
Abstract
Background
Migraine is considered a multifactorial genetic disorder. Different platforms and methods are used to unravel the genetic basis of migraine. Initially, linkage analysis in multigenerational families followed by Sanger sequencing of protein-coding parts (exons) of genes in the genomic region shared by affected family members identified high-effect risk DNA mutations for rare Mendelian forms of migraine, foremost hemiplegic migraine. More recently, genome-wide association studies testing millions of DNA variants in large groups of patients and controls have proven successful in identifying many dozens of low-effect risk DNA variants for the more common forms of migraine with the number of associated DNA variants increasing steadily with larger sample sizes. Currently, next-generation sequencing, utilising whole exome and whole genome sequence data, and other omics data are being used to facilitate their functional interpretation and the discovery of additional risk factors. Various methods and analysis tools, such as genetic correlation and causality analysis, are used to further characterise genetic risk factors.
Findings
We describe recent findings in genome-wide association studies and next-generation sequencing analysis in migraine. We show that the combined results of the two most recent and most powerful migraine genome-wide association studies have identified a total of 178 LD-independent (r2 < 0.1) genome-wide significant single nucleotide polymorphisms (SNPs), of which 99 were unique to Hautakangas et al., 11 were unique to Choquet et al., and 68 were identified by both studies. When considering that Choquet et al. also identified three SNPs in a female-specific genome-wide association studies then these two recent studies identified 181 independent SNPs robustly associated with migraine. Cross-trait and causal analyses are beginning to identify and characterise specific biological factors that contribute to migraine risk and its comorbid conditions.
Conclusion
This review provides a timely update and overview of recent genetic findings in migraine.
Introduction
Migraine is considered a multifactorial (complex) genetic disorder with a strong familial aggregation (1,2). Complex traits are typically brought about by a combination of multiple genetic variants, as well as behavioural and environmental factors, each with a small effect size. In this review, we provide an overview of the recent genetic findings with a focus mainly on common polygenic migraine. Furthermore, we discuss state-of-the-art strategies for performing genetic studies of migraine.
Rationale for genetic studies
When identifying the gene(s) involved in a disorder, different approaches are used depending on the disorder’s genetic architecture (i.e. monogenic, oligogenic, or polygenic). The more oligogenic a disease is (i.e. the lesser the number of genes involved, with monogenic being the extreme), the larger the effect size of the associated genetic variant(s) tends to be, in line with epidemiological data from disorders where rare disorders are monogenic and common disorders are polygenic.
Hemiplegic migraine
A substantial amount of our knowledge of molecular mechanisms in migraine pathophysiology came from studying rare hemiplegic migraine (HM) (3). The classical linkage method in migraine research was used to study large families with HM and revealed a clear Mendelian (monogenic) type of inheritance. This led to the identification of three undisputed HM genes; CACNA1A (FHM1), ATP1A2 (FHM2), and SCN1A (FHM3) (4–6). Detailed information on how mutations in HM can be studied in cellular and animal models and how they cause disease can be found elsewhere (see [7,8]).
In many patients with HM no pathogenic mutation has been detected in the HM genes (9,10). In recent years, whole-exome (next-generation) sequencing (WES) has been used to try and identify additional causal genes in patients without mutations in the known HM genes, but this has proven difficult and no “fourth” gene has been identified thus far (11). A study by Pelzer et al. (11) did, however, show that patients with a more severe phenotype were more prone to have a causal mutation in one of the HM genes. Patients with a causal mutation in CACNA1A, ATP1A2, or SCN1A had a lower age-at-onset, more affected family members, and had attacks more frequently. Moreover, attacks were i) brought about by mild head trauma, ii) typically with extensive motor weakness, and iii) with brainstem features, confusion, and brain oedema. Noteworthy, progressive ataxia and intellectual disability were only found in patients with a causal gene mutation (11). As no causal mutation was found in “milder” patients, it was proposed that such HM patients have the more extreme phenotype in the migraine with aura continuum (9). Illustrative of this is a Finnish polygenic risk score study that showed that patients with HM, but without a high-penetrant disease-causing mutation in a known HM gene, carry an excess of genome-wide association studies (GWAS) variants associated with common migraine compared to patients suffering from the common migraine subtypes (12), suggesting a spectrum ranging from common low-risk variants to rare high-penetrant mutations contributing to the risk for migraine. Further support for this are loss-of-function mutations in PRRT2, which do not cause HM on their own, but rather function as modifying risk factors (13). A further illustration of the complex genetic architecture of HM is a recent whole-genome sequencing (WGS) study where patients with HM were more likely to accumulate frameshift indels in multiple genes that have a role in synaptic signalling in the central nervous system compared to patients with common migraine (14).
Genetic studies in common migraine
Various twin and familial studies investigating the genetic and environmental susceptibility in migraine have shown that migraine is a multifactorial (complex) genetic disorder with a strong familial aggregation (1,2). The heritability of migraine was estimated to range from 35% to 60% (15). Population-based studies have shown that the relative risk for a first-degree relative of a migraine patient is increased by 1.5- to 4-fold in comparison to a patient in the general population (1). The risk was highest for those patients with a higher pain score and frequency of attacks, an early age of disease onset, and a migraine with aura phenotype (1,2,16). Studies of twins identified a higher genetic load in migraine with aura compared with migraine without aura (17). Migraine frequency, being the number of migraine days per month, appears mainly to be associated with a genetic predisposition in males (16). A stronger family history of migraine is also associated with migraine with aura, a lower age-at-onset and more medication days (16). For decades, identifying gene variants involved in complex disorders, such as migraine, has proven challenging.
Genome-wide association studies in migraine
As a result of the improvement in DNA technology and the advancement of cost-effective genotyping platforms, GWAS has become the method of choice to identify DNA risk variants for complex traits. Typically, in GWAS, several millions of single nucleotide polymorphisms (SNPs) are tested for association with migraine by assessing differences in allele frequencies between very large numbers of (migraine) patients and controls. Of note, only common variants with a low to high minor allele frequency (MAF ≥ 0.01) are interrogated.
Since 2010, the International Headache Genetics Consortium (IHGC; www.headachegenetics.org/) has conducted several migraine GWAS, and with increasing sample sizes, the number of associated risk variants steadily expanded.
The 2016 migraine GWAS by Gormley et al. (18) reported 44 linkage disequilibrium (LD)-independent SNPs mapping to 38 distinct genomic loci associated with migraine. Downstream bioinformatics analyses focussed on the respective functions of the likely associated genes and concluded that GWAS findings in migraine are primarily linked to arterial and smooth muscle function. This is in line with the shared polygenic risk between migraine, stroke, and cardiovascular diseases that has been described previously (19,20). Finucane et al. (21), through heritability enrichments of the GWAS summary statistics, also showed a neurological enrichment for migraine (all subtypes) and a cardiovascular enrichment for migraine without aura. In addition to vascular and neuronal mechanisms, additional mechanisms like metal ion homeostasis were indicated to contribute to migraine susceptibility (22).
The most recent IHGC migraine GWAS published in 2022 by Hautakangas et al. (23) included 102,084 cases and 771,257 controls and identified 123 distinct genomic regions associated with migraine, of which 86 loci were novel compared to the 2016 migraine GWAS (18,23). In addition, the authors reported 48 genome-wide significant variants that were LD-independent (r2 < 0.1 within either UK Biobank [UKBB; www.ukbiobank.ac.uk] or 23andMe [www.23andme.com]) of the 123 lead variants, bringing the total to 171 independent index SNPs. When examining the 171 SNPs in the 1000 Genomes Project (1kGP; www.internationalgenome.org) EUR reference panel, four SNPs were found to be in LD (rs139846391–rs2078371, r2 =0.247; rs4240603–rs4599655, r2 = 0.564; rs1416901–rs10828247, r2 = 0.958; rs7148352–rs11624776, r2 = 0.102), resulting in 167 independent SNPs (r2 < 0.1) (Online Supplementary Table 1).
Enrichment analyses in the 2022 migraine GWAS implicated the involvement of both vascular and central nervous system tissue/cell types, well in line with previous data and the concept that migraine is a neurovascular disorder. In addition, for the first time, risk loci were found that encode genes that had been identified as migraine drug targets, namely the calcitonin gene-related peptide (CGRP encoded by CALCA/CALCB) and the serotonin 1F receptor (HTR1F). An increase in CGRP has been observed in migraine patients outside and during an attack (24,25), which led to the development of CGRP (receptor)-related small molecule (gepants) and monoclonal antibodies with proven efficacy to treat migraine (26). The gene that encodes the serotonin 5-HT1F receptor is a target for which migraine drugs are being developed; these drugs are known as ditans (27).
The 2022 migraine GWAS also evaluated both shared and distinct genetic components of migraine subtypes (migraine without aura and migraine with aura) (23). A subsequent analysis of the 123 lead index SNPs using 29,679 (15,055 migraine without aura and 14,624 migraine with aura cases) clinically well-diagnosed cases, indicated with a probability above 95% three risk variants (in MPPED2, CACNA1A and HMOX2) that were specific for migraine with aura, two (one near FECH and one near SPINK2) that were specific for migraine without aura, and nine (i.e. DLST, PRMD16, SUGCT, PLEC1, MRVI1, LRP1, FHL5, near TRPM8, and near FGF6) that were associated with both migraine subtypes (23). For comparison, in the 2016 migraine GWAS, seven loci (i.e. ASTN2, PHACTR1, LRP1, FHL5, TRPM8, near FGF6, and near TSPAN2) were associated specifically with migraine without aura (in a sample of 8,348 cases vs 139,622 controls) and no gene association was found for migraine with aura (6,332 cases vs 144,883 controls) (18). In the 2022 migraine GWAS, there was no evidence that any of the seven loci were specific for migraine without aura, still, four of them (LRP1, FHL5, near TRPM8, and near FGF6) are among the nine loci that were associated with both subtypes. The fact that multiple loci are strongly associated with both migraine with and migraine without aura provides further genetic evidence for the concept that migraine subtypes in part share a genetic background (7,28,29). One of the new migraine loci identified, the CACNA1A gene, links to both monogenic and polygenic forms of migraine (7,23).
In a third recent migraine GWAS from 2021 by Choquet et al. (30), 79 independent loci were significantly associated with migraine (online Supplementary Table 2). Of note, this was an ethnically diverse study that included adult individuals (28,852 cases vs. 525,717 controls) from East Asian, African American, and Hispanic/Latino descent.
Both Hautakangas et al. (23) and Choquet et al. (30) GWAS used data from Gormley et al. (18) supplemented with additional cohorts (online Supplementary Table 3) and both compared their findings with those of Gormley et al. (18), but not with each other. Therefore, we compiled and compared the 171 (167 LD-independent with r2 < 0.1) genome-wide significant SNPs listed in Supplementary Table 1 of Hautakangas et al. (23) and the 95 (79 LD-independent with r2 < 0.1) genome-wide significant SNPs drawn from Supplementary Data 1 and Supplementary Data 4 of Choquet et al. (30). Analysis of these SNP lists resulted in a total of 178 LD-independent (r2 < 0.1) genome-wide significant SNPs across both studies, of which 99 were unique to Hautakangas et al. (23), 11 were unique to Choquet et al. (30), and 68 were found in both studies (Figure 1, Table 1). This means that 41% of the 167 Hautakangas et al. (23) index SNPs were genome-wide significant in Choquet et al. (30) and 86% of the 79 Choquet et al. (30) index SNPs were genome-wide significant in Hautakangas et al. (23). In addition, three additional (novel) loci rs1047891, s11718509, and rs10150336 (CPS1, PBRm1, and SLC25A21, respectively) specific for women with migraine, were identified by Choquet et al. (30). This means that there are now at least 181 SNPs robustly associated with migraine.
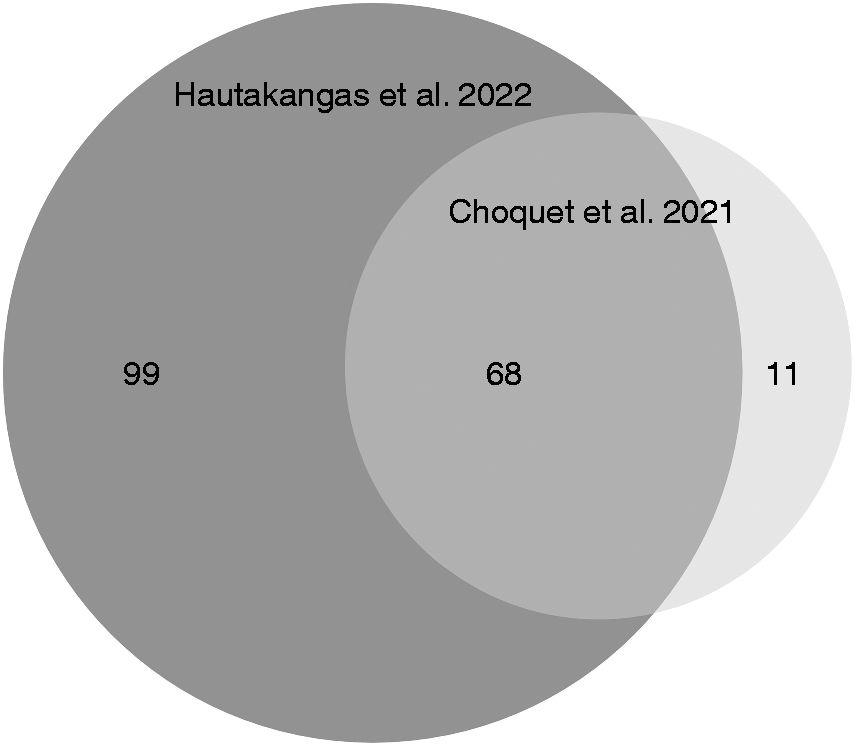
Venn diagram of the genome-wide significant index SNPs across Hautakangas et al. (2022) (dark grey) and the Choquet et al. (2021) (light grey) migraine genome-wide association studies. The size of the circles is representative of the number of SNPs.

Chr, chromosome; Pos, nucleotide position in hg19; SNP, single nucleotide polymorphism.
Clearly, an increase in sample size leads to more migraine loci all of which have a small effect size indicating that none of the identified associated variants, unlike mutations in monogenic HM, are sufficient to bring about disease. As a note of caution, the increase in sample size primarily comes from large population-based datasets, such as UK Biobank and 23andMe (online Supplementary Table 3), that make up the larger part of recent migraine GWAS; 63,990 (63%) of the 102,084 cases in Hautakangas et al. (23) and 47,997 [54%] of the 88,526 cases in Choquet et al. (30). The obvious consequence is a loss of clinical specificity. Not only are cases in these public datasets less specifically defined, the controls are often not necessarily healthy compared to those in a clinic-based cohort, so both cases and controls may be genetically confounded by comorbid conditions that share a genetic aetiology with migraine. Hence, population-based GWAS are poorly suited to identify SNPs associated with specific migraine subtypes. Regardless, including population-based cohorts still renders meaningful findings, already because headache in general is a rather easily definable phenotype. This was exemplified by Meng et al. (31), where a GWAS was performed using the UK Biobank based on a broad definition of headache. Half of the identified 28 associated SNPs were loci that previously had been associated with migraine. So even though the study population likely was rather heterogeneous clinically, there were still known migraine loci present. A possible next step could be to perform more detailed analyses comparing and combining GWAS results for headache and migraine.
Until now, the majority of migraine GWAS have been performed in individuals of European descent, hence the results are not directly transferable to other populations. The first study to investigate migraine in a non-European GWAS concluded that of the three risk variants known at the time, i.e. rs10166942 (TRPM8), rs2651899 (PRDM16), and rs11172113 (LRP1), only the first one showed association with migraine in a Han Chinese population (32,33). In a later Chinese study, only two, i.e. rs4379368 (C7orf10) and rs13208321 (FHL5), of five ‘European’ migraine risk variants showed association with migraine in the She population (34). A subsequent Chinese replication study of 581 migraine cases and 533 ethnically matched controls identified three risk loci rs2274316 (MEF2D), rs6478241 (ASTN2) and rs2651899 (PRDM16) previously identified in European samples (35). In a North Indian population, researchers performed a replication study of five migraine GWAS loci, i.e. rs1835740 (near MTDH), rs11172113 (LRP1), rs2651899 (PRDM16), rs10166942 (TRPM8) and rs7640453 (TGFBR2)), of which only the first three showed association (36) and in another study, only rs2651899 (PRDM16) was associated with the risk for migraine without aura (37).
In addition to replicating European risk loci, the first GWAS have been performed in Asian populations, albeit all with small sample sizes. For instance, a recent GWAS in the Han Chinese population (1042 patients vs 2264 controls) revealed two SNPs, i.e. rs10456100 (KCNK5) and rs7775721 (FHL5), which were significantly associated with migraine without aura (38). A separate study of 715 patients showed the first associations with age of onset of migraine in Han Chinese (39) and a GWAS in 2084 Asian patients with self-reported headache and 11,822 sex- and age-matched controls reported two migraine loci, i.e. rs10493859 (TGFBR3) and rs13312779 (FGF23) (40).
Taken together, it can be concluded that the GWAS era has come of age and already yielded many dozens of low-effect-size DNA variants; very many of which replicate in other GWAS, hence providing overwhelming proof of the robustness of the findings. Still, larger studies, also in non-European populations, have to be performed to identify additional loci and establish to what extent associated variants play a role in migraine patients across the globe. As a proof of concept, it is interesting that some SNPs point to already identified treatment targets such as CGRP and 5-HT1F. This clearly shows the potential of genetic studies to identify treatment targets, which is dearly needed as many patients are unresponsive to current treatments. Studying specific phenotypes, such as migraine with aura or chronic migraine, remains challenging as in most GWAS data only limited data are available on subtypes of migraine and, to certain extent, their characteristics.
Another important challenging aspect of GWAS that remains is to determine the exact gene that is affected by a risk SNP, as this is far from straightforward. Major complications are that the associated SNPs are typically located in intronic or intergenic regions and are merely genomic markers at a distance, so in LD with the true causal variant. An often-used method is to link a risk index SNP (i.e. the SNP with the lowest p-value) to the “nearest gene”. However, it was shown that in two-thirds of cases it is not the nearest gene that is affected by the risk SNP (41,42). Notably, there can be multiple independent association signals at the same locus (secondary SNPs) that can influence other regulatory features of the same (or nearby) gene. In the 2016 migraine GWAS (18), of the 38 distinct genomic loci, six contained such a secondary SNP.
Risk SNPs in common diseases rarely affect amino acid changes and most likely indirectly regulate gene expression, for instance by disrupting enhancer elements. Of note, the causal gene can be located even hundreds of kilobases away from the location of the risk SNP. Hence, a key challenge for the future is to effectively integrate data from GWAS and expression quantitative trait loci (eQTL) analyses (and other omics data) to prioritise likely causal genes, which is needed for meaningful functional follow-up of GWAS findings (43). The magnitude of the challenge is well-demonstrated by the functional follow-up of intronic SNP rs9349379 near PHACTR1 that surfaced not only in migraine GWAS but also in multiple GWAS of vascular diseases (e.g. coronary artery disease, fibromuscular dysplasia, hypertension and cervical artery dissection) (44). Detailed functional follow-up indicated that this SNP affects not PHACTR1, but endothelin-1 (ET-1; EDN1), a strong vasoconstrictor that acts on smooth muscle cells and is located no less than 600 kb upstream of the risk SNP (44). Still, the debate on which gene is affected rs9349379 has not been settled. More recent data have revived PHACTR1 as the causal gene based on the changed expression of PHACTR1, and not EDN1, in cells that carry the risk allele and functional human and mouse studies of PHACTR1/Phactr1 revealing a role, in arterial compliance—and not measures of dynamic arterial function—across multiple vascular beds (45).
Studies like these indicate that the road ahead to understanding the pathophysiology behind migraine risk loci is far from easy. Similar challenges apply to other disease areas (46). Clearly, we are only at the beginning of performing “wet lab” functional follow-ups of GWAS risk SNPs, which will likely require a plethora of animal- and/or cell-based assays depending on the variant/gene that is affected (47).
What about candidate gene association studies?
Before the advent of high-throughput low-cost genotyping, so-called candidate gene association studies (CGAS) were performed. These tested for association with migraine one or a few SNPs in genes from neurological, vascular, hormonal, and mitochondrial pathways that had been selected because of their perceived involvement in migraine pathophysiology. Unfortunately, findings in CGAS almost exclusively came from too small sample sets and were not robustly replicated, so the risk of a false-negative or false-positive result is very high. Details of CGAS findings can be found in other reviews (48,49). In fact, a study by de Vries et al. (50) investigated whether 27 genes from both CCAS and other genetic studies showed an association with migraine in IHGC GWAS data from a large sample of 5175 clinic-based migraine patients with and without aura (and 13,972 controls) and found none to be associated with migraine. A genome-wide significant result was found for one of the 27 genes, namely the CACNA1A gene, in the Hautankagas et al. paper (23), but gene-based analyses did not show significant association for any of the other 26 genes (data not shown), even though the sample size had considerably increased. Because of the possibility of a straightforward “look-up” of genes of interest, i.e. by gene-based analysis testing, GWAS can replace GCAS studies. One might still consider wet lab testing of specific SNPs and/or investigating specific study populations, such as response to certain medications, as long as one takes into account sample sizes and replication of promising findings.
Next-generation sequencing in migraine
A large part of the genetic variance and heritability in common diseases cannot be explained (usually referred to as “missing heritability”) with a GWAS approach alone. One reason is that rarer variants (MAF < 0.01), potentially with larger effect sizes, are not well interrogated by genotyping arrays typically utilised in the GWAS approach. Such mediate-effect-size variants can be identified using a next-generation sequencing (NGS) approach, i.e. by the simultaneous large-scale sequencing of the coding exons (whole-exome sequencing; WES) or the entire genome (whole-genome sequencing; WGS). In addition, the simultaneous sequencing of RNA transcripts (“transcriptome”; RNA-seq), either of bulk tissue or of its single nuclei can shed light on molecular mechanisms.
Exome and genome sequencing in migraine
Only a few NGS studies have been performed in migraine thus far. Until now, WES was typically applied to cohorts of patients with HM, testing several hundred cases in an attempt to either find causal mutations in known HM genes or novel HM genes patients that are negative for mutations in CACNA1A, ATP1A2, and SCN1A. Until now results have not led to additional (undisputed) HM genes (9–11,51,52). This may indicate that HM in mutation-negative patients may be oligogenic or polygenic, in line with the excess presence of common variants in such patients (12). A few recent studies in the field of migraine used NGS for a different approach. First, SNP genotyping followed by NGS was used for linkage, haplotype, and variant analyses within a single large Finnish migraine-epilepsy family and found an association between the epilepsy phenotype and the NCOR2 gene that colocalises with one of the migraine risk loci (53). Second, in a WES of 16 individuals, who developed numerous neurological- and concussion-related symptoms following minor head injuries of which seven had developed migraines following the injury, revealed possible mutations in various ion channel, neurotransmitter, and ubiquitin-related genes, but causality of any of them needs to be established (51). Third, in an attempt to investigate whether certain genetic variants determine treatment response to verapamil prophylaxis, WES was applied to a set of 21 definitive responders and 14 definitive non-responders (discovery cohort) and promising data were further analysed in 185 verapamil-treated patients (replication cohort) to yield 39 ‘possible variants’ (54). Subsequent bioinformatics analyses revealed the possible involvement of myo-inositol biosynthetic and phospholipase-C second messenger pathways in verapamil responsiveness (54).
The added value of the WGS approach, when combined with GWAS data, was shown in a recent Danish study of extended migraine pedigrees (encompassing 1040 individuals from 155 families) (55). An excess of rare segregating variants in regulatory regions (one CpG island and three polycomb group response elements) was identified near, but independent of, four migraine GWAS risk loci with replication in an independent case-control cohort (encompassing 2027 migraineurs and 1650 controls). Another study of the same Danish group combined RNA sequencing data from brain and vascular tissues relevant to migraine with WGS data of over one hundred migraine families in a ‘systems genetics approach’ and identified a “visual cortex module” with rare functional gene variants implicated in migraine pathophysiology (56). Their approach suggested the involvement of glutamate, serotonin, G-protein signalling pathways and hormonal pathways in migraine pathophysiology.
Gene expression analysis in migraine
An alternative approach to understanding molecular mechanisms involved in migraine pathophysiology is to study gene expression profiles. Contrary to genetic variation, gene expression is not fixed through life and expression is driven by both genetic and environmental factors (57). Typically, an RNA-seq approach (i.e. simultaneous sequencing of coding (messenger) and non-coding RNAs in a sample) is for instance used to identify differences in expression between individuals with and without migraine or over the course of a migraine attack. Various RNA-seq studies have been performed in migraine, but the results are not unambiguous not in the least because of potential caveats of using peripheral blood, the main source of biomaterial for such studies in the case of migraine (58). First, an Australian genome-wide expression study comparing whole blood expression profiles of 83 migraine cases and 83 non-migraine controls revealed that immune-inflammatory pathways, including multiple pathways expressed in microglial cells, seem to play a role in migraine (59). Second, RNA-seq-based expression profiles in the blood of 24 Hungarian migraine patients, collected during both the headache and the headache-free (interictal) period, were compared with that of 13 sex- and age-matched healthy controls (60). Some 144 differentially expressed genes (DEGs) were identified when comparing the headache and headache-free samples in patients and 163 DEGs were identified between the interictal and control samples. Subsequent network analysis, revealed enrichment of inflammation, like in the study mentioned above, cytokine activity and mitochondrial dysfunction pathways between patients (both headache and headache-free) and controls. On the contrary, no overt differences in RNA-seq expression profiles were observed when comparing interictal blood samples of 26 Danish migraine patients with those of 20 age- and sex-matched controls (61). However, a comparison of gene expression profiles of blood samples from 27 Danish migraine patients taken during the attack, two hours after treatment, and on a headache-free day, revealed 29 DEGs between “attack” and “after treatment” (62). Among the results of their gene network analysis appeared the “5HT1-type receptor mediated signalling pathway”, which is not that surprising given the sumatriptan treatment patients had received. Finally, a study by LaPaglia et al. (63) used RNA-seq to assess gene expression in sub-regions of 16 human post-mortem trigeminal ganglia, as to establish the human transcriptome from a brain region highly relevant to migraine pathophysiology, already because trigeminal neurons release neuropeptides such as CGRP (64).
Single-cell genomics in migraine
Single-cell genomics, i.e. RNA sequencing of single cells (scRNAseq)/single nuclei (snRNAseq), provides unique opportunities to elucidate in greater detail not only which genes but also in which cell types migraine-associated genes are expressed. The latter is especially relevant to better understand how migraine susceptibility genes, for instance from recent migraine GWAS, may affect migraine pathophysiology.
A major advancement in single-cell sequencing in migraine came from a study by Renthal (65), which used single-brain cell RNA sequencing data from healthy human cortex cells (astrocytes, oligodendrocytes, neurons, microglia and endothelial cells) and revealed that over 40% of migraine-associated genes showed a preference for a specific brain cell type. Strikingly, some 70% of the neuronal migraine-associated genes were significantly enriched in inhibitory neurons compared to 30% in excitatory neurons (65). In a follow-up scRNAseq mouse study encompassing more cells (∼500,000 vs ∼3000) and cell types across more nervous system structures implicated in migraine, it was shown that expression of 52% (28/54) of the investigated migraine-associated genes was enriched in both the central (CNS) and peripheral (PNS) nervous system, 20% was enriched in the CNS, PNS and neurovascular cell types, and some 17% showed tissue-selective enhancement (66). The study indicated that most migraine-associated genes are widely expressed, with functional changes that likely affect multiple cell types. A study like this i) may guide future mechanistic studies into the function of migraine-associated genes and ii) holds the promise that it may be feasible to stratify patients based on a predominant CNS or PNS cell type polygenic migraine risk. Finally, the same group created a transcriptional and epigenomic cell atlas of the mouse and human trigeminal ganglion (TG) at single-cell resolution and revealed species-specific gene expression and evolutionarily conserved in distinct TG cell types (67). The TG atlas will be an enormous asset to migraine research given the relevance of TG neuron sensitisation in migraine, while the relative contribution of individual neuronal and non-neuronal TG subtypes still is unknown. It is to be expected that the TG atlas will also guide the search for cell types most relevant to migraine-associated variants. Of note, the expression of CALCA, CALCB, and HTR1F, which are among the targets of current migraine treatments (23), as well as various other genes implicated by GWAS, exhibit species-specific and cell-type-specific expression patterns. To assess the TG cell types involved in migraine, two experimental mouse migraine models, i.e. the inflammatory soup (IS) model (relevant for activating and sensitizing trigeminal meningeal nociceptors) and the cortical spreading depolarization (CSD) model (the neurophysiological correlate of the migraine aura) were used to identify activated TG cells, based on the expression of immediate early genes. Using IS as a trigger some 5% of TG cells were activated, whereas the number was slightly lower (3.5%) when the less profound trigger CSD was induced. It is reassuring that at least some convergence was present at the TG cell type activation level between both experimental models. For instance, a comparison of activated vs non-activated nuclei revealed 96 upregulated genes in the IS model and 72 genes in the CSD model, and within both models, genes were involved with axon guidance and inflammation and several were near to or predicted to be influenced by migraine-associated GWAS variants. Of note, the study also has clear epigenomic implications but these are discussed in the parallel review in this issue by Gallardo et al. (68).
Genetic relation with comorbid disorders
Migraine is associated with a high prevalence of stroke and psychiatric comorbidities, such as anxiety, depression, and post-traumatic stress disorder (69–72). Although the exact relationship remains elusive, twin and family studies have revealed a strong heritable factor in migraine and various other brain disorders (15). Comorbidity between disorders can be due to a causal relationship and/or may be due to shared genetic and/or environmental factors. Epidemiological comorbidity and sharing of symptoms can stem from aetiologic overlap. To investigate shared genetic architecture between migraine and other traits correlation analyses using GWAS data can be performed.
Genetic correlation with psychiatric disorders
In correlation studies, the proportion of variance two traits share due to genetics, defined as the genetic correlation rG, is measured. A recent example that shed important light on the genetic architecture underlying migraine comorbidities came from the Brainstorm Consortium in which correlations were calculated to quantify the degree of genetic overlap between 25 brain disorders using the GWAS data of over 1 million individuals (265,218 patients and 784,643 controls) (73). A major finding of the study was that psychiatric disorders seem to share more common genetic risk variants, whereas neurological disorders seemed more distinct from each other and from psychiatric disorders. The notable exception was migraine, which was significantly correlated to major depressive disorder (MDD), attention deficit hyperactivity disorder (ADHD), and Tourette syndrome (73).
The genetic correlation of migraine with MDD was confirmed in a separate study that showed a significant genetic correlation between the two disorders (rG = 0.25; P = 0.04) (74). In that study, a meta-analysis of the combined GWAS summary statistics of migraine and MDD revealed three novel risk loci associated with both disorders, and a subsequent gene-based association analysis suggested the involvement of ANKDD1B and KCNK5 (74). More recently, it was shown that most migraine-associated variants also influence schizophrenia and depression, i.e. 36 loci were found to be associated with migraine and schizophrenia and 14 loci were associated with both migraine and depression (75).
Genetic correlation between migraine and other disorders
Several disorders are comorbid with migraine and show at least some genetic overlap. First, epidemiological studies have demonstrated repeatedly an increased risk of ischemic stroke in migraine patients, particularly for those with migraine with aura (76,77). Evidence for an overlap at the genetic level between migraine and ischemic stroke was shown by Malik et al. (20). Second, migraine and endometriosis share epidemiological characteristics and risk factors, such as female dominant prevalence and the involvement of the menstrual cycle (78). Although co-occurrence of migraine and endometriosis already suggested that both traits may also be genetically correlated (79), only recently it was demonstrated that there is a significant overlapping genetic architecture (rG = 0.38, P = 2.30 × 10−25) between endometriosis and migraine, with evidence for the involvement of various genes, among which TRIM32 and SLC35G6 (80). Third, sleep disturbances and sleep-related disorders have also been reported in migraine patients (81). One study showed a genetic overlap of seven sleep traits with migraine. Insomnia was the most significantly correlated with migraine (rG = 0.29, P = 1.87 × 10−32); weaker correlations were found for short sleep duration (rG = 0.18, P = 1.69 × 10−9), difficulty awakening (rG = 0.11, P = 2.02 × 10−5), daytime napping (rG = 0.11, P = 1.31 × 10−5), daytime sleepiness (rG = 0.09, P = 1.21 × 10−4), long sleep duration (rG = 0.12, P = 7.60 × 10−4), and sleep duration in general (rG = −0.08, P = 1.56 × 10−3) (82). Fourth, fibromuscular dysplasia was found positively correlated with migraine (rG = 0.28, P = 8 × 10−4) (83). Finally, a broad cross-trait correlation analysis of migraine reported genetic correlations with various disorders, including heart disease, type 2 diabetes, as well as various autoimmune and psychiatric phenotypes (84).
Genetic correlation between migraine and cluster headache
Migraine and cluster headache both share pathophysiological features, both show involvement of the trigeminovascular system, and both can be effectively treated with CGRP monoclonal antibodies and triptans (85). Recently, two parallel GWAS papers on cluster headache showed genetic overlap between migraine and cluster headache (86,87). Of note, a co-localization analysis showed that one of the top cluster headache loci (UFL1/FHL5) was the same as in migraine (87), which seems to reflect a partially shared genetic architecture underlying both migraine and cluster headache.
Genetic correlation between migraine and metabolites
In clinical practice, serum levels of chemical compounds are often utilised for diagnosing disease and monitoring the body’s function. As migraine is associated with an increased risk of cardiovascular disease (CVD) and stroke (77,88), routine chemistry tests and serum markers for these traits might be associated also with migraine. A recent study by Tanha et al. (89) shows that shared genetic factors between migraine risk and markers for CVD risk, iron deficiency and liver dysfunction exist. The shared genetic architecture implies an alteration of metabolite concentrations in individuals with migraine. Moreover, an additional study investigating the genetic underpinning and causality of the 316 blood metabolites and migraine risk revealed a significant correlation between migraine and 44 metabolites, largely organic acid and lipid metabolic traits (90). Changes in metabolite levels are in line with recent metabolomics studies in the blood of migraine patients (91,92).
Causality in migraine
Clear pleiotropy exists across many traits and one way of entangling this is to use Mendelian randomisation (MR). In an MR analysis, genetic variants associated with an exposure are identified and regressed upon an outcome measurement to infer causality (i.e. direction) of the association. Given the random assortment of alleles at gametogenesis in early life, this method is less likely to suffer from issues of confounding and reverse causation than methods used in conventional observational epidemiological studies (93). For a successful MR analysis, three assumptions need to be fulfilled (94). i) Variants used as instrumental variables (IVs) need to be associated with the exposure. ii) The IVs only affect the outcome through the exposure, not through any other causal pathway. Factors that may lead to violation of this assumption include population stratification, LD and horizontal pleiotropy, the latter means that there is an (in)direct independent association of the IV (or another SNP in LD with the IV) with another trait that is not in the causal pathway of the investigated relation. iii) The IVs must not be associated with confounders. Essentially, MR studies are still in their infancy, hence it is important and necessary to replicate the results of different studies to reach robust conclusions. In one-directional MR the possible causal relation between trait X on trait Y is investigated, in bidirectional MR studies the directional effect from trait Y on trait X is also investigated. An increasing number of MR studies related to migraine have already been performed (Table 2).
Papers describing a Mendelian randomisation analysis with migraine.
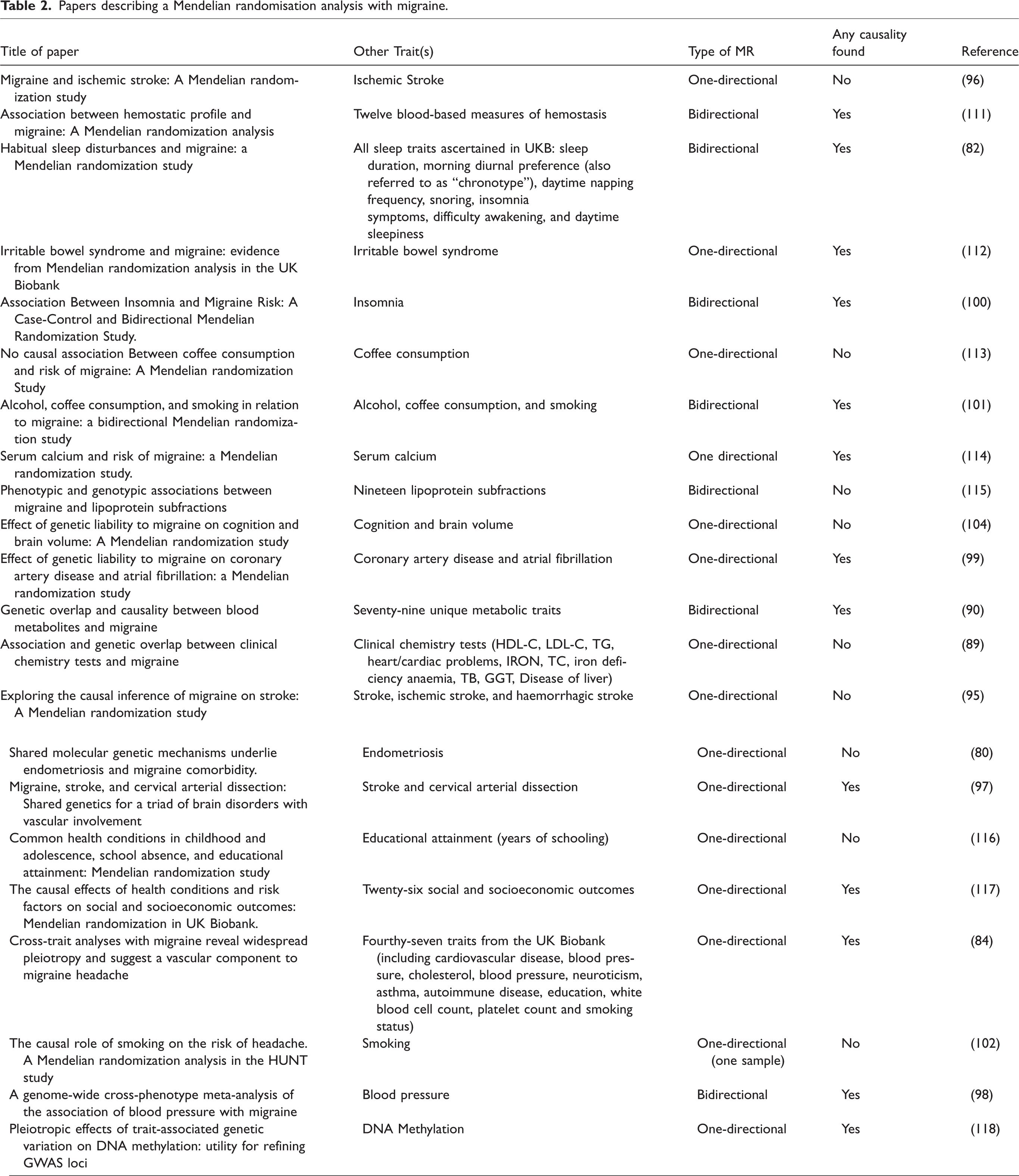
Causality between migraine and different traits
Of the many published MR studies that examine the relationship between migraine and a given disorder/trait, only some are discussed below. First, the causality between migraine and stroke has been investigated in three studies, two of which do not support a causal relationship between migraine and stroke (95–97), whereas one found a protective influence of large artery stroke (OR [95% CI] = 0.86 [0.76–0.96], P = 0.0067) on migraine, but no relation with stroke subtypes (97). Second, several other cardiovascular traits/risk factors and their possible causal relationship with migraine have been investigated, one showing that an increase in blood pressure directly contributes to migraine risk (84). A similar result was observed by Guo et al. (98) where an increase in both diastolic blood pressure (OR [95% CI] = 1.20 [1.15–1.25]/10 mmHg; P = 5.57 × 10−25) and systolic blood pressure (1.05 [1.03–1.07]/10 mmHg; P = 2.60 × 10−7) increased migraine susceptibility. In addition, liability to migraine leads to an increased coronary artery disease risk (99). Third, two studies explored the relationship with sleep-related comorbidities and provided evidence for a significant causal effect of difficulty awakening (OR [95% CI] = 1.37 [1.12–1.68], P = 0.002) (81) and insomnia (OR [95% CI] = 1.24 [1.11–1.38], P = 1.01 × 10−4) (100) on the risk of migraine. Fourth, various MRs exploring behavioural traits, such as smoking, alcohol and coffee consumption, have shown that a higher alcohol or coffee consumption leads to a reduced migraine risk (101). Although initially no causal relationship was found between smoking and migraine (102), the bidirectional MR study by Yuan et al. (101) showed that a decreased prevalence of smoking initiation led to reduced migraine risk. However, the reverse hypothesis that migraine leads to a reduced smoking initiation, was not found in this study (101). Although from a clinical perspective that might have been expected. Finally, despite the above-mentioned genetic correlation between migraine and endometriosis, there was no evidence for a causal relationship between both disorders (80).
In addition to hypothesis-driven MR studies, a recent hypothesis-generating phenome-wide analysis examined 1504 phenotypes and identified 17 potential causal relationships with self-reported migraine and traits related to e.g. stressful life events, vasoconstriction, platelet clumping, and occupational exposure to paints, glues or thinner, and diesel exhaust (103). Studies are also starting to emerge that examine other health-related markers such as brain volume (104), blood metabolites (90), and blood proteins (105). In addition to the caution that replication of positive MR findings is urgently needed, it is also important to note, that the power of a MR study is dependent on the size of the original GWAS. This means that when no causality is found at this moment, it can be causal in the future when there are more loci found.
Polygenic risk score in migraine
One way of making better use of the large number of small effect variants identified in migraine GWAS to have clinical benefit is the calculation of polygenic risk scores (PRSs). A PRS is the combined effect of many common risk variants of genetic load for the discovery trait that can be used to estimate risk for a certain trait/phenotype in individuals in a target sample (106). This is done by testing whether a higher PRS based on the discovery sample is associated with case status or a specific trait in the target sample via regression models. A PRS provides a promising possibility to investigate the shared genetic architecture between migraine with known and hitherto unknown co-morbidities or traits.
The aggregation of migraine in families and the earlier age of onset of migraine can to some extent be contributed to common polygenic variations, where the PRS explained a larger part of the phenotype variance in familial cases, especially those with migraine with aura and hemiplegic migraine compared to population cases (12). Other PRS research shows a correlation between the current ICHD-3 criteria and the PRS of migraine, where the migraine phenotype progressively follows the polygenic burden (107). A migraine PRS is also able to identify patient groups more likely to respond to triptans (108). On the other hand, no association between the migraine PRS and individual cerebral blood flow (CBF) measurements was found, although one individual SNP (rs67338227) was associated with CBF measures (109). An inverse association was seen between the coronary artery PRS and migraine headaches (OR [95% CI]: 0.94 [0.93–0.96]), where the signal was due to a large number of genetic variants with opposing effects on the two diseases (110). The continued discovery of new risk loci for common migraine based on ever-growing size GWAS will only further improve risk prediction by PRS.
Future of omics
GWAS is considered part of the most mature omics field, namely genomics and as presented in this review its data can be utilised to assess the correlation between different traits, assess causality and calculate PRS. However, multiple additional omics fields have arisen (e.g. epigenomics, transcriptomics, metabolomics, and proteomics). Integration of all different types of omics data will further our understanding of the underlying biology of migraine.
Key findings
There is an overlap of 68 genetic loci in between the two latest GWAS studies in migraine. Based on these studies there are in total 181 independent SNPs associated with migraine, of which 3 are female specific. There are multiple genetic methods to characterise genetic risk factors and architecture of migraine.
Supplemental Material
sj-xlsx-1-cep-10.1177_03331024221145962 - Supplemental material for Migraine genetics: Status and road forward
Supplemental material, sj-xlsx-1-cep-10.1177_03331024221145962 for Migraine genetics: Status and road forward by Aster VE Harder, Gisela M. Terwindt, Dale R Nyholt and Arn MJM van den Maagdenberg in Cephalalgia
Supplemental Material
sj-xlsx-2-cep-10.1177_03331024221145962 - Supplemental material for Migraine genetics: Status and road forward
Supplemental material, sj-xlsx-2-cep-10.1177_03331024221145962 for Migraine genetics: Status and road forward by Aster VE Harder, Gisela M. Terwindt, Dale R Nyholt and Arn MJM van den Maagdenberg in Cephalalgia
Supplemental Material
sj-xlsx-3-cep-10.1177_03331024221145962 - Supplemental material for Migraine genetics: Status and road forward
Supplemental material, sj-xlsx-3-cep-10.1177_03331024221145962 for Migraine genetics: Status and road forward by Aster VE Harder, Gisela M. Terwindt, Dale R Nyholt and Arn MJM van den Maagdenberg in Cephalalgia
Footnotes
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: GMT reports consultancy or industry support from Novartis, Lilly and Teva, Abbvie, and Lundbeck and independent support from the Dutch Heart and Brain Foundations, IRRF, Dioraphte and the Dutch Research Council. AMJMvdM. reports research support from Praxis Precision Medicine and Schedule 1 Therapeutics and consultancy support from AbbVie. All other authors declare no conflict of interests.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.