
Abstract
Introduction:
This review aims to model migraine nociception.
Methods:
Personal experience and litterature.
Results:
Genetic and environmental factors in combination decide whether a person suffers from migraine. Endogenous and/or exogenous factors precipitate the individual attacks. Nociception takes place around blood vessels. There is a growing understanding of the molecular pathophysiological mechanisms of migraine from human provocation studies. Rodent models of migraine are necessary to understand the complex interrelation between the many putatively involved molecules and tissues but their relevance for human migraine is uncertain. The crucial element in migraine nociception is a unit consisting of endothelial cells, vascular smooth muscle cells, perivascular nerve fibers (trigeminal, parasympathetic and sympathetic) and mast cells. Attacks may start outside the brain by humoral or neurogenic activity releasing nociceptive substances around blood vessels. They may also (perhaps more often) start by the brain generating efferent activity in autonomic and somatic nerves.
Conclusion:
Human and rodent studies can quickly uncover the “mystery of migraine”.
Introduction
Human models have revealed several signaling molecules and ion channel openers that can induce attacks in migraine sufferers (1–3). Basic animal experimental research has established that the trigeminovascular system has a fundamental role in migraine pathophysiology (4,5) and validated rodent models have further contributed to our understanding of migraine mechanisms (6,7). Despite all this new information, it is still debated whether migraine is a central nervous system disorder or a vascular disorder or, perhaps as recently indicated by genetic studies, both (8,9). There are many excellent reviews about the mechanisms of migraine but only few attempts to present an overarching view of migraine nociceptive mechanisms (10). Based on five decades of headache research the present author now proposes models of migraine without aura at different conceptual and anatomical levels. The models may seem too simplistic, but simplicity is necessary to get an overview of migraine mechanisms, and the models may hopefully serve as a vehicle for further development of our concepts of migraine.
Why is migraine expressed and why does it occur in attacks?
We know from studies in monozygotic twins that genetic disposition by itself is not enough for migraine to be expressed. Only half of monozygotic couples are concordant (11). Thus, while one twin has migraine the other does not, despite their identical DNA (excepting somatic mutations and epigenetics). This illustrates the importance of environmental factors. In twin studies approximately 50% of the risk of migraine is genetic and 50% is environmental (12). It can be assumed that the relative proportion of risk varies between cases and families ranging from almost zero genetic disposition with strong environmental exposure to 100% genetic disposition without environmental influence such as seen in familial hemiplegic migraine (FHM)with dominant inheritance and complete penetrance. Figure 1 illustrates the importance of genetic and environmental risk factors as a background setting for the expression of the migraine disease. Such factors also decide attack frequency and severity over long periods of time (years). The threshold may change over the years and is, for example, high in old people and generally lower in females than in males. Thus, the threshold explains why migraine is present in some individuals and not in others and why it is present during certain periods of a person’s lifetime (13).

Lifetime trajectories of migraine.
But why does migraine occur in attacks rather than being constantly present? Why are attacks initiated and why do they stop? This is where attack precipitating factors come into play (Figure 2).

Attack precipitating factors may be additive.
One strong precipitating factor may occasionally be enough to cause a migraine attack. This is illustrated by those rare patients with pure menstrual migraine who have attack at every menstruation (and at no other time). Most attacks are, however, better explained by a coincidence of several endogenous and/or exogenous precipitating factors. This is illustrated by females who often, but far from always, have attack at menstruation (14). In these women one or more additional factors such as stress, strong light etc. must add to menstruation to precipitate an attack (15). In most other migraine patients, more than two coinciding factors are probably necessary to precipitate attack. Several independent but simultaneous factors would result in the fluctuating unpredictable attack frequency according to a Poiseuille distribution seen in most migraine patients. Attempts at experimental induction of attack using strong light or vigorous exercise failed, probably because more factors were necessary (16). The model regarding several precipitating factors might perhaps also explain why the attacks stop. That could happen when one or more of the precipitating factors spontaneously disappears.
Cerebral and extracerebral precipitating factors
There are cerebral, endogenous extracerebral and external environmental factors that may precipitate/generate migraine attacks (15). The division into these three groups is not absolute. Stress, for example, is thought to precipitate attacks via brain mechanisms, but it can perhaps also increase plasma level of substances that act peripherally (Figure 3).

Cerebral and extracerebral attack precipitating factors.
Hunger causes systemic changes but may also affect the hypothalamus. A precipitating factor may thus have more than one site of action. The different attack precipitating factors indicate that a migraine attack can start in the brain, for example in association with stress or anxiety, but it can also start because of endogenous extracerebral factors such as swings in female hormones or hunger (14,17). Finally, attacks can be initiated by external environmental factors such as umbellulone (18) and by several substances used experimentally to induce attacks (19). The last-mentioned factors usually have effect outside of the brain, but some may also influence the brain either by direct chemical action or by inducing afferent sensory input.
Migraine pain originates around dilated cephalic blood vessels
While the migraine attack may start in the brain (10,20) the origin of painful afferent input is different. There is an abundance of arguments supporting that pain (nociception) in migraine originates around cephalic blood vessels. The pain is pulsating and pulsations as well as pain are aggravated by physical activity. One paper has suggested that there is no synchrony between pulsations and heartbeat but this unlikely finding needs replication (21). All substances that can induce a migraine attack in humans are vasodilators even though the temporal relation to migraine induction is variable (19) The most effective attack treatments (triptans) are vasoconstrictors although also non-constricting drugs are effective (22), headache is a cardinal symptom of many vascular diseases and blood vessels are dilated on the painful side of half-sided migraine attacks (22). Furthermore, most migraine inducing substances do not cross the blood-brain barrier (23). Genetic variants recently found are preferentially expressed in vascular smooth muscle (8). Figure 4 shows a cephalic blood vessel with its innervation from the sympathetic, parasympathetic, and trigeminal sensory nerves.
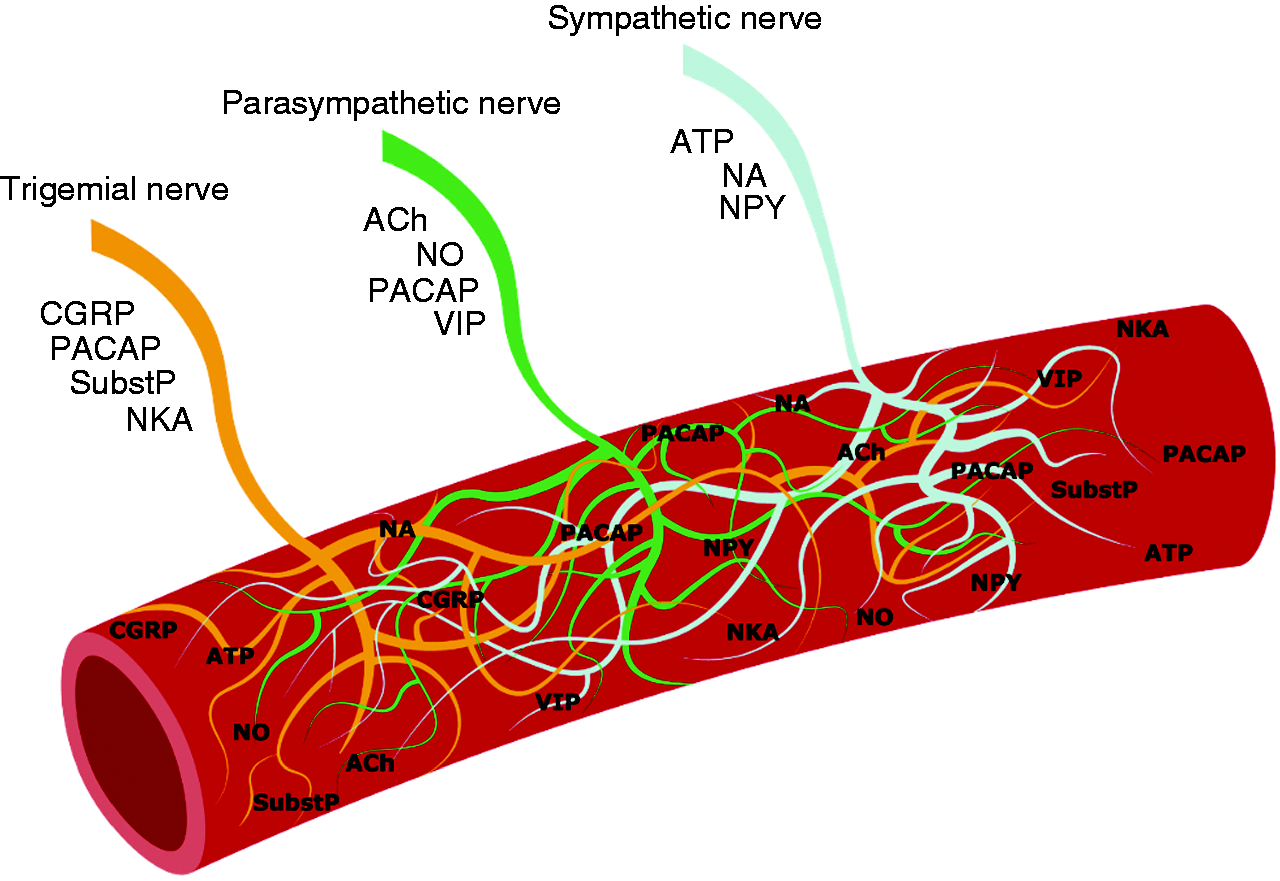
Innervation of cephalic arteries and their signalling molecules.
The signaling molecules from these nerve systems have been known for decades and many induce attacks when infused into migraine patients (19). They are discussed in more detail below. Although brain mechanisms in recent years have been in focus as the possible site of origin of the migraine attack (10), evidence for this is still missing while evidence for vascular involvement and (peri)vascular origin of migraine pain is gaining support (24,25).
A model of intracellular events
We do not know for sure in what type of cells the most important nociceptive migraine mechanisms take place. Probably several different types of cells are involved. A vascular smooth muscle cell is chosen because we know that it is responsible for the vasodilatation of migraine. Signal conduction may have similarities but also differs in endothelial cells, neurons and mast cells. This is an issue in need of further investigation. Figure 5 illustrates some of the known migraine inducing mechanisms.

Possible cellular migraine chemistry.
Calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase activating peptide (PACAP) act on receptors in the cell membrane that stimulate adenylate cyclase which converts adenosine tri-phosphate (ATP) to cyclic adenosine mono-phosphate (cAMP) (19). It phosphorylates protein kinase A (PKA) which again phosphorylates ATP sensitive potassium channels and calcium sensitive potassium channels causing vasodilatation (3,26). Cilostazol inhibits the breakdown of cAMP and activates that pathway (27). Histamine and prostaglandins also act via cAMP signaling. Glyceryl tri-nitrate (GTN) liberates nitric oxide (NO) which stimulates soluble guanylyl cyclase (28). It catalyzes the conversion of guanylyl tri-phosphate (GTP) to cyclic guanylyl mono phosphate (cGMP) which phosphorylates PKG. It again phosphorylates the potassium channels and the effect is vasodilation (29). Sildenafil blocks the break-down of cGMP causing its accumulation and increased activity in the pathway (30).
Are blood vessels dilated during a migraine attack?
High resolution magnetic resonance (MR) angiography demonstrated that major cerebral arteries are dilated approximately 10% compared to outside of attack and when the painful side was compared to the non-painful side in patients with strictly half sided headache (22). A similar result was obtained for the middle cerebral artery many years earlier using transcranial doppler imaging (31). In the MR study there was no dilatation of extracerebral arteries, but an older study using high resolution ultrasound imaging showed a difference in diameter of the superficial temporal artery between the painful side and the non-painful side (32). Other studies of induced attacks were negative but used less sensitive techniques (33). Thus, arterial dilatation does exist, but it is slight and certainly not enough to cause pain by simple mechanical distention. It might play a role, however, when combined with sensitization of trigeminal nerve terminals.
The diameter of arteries is tightly controlled. Therefore, an observed slight dilatation can be the resultant of opposing effects. In other words, there may be a considerable leakage of vasodilating nociceptive substances around the blood vessels counteracted by sympathetic activity and release of vasoconstrictive agents. The resultant vasodilation would be small or absent but the sensitizing effect could be considerable. The importance of compensatory mechanisms is illustrated by the fact that blockade of the effect of CGRP, the strongest vasodilatory substance known, has no effect on blood flow or arterial diameter (34). Other mechanisms must compensate 100%. Similar mechanisms may explain that extracerebral arteries were not dilated during migraine but sumatriptan constricted these arteries simultaneously with relief of migraine pain (22).
Vascular/perivascular nociception
A grossly magnified segment of a blood vessel and its innervation is shown in Figure 6. The relevant cellular components of this segment, which we call the migraine nociceptive unit, are the endothelial cells, the vascular smooth muscle cells (VSM), the perivascular nerves and the mast cells.

The hypothetical migraine nociceptive unit.
Varying between blood vessel territories, endothelial cells contain NO, endothelin, vasoactive intestinal peptide (VIP) and other signaling molecules, many of which are known to induce migraine attacks. In brain the endothelial cells are linked closely together with adhesion molecules and so-called tight junctions. Water soluble molecules cannot diffuse across the blood-brain barrier but may be actively transported in and out of the brain (35). The main possibility for water soluble molecules to affect the cerebral arteries is, therefore, to bind to receptors in the endothelial cells. In extracerebral arteries signaling molecules may cross the endothelial lining to reach VSM causing relaxation and vascular dilation. Dilatation causes stretching of perivascular nerves. Why vasodilatation may cause pain in the cephalic circulation and not in other parts of the body is a mystery in need of future investigation. Further down the figure, the perivascular nerve fibers are shown. There are trigeminal fibers with a content of signaling molecules that can be liberated. The parasympathetic nerves may liberate several signaling migraine inducing molecules by efferent activity. The efferent sympathetic nerve fibers liberate several substances but only ATP is known to be nociceptive. Finally, mast cells contain several substances with nociceptive properties many of which are included in a variant of the so-called inflammatory soup (36). In the dura mater mast cells are abundant and in close proximity to blood vessels. Degranulation of mast cells leads to liberation of several substances that are both vasodilating, pro-inflammatory and nociceptive. Therefore, these cells have been a focus of interest for several decades. Unfortunately results remain inconclusive. Some studies support a role in migraine, but others are negative.
Extracerebral induction of attack
We know from experimental provocation that a migraine attack including both pain and associated symptoms can be precipitated by substances that do not cross the blood-brain barrier (19). How endogenous extracerebral factors such as hunger and hormonal swings can induce a migraine attack is, however, uncertain. Studies of the chemical composition of blood have not revealed reproducible attack related changes in known migraine provoking agents. Even the previous finding of increased CGRP in external jugular venous blood during attack (37) could not be reproduced (38). There seems to be some hope, however, that a combination of genetics and chemistry may help in the future (39). Migraine attacks might also be precipitated, for example from the GI tract, due to afferent neuronal activity that via the brain stem and hypothalamus can induce efferent migraine activity. In that situation the mechanisms would be like attacks that are initiated by cerebral mechanisms (see later). Another possibility is that circulating factors not yet identified can directly affect cephalic blood vessels and their sensory innervation (Figure 7a).

(a) Cephalic blood vessel with its innervation and an offending molecule. Early phase of attack and (b) Cephalic blood vessel with its innervation and an offending molecule. Late phase of attack.
Best understood is attack-induction by experimental infusion of migraine provoking agents (Figure 7a). Most of these do not dilate cerebral blood vessel protected by the blood-brain barrier, but they all dilate extracerebral blood vessels in the dura mater and in the cranial and extracranial territories. Every single substance (and by now there are many) that can induce a migraine attack is a vasodilator (19). Human provocation studies with vasoconstrictors prostaglandin F2 alpha, noradrenaline or endothelin (40–42) resulted in no headache at all compared to placebo despite some indication in the literature that they may sensitize nerve endings (41). Most likely extracerebral induction of migraine attacks must therefore take place in and around dilated blood vessels of the head (Figure 7B).
A lot is known about the molecular mechanisms of migraine but the exact territory and tissue type where nociception takes place remains unknown. Is it in the dura mater as most basic scientists believe (43–45) or is dura a focus of interest because it is so easily accessible and without a blood-brain barrier? Could it perhaps be intracerebral arteries activated by endothelial mechanisms (45,46)? This is possible because even if cerebral arteries do not dilate during most migraine provocations, they do so during spontaneous attacks (22). Or is it blood vessels in the cranium or outside of the cranium? As mentioned above the answer to this question remains unknown but it is possible that all cephalic arteries including their innervation are involved (23). Figure 7b therefore depicts a dilated blood vessel somewhere in the head or brain. These vessels all have largely the same innervation which permits the use of a single general model. Vasodilatation is likely to induce activity in sensory nerve fibers. They may act like a network of strain gauges although this has not been experimentally proven (Figure 4). Activity may perhaps also be induced in parasympathetic or sympathetic nerve fibers as an autoregulatory response. Therefore dilation may be small or in some cases unmeasurable. Unknown circulating migraine provoking substances may perhaps also directly activate receptors on extracerebral perivascular nerve terminals. Spontaneous retrograde (efferent) activity in sensory nerves generally or in the trigeminal nerve does not exist, but it may occur in peripheral branches as part of axonal reflexes. Activation of some trigeminal nerve terminals may therefore spread to others and liberate CGRP, PACAP, neurokinin A (NKA) and substance P (Figure 8).
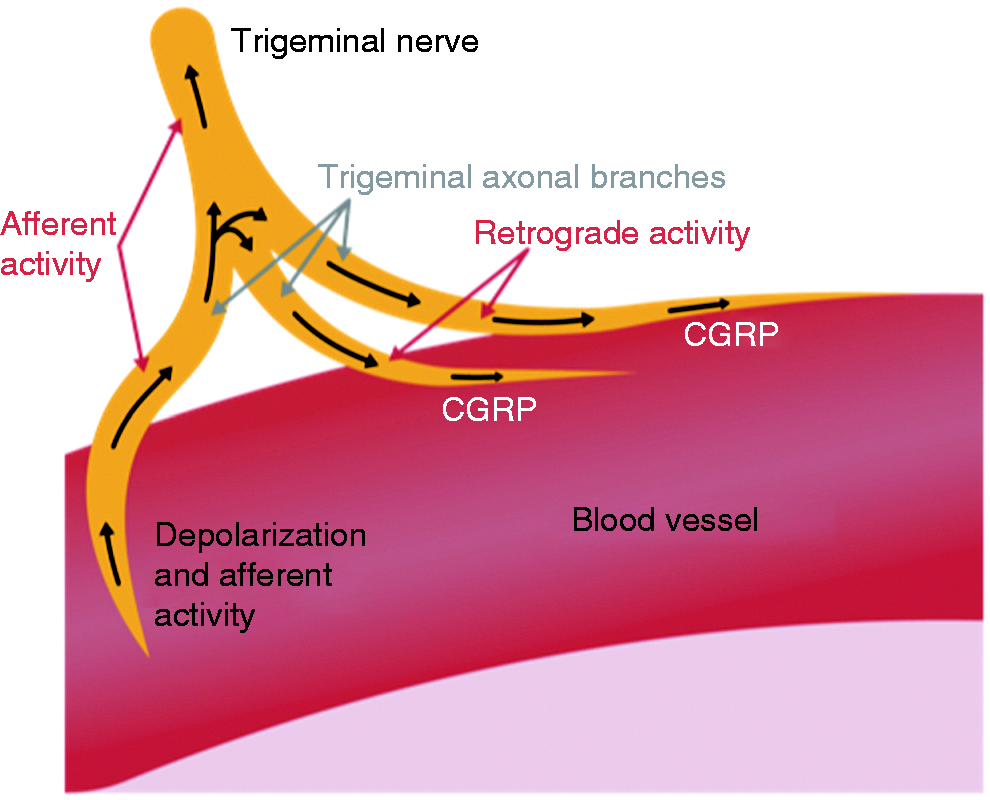
Axonal reflex in trigeminal terminals of possible importance for migraine.
These substances may theoretically affect the parasympathetic and sympathetic nerve fibers. The results of the above actions may be a mixture of nociceptive signaling molecules in the perivascular space (36). Finally, extracerebral factors such as CGRP may also act directly on the trigeminal ganglia which have no barrier to diffusion. How circulatory migraine provoking factors act on trigeminal psudounipolar ganglion cells is, however, poorly understood and worthy of future study.
Cerebral induction of attacks
It is a well-known fact that painful impulses undergo significant modulation in the central nervous system. All known painful diseases depend, however, on the existence of nociceptive input. Even when migraine begins with alterations in brain function it must therefore lead to nociception before migraine pain can be felt. But how can brain, which by itself is insensitive to pain, generate painful afferent input? Neurogenic inflammation is a model of migraine that previously received a lot of interest. It is induced experimentally by vigorous electrical stimulation of the trigeminal ganglion leading to retrograde activity in the trigeminal nerve and leakage of signaling substances such as substance P and CGRP from the trigeminal nerve terminals around blood vessels (47). This causes peripheral inflammation and increased sensitivity of nerve fibers (5). While this model was sensitive to migraine specific drugs it was far from specific. In other words, several drugs that have no effect in migraine were effective in blocking neurogenic inflammation (48). Another difficulty with the neurogenic inflammation model was, as mentioned above, that spontaneous efferent activity has never been recorded in purely sensory nerves apart from axonal reflexes in peripheral branches. Therefore, the brain cannot send out signals via the trigeminal nerve, it can only receive input.
Figure 9A depicts the early phase of brain-initiated attacks.
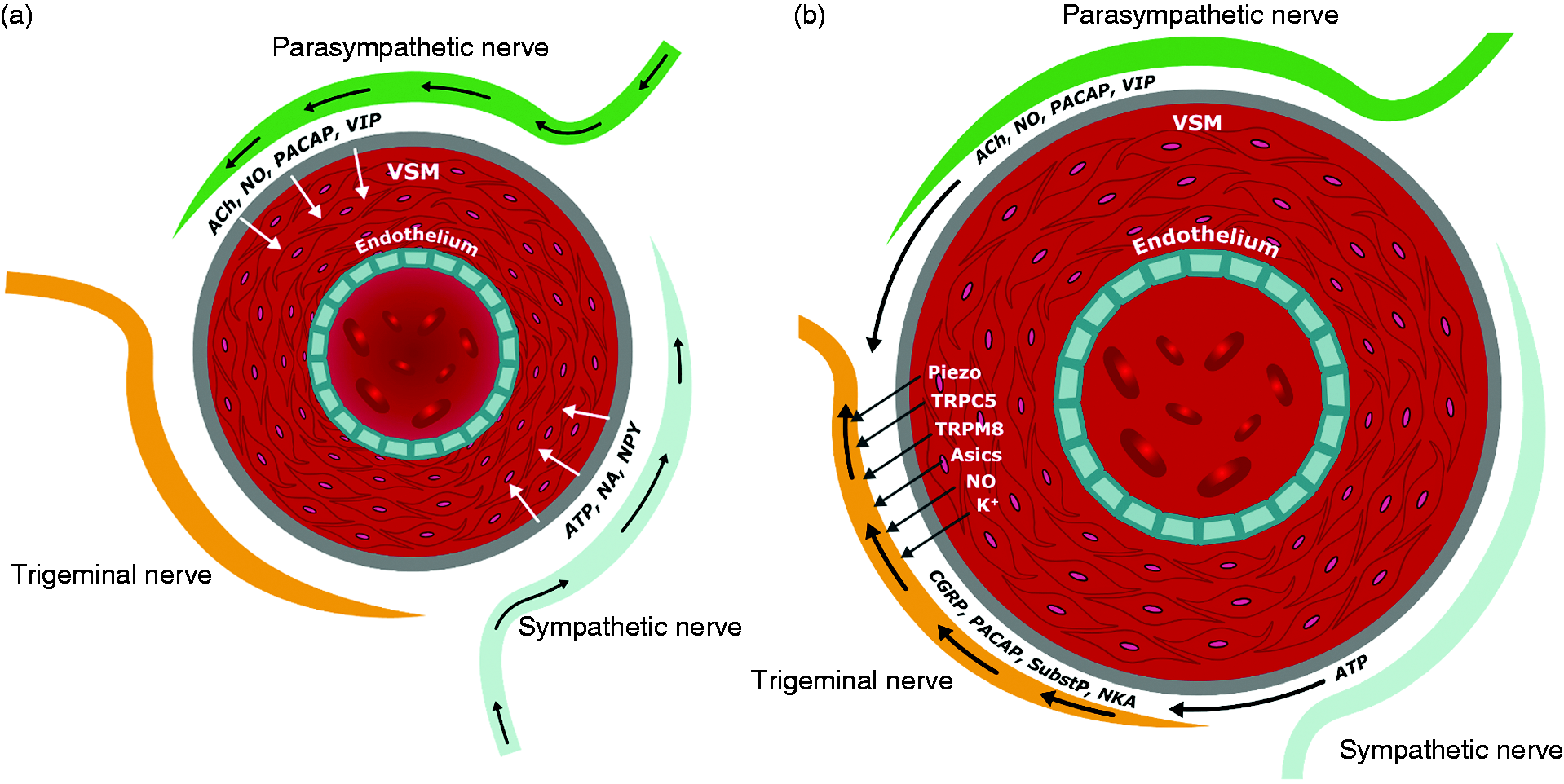
(a) Cephalic blood vessel with its innervation and neurogenic output. Early phase of attack and (b) Cephalic blood vessel with its innervation, neurogenic output and release of signaling molecules. Late phase of attack.
The brain influences the perivascular space via efferent output in the parasympathetic and sympathetic nervous systems. These nerves contain several substances that are known to induce migraine attacks. In the parasympathetic nervous system, it is acetylcholine, NO, PACAP and VIP. In the sympathetic nervous system ATP can induce pain (36) while noradrenaline cannot induce migraine (49) and neuropeptide y (NPY) has never been tested. Efferent activity in parasympathetic nerves with liberation of migraine provoking signaling molecules may affect trigeminal sensory nerve fibers directly. Parasympathetic activity also dilates blood vessels and thereby may mechanically activate trigeminal nerve fibers (Figure 9B). There is a need for studies of neurogenic liberation of substances around blood vessels in experimental models.
The blood vessel diameter is tightly controlled. Any dilatation will at least partly be counteracted by sympathetic constrictive activity. This may lead to liberation of ATP, a nociceptive substance, which adds to the pro-nociceptive environment created in the perivascular space. Thus, a situation of peripheral sensitization, vasodilatation and activation of the trigeminal nerve fibers has been created.
The brain can also send efferent activity to pericranial muscles (Figure 10). That might create tender points and/or activate existing tender points. During migraine attack there is increased myofascial tenderness pericranially (50). Furthermore, blocking tender spots with lidocaine or saline infiltration is a highly effective treatment of migraine attacks (51). To what extent efferent nervous output to pericranial muscle is involved in the migraine attack is an issue in need of further study.
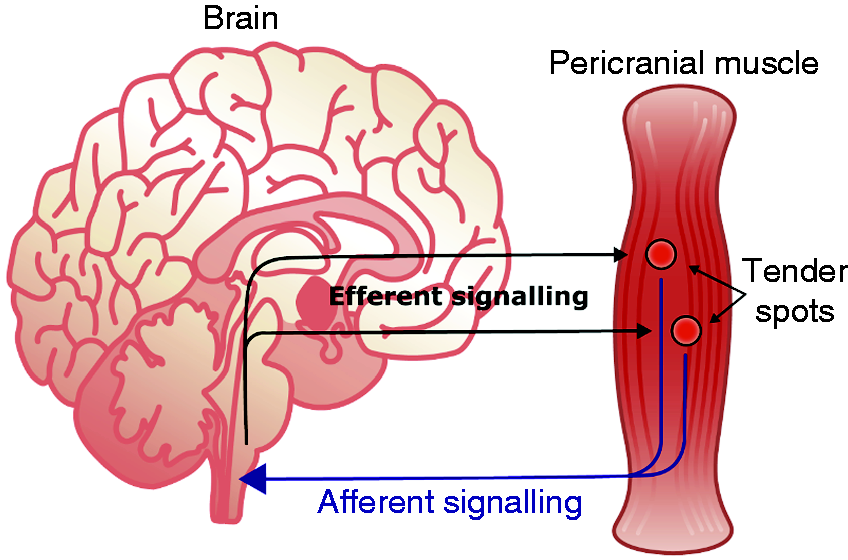
Possible myofascial pain generation during migraine.
An alternative hypothesis to peripheral nociception has been proposed but seems highly unlikely. It claims that there is no abnormal nociceptive input in the trigeminal nerve, but that the attacks are caused by normal input combined with a lack of tone in the descending central nervous system pain inhibiting system or an increased activity in the descending pain activating system (10,52). No other painful conditions are known where peripheral nociception is absent (apart from central nervous system deafferentation) and there is little experimental evidence to support the hypothesis. While attacks may not be caused by disturbance of the CNS pain control system, that system probably plays a role in setting the migraine threshold. Anxiety or depression are thus likely to lower the migraine threshold via such mechanisms (Figures 1 and 2).
Concluding remarks
While a lot is known about the machinery of migraine ranging all the way from genetics, environment, organ systems, different kinds of innervation and cellular responses, we are just at the beginning of a real understanding of the complexity of the migraine attack. The field is now open to molecular biologists and other basic scientists who wish to characterize the many different cellular elements that may be involved in the migraine cascade. Brain initiated parasympathetic and sympathetic activity in relation to perivascular nociception also needs study. Endothelium and smooth muscle in blood vessels in the head need to be compared to blood vessels in other parts of the body. While incomplete understanding is annoying, it is on the other hand exiting that the black box of migraine mechanisms has been widely opened. Resource is the only limiting factor for further progress in our understanding of migraine. It is not possible at present to construct one model that explains all the complex mechanisms of migraine. However, a series of models as presented here give together some understanding of migraine. These models can easily be revised as new scientific information becomes available.
Article highlights
Genetic and environmental factors cause expression of the migraine disease and precipitate attacks. Precipitation may be central or peripheral. Arteries and their surrounding space are crucial for nociception. Molecular nociceptive mechanisms are known. Complex interaction of arterial dilatation and pain signaling is poorly understood.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.