortical spreading depression was discovered as a side product of experimental epilepsy research (26). The tight relationship between spreading depression and seizures in the hyper-excitable cortex of humans (25) and animals (32,33) has been previously shown. It is accepted that cortical hyper-excitability leads to either spreading depression or seizures in an unpredictable way (32). Our experiments in awake rats exhibiting gradual development of cortical hyper-excitability have shown that spreading depression began to be triggered in the cortex long before its epileptic activation. Such early appearance of spreading depression is likely to reflect first changes in cortical excitability during kindling and suggests that in the cortex a threshold required for the synaptic initiation of spreading depression is lower than that required for triggering seizure activity. Since growing evidence from brain-imaging studies points at involvement of the brainstem in expression of generalized seizures in epileptic patients (13,21,22), the present experimental data may be useful for understanding pathophysiological mechanisms of migraine/epilepsy co-morbidity and frequent occurrence of post-ictal headache in patients with idiopathic generalized epilepsy (4,5).
On the other hand, the data obtained in a subpopulation of rats resistant to audiogenic kindling may be relevant to understanding pathophysiological mechanisms of migraine. In these rats, sound evoked only brainstem seizures (brief episodes of running) and unilateral spreading depression was the only and extremely reproducible response of the cortex to its repeated upstream activation from the brainstem. Actual trigger(s) of spreading depression (migraine aura) in the intact cortex of migraine patients remains unknown. It is accepted that spreading depression may occur as a result of aberrant neuronal firing in the hyper-excitable cortex (11). But so far, the nature of brain dysfunction that can lead to such excitation of cortical neurons and spontaneous initiation of spreading depression remains unknown. In experimental models, spreading depression is induced by local direct stimulation or injury to the cortex and there is yet no evidence for spontaneous triggering spreading depression by only synaptic mechanisms. Our studies in the audiogenic seizure model have demonstrated that a unilateral cortical spreading depression wave can be synaptically triggered by a brief activation of ascending inputs to the cortex during episodic sensory-induced dysfunction of the brainstem.
It is well-known that both the cortex and brainstem are involved in the pathophysiology of migraine (23,34). Although the migraine aura has certainly a cortical origin, imaging studies have demonstrated profound activation of the dorsal midbrain and rostral pons at the earliest phase of migraine attack (23,34–36). Based on the imaging data, it has been hypothesized that pathogenesis of migraine depends on episodic dysfunction of brainstem pathways (23). However, a causative link between cortical and brainstem alterations and a site of primary brain dysfunction are still debated. One theory suggests that the brainstem activation is secondary to activation of meningeal nociceptors by cortical spreading depression (28–31). The alternative view considers migraine as a subcortical disorder in which brainstem dysfunction is a primary event leading to secondary activation of cortico-subcortical networks and development of migraine symptoms, including aura (23,36,37). The results of our experimental studies showing easy synaptic initiation of unilateral cortical spreading depression by episodic excitation of the brainstem suggest that migraine aura (cortical spreading depression) may be a secondary event produced by abnormal upstream activation of the cortex.
In addition to a drive that ascends from the activated brainstem, triggering spreading depression in the cortex of rats with innate acoustic hypersensitivity certainly involves cortical excitability changes. Spreading depression was never induced by first brainstem audiogenic seizures and propensity to trigger cortical spreading depression increased with the seizure number. Moreover, spreading depression appeared more rapidly in rats expressing cortical seizures as compared to those resistant to cortical epileptogenesis. Because it is known that repeated audiogenic seizures lead to gradual development of cortical hyper-excitability (15,17) and occurrence of spreading depression increases with raising excitability of the cortex (32,38), the present findings suggest that elevated cortical excitability is the prerequisite for the spreading depression initiation by brainstem seizures.
Mechanisms of triggering cortical spreading depression by brainstem audiogenic seizures certainly involve synaptic processes leading to intense neuronal depolarization either due to direct excitation of cortical neurons or via a cascade of events (changes in ionic balance etc.) activating a regenerative process of spreading depression. It is known that locally triggered spreading depression propagates independently of synaptic activity at the slow velocity of 2–6 mm/min (19,26,38). The non-synaptic propagation of spreading depression over the grey matter occurs in all directions and involves subsequently distant parts of the cerebral cortex. In humans, neuroimaging data have shown that the spreading depression-like process during migraine aura is triggered at the extrastriate visual cortex of one hemisphere (27). In the gyrencephalic human brain, spreading depression propagation is restricted to the occipital lobe due to significant cortical convolutions preventing long-distance non-synaptic spread of spreading depression (27). In the lisencephalic cortex of rats, locally triggered spreading depression easily spreads across all the cortex of the hemisphere, although never crosses the midline (26,38). In our experiments, unilateral spreading depression appeared in the motor, somatosensory, occipital and auditory areas of the rat cortex with a long time delay (1–2 min) after a brainstem seizure. The delayed appearance of unilateral spreading depression in the neocortex indicates its local triggering in a restricted cortical area of one hemisphere and subsequent non-synaptic propagation over the cortex.
Previously, we have shown a close relationship between lateralization of the unilateral cortical spreading depression and asymmetry of brainstem audiogenic seizures triggering spreading depression (39). It has been found that cortical spreading depression is always ipsilateral to brainstem epileptic excitation, suggesting lateralized activation of ascending brainstem-to-cortex pathways. It is interesting to note that migraine patients also exhibit lateralized brainstem activation tightly correlating with lateralization of headache (40).
The results of the present study have shown that development of mild cortical seizures does not affect the long-latency unilateral pattern of spreading depression waves triggered by brainstem audiogenic seizures. However, intense epileptic activation of the cortex (at the late kindling stages) leads to bilateralization of spreading depression and great shortening of its latency. Immediate post-seizure appearance of spreading depression in all sensory cortical regions suggests that, in the highly hyper-excitable sensory cortex, initiation of spreading depression occurs in multiple sites as a result of generalized lowering of the spreading depression thresholds. However, such expansion of the spreading depression initiation area during severe cortical seizures does not seem to involve the motor cortex. In this cortical region, spreading depression appears with a 40-s delay after the end of the seizures, indicating that spreading depression is not generated but propagates here from other cortical regions. Given identical seizure patterns in sensory and motor areas of the neocortex, this result indicates a regional difference in sensitivity to triggering spreading depression by seizures. The sensory cortex seems to be more inclined to spreading depression generation than the motor cortex.
Limitations of the experimental study are due to: (i) structural and functional differences between the human and rodent cortices; (ii) a difference in neuroanatomical substrates between human epilepsy and rodent audiogenic seizures. The cortex is thought to play the leading role in human epilepsy whereas audiogenic seizures have brainstem origin. Nevertheless, evidence is accumulating that the brainstem is tightly implicated in expression of generalized seizures (13,21,22) and the last concept for International League Against Epilepsy classification of epilepsies postulates that seizures may originate not only in the cortex but in subcortical sites too (41).
To sum up, the results of the present experimental study suggest a very low threshold for synaptic initiation of cortical spreading depression by ascending drive from subcortical structures involved in sensory perception. The study describes a subpopulation of rats in which a unilateral spreading depression is the only manifestation of cortical hyperexcitability produced by repeated episodes of brainstem dysfunction (audiogenic seizures).
Footnotes
Article Highlights
This basic research study shows that spreading depression, the electrographic correlate of migraine aura, is easily triggered by upstream synaptic activation of the cortex, irrespective of its epileptic excitation.
Experiments were performed in awake rats with innate acoustic hypersensitivity (reflex audiogenic epilepsy) in which sound induces epileptic activation of the brainstem. Repetition of brainstem seizures can lead to secondary epileptic activation of the cortex as a result of seizure propagation along ascending pathways. The present study has shown that spreading depression is the most frequent and early event triggered in the cortex by repeated brief brainstem seizures.
Unless prolonged and severe seizure activity developed in the cortex, brainstem seizures were followed by a unilateral cortical spreading depression wave.
The study provides experimental evidence to suggest that a dysfunction of the brainstem may be a causative factor for triggering unilateral cortical spreading depression (migraine aura).
Funding
Supported by Russian Fund of Basic Research (14-04-01184).
Conflict of interest
None declared.