
Abstract
Background
Reactive nitroxidative species, such as nitric oxide but particularly peroxynitrite, have been strongly implicated in pain mechanisms. Targeting peroxynitrite is anti-nociceptive in pain models, but little is known about its role in migraine mechanisms. Given the need to validate novel targets for migraine headache, our objective was to study the potential of reactive nitroxidative species, particularly peroxynitrite, as novel targets for drug discovery and their role in migraine mechanisms.
Methods
We recorded neuronal activity in rats with extracellular electrodes and examined the effects of targeting nitric oxide or peroxynitrite on ongoing and cranial-evoked firing rates of central trigeminocervical neurons. We injected calcitonin gene-related peptide (which produces migraine-like headache in migraineurs) and characterized neuronal responses to cranial stimulation and on behavioral responses to nociceptive periorbital stimulation and determined the effects of targeting reactive nitroxidative species on the mediated changes.
Results
L-NAME (nitric oxide synthase inhibitor) and Fe(III)5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato chloride (FeTPPS; peroxynitrite decomposition catalyst) inhibited ongoing and dural-evoked responses of trigeminocervical neurons, without affecting normal facial-cutaneous responses. Calcitonin gene-related peptide caused activation and sensitization of dural-responsive trigeminovascular neurons with hypersensitivity to intracranial and extracranial stimulation, and reduction of periorbital withdrawal thresholds. Only the peroxynitrite decomposition catalyst prevented these neuronal and behavioral nociceptive responses.
Discussion
The data support that calcitonin gene-related peptide mediates the underlying neurobiological mechanisms related to the development of migraine-like headache. They also confirm the role of nitric oxide and implicate peroxynitrite production along the trigeminovascular migraine pathway in these mechanisms. The data also support peroxynitrite as a novel and potentially effective target for migraine treatment. The current drug development focus on peroxynitrite decomposition catalysts for chronic pain disorders should therefore extend to migraine.
Introduction
Migraine headache is a complex brain disorder that is thought to be the result of abnormal activation and sensitization of trigeminal primary afferents that innervate the nociceptive-specific dural vasculature and their central projections to the medullary dorsal horn and upper cervical spinal cord (1,2). While current therapeutics are effective in some patients, it is estimated that over 50% of patients gain little benefit from these established therapies (3). There is therefore an urgent need to identify and validate novel targets for treatment, which should also benefit our understanding of the underlying neuropharmacological mechanisms of migraine.
Reactive nitrogen and oxygen species (reactive nitroxidative species; RNOS) are a group of molecules that produce oxidative, nitrosative, and nitroxidative stress. These include nitric oxide (NO), superoxide (O2.−; SO), and also peroxynitrite (ONOO-; PN), which is the downstream reaction product of NO and SO (4,5). The last three decades have seen nitric oxide (NO) be strongly associated with mechanisms of migraine headache, from NO donors triggering migraine-like headache and associated symptoms that respond to migraine abortives (6,7), through to blocking NO production as a potential therapeutic (8). Although little is known about whether PN and SO are involved in migraine mechanisms, several clinical studies have highlighted that platelet levels of PN and SO-related proteins are disrupted in migraine patients (9,10) suggesting a possible role. PN is also thought to be critical to the development and maintenance of spinal sensitization associated with persistent neuropathic and inflammatory pain (11–15). Further, targeting PN production both prevents and aborts nociceptive responses, providing compelling evidence as a novel target for pain management (4,15).
While both PN and SO may offer novel opportunities for discovery efforts in migraine, PN is considered the best approach for drug development (15). It has no inherent physiological role, while SO is important in learning and memory development (16). Sparing SO, while directly targeting PN, is considered the best approach for potential drug development (15). Therefore, in this study our aim was to validate PN as a novel target for migraine, using a PN decomposition catalyst (PNDC) that spares SO in validated preclinical approaches for drug screening (17–19). We also wanted to determine whether PN is involved in the underlying neurophysiology of migraine-like mechanisms. We used CGRP injection (which produces migraine-like headache in migraineurs) (20) and measured changes in trigeminocervical neuronal firing in response to intra and extracranial stimulation. We also measured changes in migraine-like peri-orbital mechanical hypersensitivity in response to CGRP. This is similar to observations and measures made previously with NO donors (7,21,22). To dissect PN-related mechanisms, we determined if there was a differential response to CGRP-mediated changes, after targeting either PN or NO production.
Material and methods
All experiments were conducted in compliance with research protocols approved by the University of Maryland Baltimore or New York University (NYU) Institutional Animal Care and Use Committees (IACUC) and conformed to the National Institute of Health Guide for the Care and Use of Laboratory Animals, adhering to both ARRIVE guidelines and those of the Committee for Research and Ethical Issues of the International Association for the Study of Pain (IASP) (23).
Animals-experimental design
Adult male Sprague-Dawley rats (250–390g: Charles River, MA, USA) were used throughout. They were housed in temperature and light controlled rooms for at least 7 days before use, with access to food and water ad libitum. Using male rats has proven a reliable and translational approach to study migraine-like mechanisms (7,24–26) in validated preclinical models that translate to the human migraine pathway, and to predict the efficacy of treatments (18,27,28) that are used by both women and men. This also allows direct comparison with our and others’ previous data (18,27–32). Given the likely role of menstrual hormones in a significant proportion of migraine in women (33) this approach also allows us to circumvent the complexity of the estrous cycle, which is still not fully understood in migraine but undoubtedly plays an important role in the complex mechanisms related to triggering migraine in women. Animals were assigned to experimental groups randomly and were operator blinded with respect to treatment and during analysis. At the end of each experiment all animals were euthanized with 200 mg/kg Euthasol (IV).
Animal preparation and electrophysiological recording
The surgical preparation, physiological monitoring, electrophysiological recording methods and analyses are the same as those reported in detail previously (34). Briefly, rats were anesthetized, surgically prepared for continuous measurement of vital physiology, including blood pressure, temperature, and expired CO2 and prepared for electrophysiological extracellular recording of dural-responsive neurons in the trigeminocervical complex (TCC), with aseptic techniques. Rats were paralyzed with pancuronium bromide (Pavulon®, Organon) during electrophysiological recording. Appropriate depth of anesthesia was judged by the absence of paw withdrawal and corneal blink reflex, and during muscular paralysis by fluctuations of blood pressure and changes to expired CO2.
Characterization of neurons
These methods have been described in detail previously (34,35). We used a tungsten recording electrode (0.5–1 MΩ) and characterized neuronal responses in the TCC region by assessing cutaneous noxious and innocuous responses through all three trigeminal territories (Figure 1(a)–(b)), ipsilateral to recording side, as the electrode was lowered into the medullary region. Cutaneous receptive fields (RF; spikes/second) within the ophthalmic dermatome were then tested for convergent nociceptive inputs to noxious electrical stimulation of the ipsilateral dura mater (100–200 μs pulse, 0.25 Hz and 8–15 V). Electrode position and stimulation parameters were optimized and were capable of activating both Aδ (‘fast’ responses, 3–20 ms latency range) and C-fiber neurons (‘slow’ responses, 20–80 ms latency range) (24). The exact latency of neuronal discharges was established separately in each experiment, with the data collected representing the number of action potential spikes that fired within a latency window per stimulation, averaged over 20 stimulations (sweeps; spikes per sweep). Innocuous cutaneous responses were assessed by brushing the RF; noxious responses by pinch with forceps (Figure 1(c)). Neurons responsive to both noxious and innocuous stimulation were classified as wide dynamic range (WDR), whereas those responsive to only noxious stimulation were classified as nociceptive specific (NS). Spontaneous activity (spikes/second) was recorded throughout and measures for analysis taken for 300 sec preceding the dural stimulation. Post and peri-stimulus time histograms of neural activity were displayed and analyzed with Spike2 v8.

Experimental set-up for electrophysiological and behavioral studies. (a) Neurons of the trigeminocervical complex (TCC) were recorded in response to dural electrical stimulation, and innocuous and noxious stimulation of the cutaneous facial RF (shaded area). (b) Somatotopic representation of the trigeminal territories for RF characterization; V1, ophthalmic; V2, maxillary; V3, mandibular. (c) Original example of an electrophysiological recording in response to innocuous brush and noxious pinch of the cutaneous V1 RF. Top panel is original electrophysiological output, bottom panel is the peri-stimulus time histogram of neural activity (d). Representation of the left (L) and right (R) cutaneous periorbital region used to determine somatosensory thresholds via mechanical von Frey probing.
Periorbital mechanical threshold testing
In a separate group of rats, we studied changes in periorbital mechanical thresholds in response to CGRP (6 μg/kg, IP, which is equivalent to the total dose given via IV infusion over 20 min in the electrophysiological studies), and determined whether Fe(III)5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato chloride (FeTPPS, 30 mg/kg, IP) could prevent any mediated changes. Rats were initially acclimated to the testing environment (room and chambers) for at least 1 h/day, and by handling continuously for 5 min, on 3 separate days during the week prior to testing. Food and water were freely accessible in the chambers throughout. Stable baseline sensory nociceptive thresholds were obtained prior to drug administration by probing the periorbital area. Sensory nociceptive thresholds were determined with von Frey filaments using the ‘up-and-down’ method (36,37). Testing began by using the 1 g filament, in the area highlighted in Figure 1(d); just rostral to the forehead, caudal to and between the eyes, and lateralized. This was increased to a maximum of 8 g. Only rats that met a facial baseline of 6–8 g were included in the study. A positive response was indicated when the stimulus induced a withdrawal of the head, sometimes accompanied with facial rubbing. After CGRP/FeTPPS administration, rats were tested hourly for 5 h.
Drugs
FeTPPS (PNDC, Cayman Chemicals, MI, USA), a characteristic metalloporphyrin catalyst that increases non-reactive nitrate formation as a consequence of PN decomposition (38,39), was dissolved in normal saline. Also, both L-NG-Nitroarginine methyl ester hydrochloride (L-NAME; a non-selective NO synthase (NOS) inhibitor, Sigma-Aldrich) and CGRP (Tocris Cookson Ltd, USA) were dissolved in normal saline. Doses are based on our previous studies, which show responses in behavioral pain (11) or trigeminovascular nociception (40) assays. CGRP was aliquoted and frozen until required. FeTPPS and L-NAME were made up fresh each day. Normal saline was the vehicle control throughout.
Data and statistical analysis
Justification of group sizes and statistical analyses used are similar to previous studies (7,20) and group size calculations that typically sees difference in means of 25–30% (standard deviation = 15–20%) with a two-sided alpha of 0.05 and power of 90%, calculated by GPower 3.1 software. This requires sample size of 9–12 animals to measure time points up to 3 h for electrophysiology studies, and 5–6 animals per group to measure up to five time points in behavioral nociceptive studies. All data are expressed as mean ± standard error of the mean (SEM) and all statistical analyses were conducted on raw data and normalized for presentation purposes only. A neuron was considered activated using the critical ratio test (41), which is based on the count data having a Poisson distribution and employs the standard normal deviate to determine at the 5% level whether two firing rates differ. In practice, this very closely approximates to a 30% change from baseline. Therefore, we would anticipate that no more than one time point would pass this test by chance, and as a consequence required at least two time points to pass this test over the course of the study to be considered activated, with 10–30% change unclassified. A neuron was considered sensitized if it exhibited enhanced responses to at least three of five parameters: Dural electrical stimulation, brushing or pinching of the cutaneous facial RF, or expansion of dural or cutaneous facial RF (42).
Analysis of variance for repeated measures was used to assess the effects of drug over time. If Mauchly’s test of sphericity was violated, we made appropriate corrections to degrees of freedom according to Greenhouse-Geisser. Student’s paired two-tailed t-test for post-hoc analysis was used to test for the time points of significance against the average of the baselines, by using the criteria of Bonferroni correction for multiple testing. A two-way between subjects’ mixed design repeated measures ANOVA was used to compare responses between groups, with treatment as the between groups factor. If data were not normally distributed, we performed equivalent non-parametric tests by using Friedman’s ANOVA and post-hoc Wilcoxon’s test, where applicable. Statistical significance was set at p < 0.05 (using IBM-SPSS 25.0 throughout).
Results
Electrophysiological studies – neuronal identification and properties
Electrophysiological recordings were made from 67 neuronal responses in 67 rats (one group per rat, with no replicates) that responded to electrical stimulation of the dura mater, and with cutaneous facial RFs that included predominantly the 1st division of the trigeminal nerve, and on occasions also the 2nd and 3rd divisions (Figure 1(a)–(b)). Of these 67, 48 (all WDR) were classified as receiving both Aδ (discharge range 3–20 ms) and C-fiber inputs (20–80 ms), 19 (18 WDR and 1 NS) were classified as receiving only Aδ-fiber inputs. Neurons were located in mainly nociceptive-specific superficial (laminae I–II) and deeper layers (laminae V and VI) of the dorsal horn of the TCC at range of depth of 50–1075 µm.
Blood pressure responses
L-NAME (100 mg/kg) caused a significant increase in blood pressure (66.4 ± 2.9 mmHg, U = 1.0, p < 0.001, n = 19) that peaked after 157.6 ± 13.6 sec. FeTPPS (10 mg/kg) did not significantly alter blood pressure (t8 = 1.4, p = 0.19, n = 9), but FeTPPS (30 mg/kg) caused a significant increase in blood pressure (19.0 ± 2.9 mmHg, t20 = 2.75, p = 0.009, n = 21) that peaked after 63.8 ± 9.2 sec. These changes returned to baseline within 10 min of administration and therefore are unrelated to any neuronal changes. CGRP caused a transient reduction in blood pressure during infusion that returned to baseline levels within minutes of the end of infusion.
Studies of acute dural-nociceptive trigeminovascular activation – summary of pretreatment (baseline) responses
Through all animals/groups studied the baseline spontaneous firing rate was 23.2 ± 2 (spikes/sec), and there was no significant difference across the different treatment groups (F3,35 = 0.62, p = 0.61). There was also no significant difference at baseline between all groups for dural-evoked Aδ-fiber units (F3,35 = 2.4, p = 0.084), or C-fiber discharges (F3,25 = 1.1, p = 0.38), with an average baseline of 7.7 ± 0.2 and 2.1 ± 0.4 action potential spikes per sweep, respectively. There was also no significant difference at baseline between groups in response to both innocuous (F3,35 = 1.1, p = 0.37) and noxious (F3,35 = 2.7, p = 0.063) somatosensory stimulation of the facial cutaneous RF. The physiological characteristics of this neuronal population are similar to previous studies (7,20,21,24).
Effects of L-NAME on dural-evoked second-order trigeminocervical neurons
L-NAME (100 mg/kg, IV, n = 9) significantly inhibited spontaneous trigeminal neuronal firing across 120 min, specifically after 30 min, and maximally after 90 min by 43% (Figure 2(a)). Neuronal responses within the Aδ-fiber range were also significantly inhibited, specifically after 15 min, maximally after 30 min by 24% (7.0 ± 0.4 to 5.4 ± 0.6 spikes per sweep, Figure 2(b)). Unitary discharges within the C-fiber range (n = 6) were inhibited, specifically after 90 min by 47% (3.2 ± 1.2 to 1.7 ± 1.1 spikes per sweep; Figure 2(c)). L-NAME had no effect on trigeminocervical neuronal firing in response to innocuous brush (F2.0,15.9 = 0.56, p = 0.58; Figure 2(d)) and noxious pinch (F4,32 = 0.23, p = 0.92; Figure 2(e)). Data with statistics are summarized in Table 1.

Targeting reactive nitroxidative species (RNOS) inhibits dural-evoked trigeminocervical neuronal responses. Time course changes in (a) spontaneous trigeminocervical neuronal firing, (b) intracranial dural-evoked neuronal responses with inputs in the Aδ-fiber range (“fast” neuronal responses), and (c) C-fiber latency range (“slow” neuronal responses), in response to L-NAME (100 mg/kg, IV) and FeTPPS (10 and 30 mg/kg, IV). Only L-NAME and FeTPPS (30 mg//kg) caused a significant inhibition of spontaneous and dural-evoked trigeminocervical neuronal responses, FeTPPS (10 mg/kg) had no effects. These data have been normalized to represent the percentage change from baseline and are expressed as mean ± SEM. Response magnitude to (d) innocuous and (e) noxious somatosensory-evoked stimulation of the cutaneous facial RF, following L-NAME or FeTPPS. Neither drug, at any dose, had an effect on neuronal responses to cutaneous stimulation. In all panels, vehicle control (normal saline) had no significant effects on neuronal responses.
Summary of trigeminocervical neuronal data over 2 hours with responses at baseline and at time of maximal inhibition, in response to L-NAME or FeTPPS.

*p < 0.05 significance from baseline.
Effects of FeTPPS on dural-evoked second-order trigeminocervical neurons
At 10 mg/kg FeTPPS (n = 9) had no effect on spontaneous trigeminal neuronal firing, or in the same neuronal population, responses within the Aδ-fiber and C-fiber range to dural electrical stimulation, or responses to innocuous (F4,32 = 0.65, p = 0.63) and noxious (F4,32 = 0.39, p = 0.81) stimulation of the V1 cutaneous RF, over 120 min. Conversely, FeTPPS (30 mg/kg, n = 9) significantly inhibited spontaneous trigeminal neuronal firing across 120 min, specifically after 15 min, and maximally after 120 min by 48% (27.1 ± 4.2 to 14.8 ± 3.0 (spikes/second); Figure 2(a)). It also inhibited dural-evoked trigeminocervical neurons within the Aδ-fiber range that was significant after 15 min, maximally after 60 min by 20% (8.2 ± 0.5 to 6.6 ± 0.7 spikes per sweep; Figure 2(b)) that remained significant until at least 120 min. It also significantly inhibited neuronal responses within the C-fiber range (n = 9). These were significant after 60 min that continued and were maximally inhibited by 52% after 2 h (2.1 ± 0.6 to 1.1 ± 0.4 spikes per sweep, Figure 2(c)). FeTPPS had no effect on trigeminocervical neuronal firing in response to innocuous brush (F1.6,12.7 = 0.44, p = 0.61; Figure 2(d)) and noxious pinch (F1.8.14.5 = 0.48, p = 0.61; Figure 2(e)). Data with statistics summarized in Table 1.
Vehicle control (saline) had no significant effect (n = 9) on all parameters measured.
Studies of CGRP-evoked dural-trigeminovascular changes – summary of pretreatment (baseline) responses
Through all animals/groups studied the baseline spontaneous firing rate was 21.6 ± 1.7 (spikes/second), and there was no significant difference across the different treatment groups (F3,39 = 0.61, p = 0.61). There was also no significant difference at baseline between all groups for dural-evoked Aδ-fiber units (F3,39 = 2.3, p = 0.098), or C-fiber discharges (F3,23 = 1.04, p = 0.40), with an average baseline of 8.5 ± 0.3 and 1.5 ± 0.2 action potential spikes per sweep, respectively. There was also no significant difference at baseline between groups in response to both innocuous (F3,39 = 0.7, p = 0.56) and noxious (F3,39 = 0.2, p = 0.89) somatosensory stimulation of the facial cutaneous RF. The physiological characteristics of this neuronal population are also similar to previous studies.
Effects of CGRP on dural-responsive second-order trigeminocervical neurons
Overall CGRP (300 ng/kg/min for 20 min, IV) significantly increased spontaneous trigeminal neuronal firing over 3 h, specifically after 60 min (25.0 ± 4.7 to 36.1 ± 6.7 spikes/sec), which remained significant until 150 min (up to 36.6 ± 4.0 spikes/sec; Figure 3(a)). Based on the critical ratio test 7/9 were considered activated over 3 h. This activation manifested as hypersensitive responses of the same trigeminocervical neurons to dural stimulation, with a significant increase over three hours in the number of dural-evoked action potential spikes within the Aδ-fiber range. This was significant after 30 min (9.4 ± 0.6 to 10.6 ± 0.6 spikes per sweep) and remained significant until at least 3 h (11.1 ± 0.7 spikes per sweep; Figure 3(b)). However, there was no effect on responses within the C-fiber range (n = 8, Figure 3(c)). Data with statistics are summarized in Table 2.
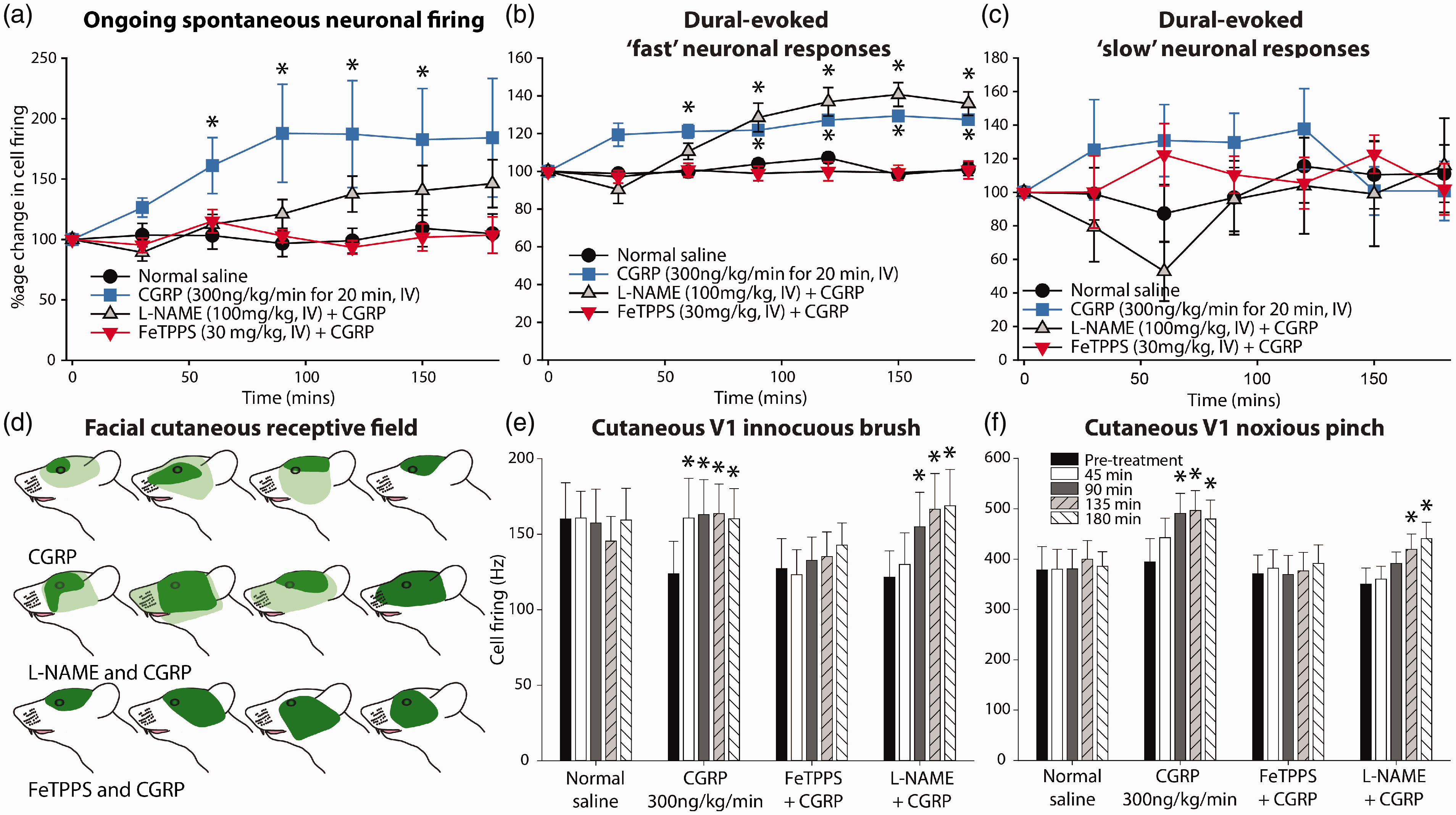
Targeting reactive nitroxidative species (RNOS) differentially affects CGRP-evoked changes to trigeminocervical neuronal responses. Time course changes in (a) mean spontaneous trigeminocervical neuronal firing, (b) intracranial dural-evoked neuronal responses with inputs in the Aδ-fiber range (“fast” neuronal responses) and (c) C-fiber latency range (“slow” neuronal responses). In each figure, the data have been normalized to represent a percentage change from baseline and are expressed as mean ± SEM. (d) Examples of cutaneous RFs in rats from each treatment group. Dark green represents the original RF, and light green is the expanded RF (including the darker green region), which is present in both CGRP-alone and L-NAME-CGRP groups. Response magnitude to (e) innocuous and (f) noxious somatosensory-evoked stimulation of the cutaneous facial RF. Observations are in response to vehicle control (normal saline, IV), CGRP (300 ng/kg/min for 20 min, IV), CGRP and L-NAME (100 mg/kg, IV), or CGRP and FeTPPS (30 mg/kg, IV). CGRP caused delayed activation and sensitization of trigeminal neurons. Pre-treatment with L-NAME partially prevented the increase in spontaneous neuronal firing, but it did not effect hypersensitive neuronal responses to somatosensory cranial stimulation or expansion of the cutaneous facial RF. However, FeTPPS prevented increases in spontaneous firing, all hypersensitive neuronal response to evoked cranial stimulation, and expansion of the cutaneous facial RF.
Summary of trigeminocervical neuronal data over 3 h with responses at baseline and after 120 min, in response CGRP and with L-NAME or FeTPPS.

*p < 0.05 significance from baseline.
In the seven animals where neurons responded to CGRP there was expansion of the cutaneous facial RF (Figure 3(d)). There was also a hypersensitive response to cutaneous facial stimulation after CGRP, with increased neuronal firing to innocuous brush (F4,32 = 12.4, p < 0.001) and noxious pinch (F2.0,15.7 = 15.7, p = 0.001). Neuronal responses to cutaneous innocuous brush (t8 = 4.3, p = 0.002; Figure 3(e)) and noxious pinch (t8 = 6.0, p = 0.001; Figure 3(f)) were significantly increased compared to baseline after 45 and 90 min, respectively, and continued until at least 3 h. Vehicle control (normal saline; data not shown) had no significant effect on all parameters measured over 3 h.
Effects of L-NAME on CGRP-evoked changes to trigeminocervical neuronal responses
When L-NAME (100 mg/kg, IV) was given prior to CGRP infusion, there was not a statistically significant change in spontaneous trigeminal neuronal firing over 3 h. The critical ratio test (41) in 6/10 rats showed that there was an increase in spontaneous firing at at least two time points, over the course of the 3 h. When the response to treatments of CGRP and L-NAME-CGRP groups were directly compared, there was no significant difference between the two (F1,17 = 2.6, p = 0.12; Figure 3(a)). It suggests that L-NAME only partially prevents the effects of CGRP in a subpopulation of neurons. There was still a significant increase in the number of dural-evoked action potential spikes firing within the Aδ-fiber range over 3 h. This was significant after 90 min (7.6 ± 0.4 to 9.7 ± 0.7 spikes per sweep) and remained significant until at least 3 h (10.2 ± 0.6 spikes per sweep; Figure 3(b)). There was no significant effect on action potential spikes firing within the C-fiber range (n = 5; Figure 3(c)), similar to CGRP alone. Data with statistics are summarized in Table 2.
In the six animals where neurons responded to CGRP after L-NAME there was expansion of the cutaneous facial RF (Figure 3(d)). There was also a hypersensitive response to cutaneous facial stimulation after CGRP, with increased neuronal firing to innocuous brush (F4,36 = 10.5, p < 0.001) and noxious pinch (F4,36 = 7.8, p < 0.001). Neuronal responses to cutaneous innocuous brush (t9 = 3.1, p = 0.014; Figure 3(e)) and noxious pinch (t9 = 4.6; p = 0.001; Figure 3(f)) were significantly increased compared to baseline after 90 and 135 min, respectively, and continued until at least 3 h.
Effects of FeTPPS on CGRP-evoked changes trigeminocervical neuronal responses
When the maximal dose of FeTPPS (30 mg/kg, IV) was given prior to CGRP infusion, there was not a statistically significant change in spontaneous trigeminal neuronal firing over 3 h. Based on the critical ratio test, in only 3/12 rats was there an increase in spontaneous firing of >30% over the 3 h in at least two time points, and when FeTPPS-CGRP was directly compared to the CGRP-alone group, with treatment as the between subjects factor, it was significantly different (F1,19 = 6.8, p = 0.017; Figure 3(a)). As a consequence, this did not manifest in hypersensitive neuronal responses to dural stimulation. There was no change in neuronal responses within the Aδ-fiber range (Figure 3(b)) or discharges within the C-fiber range (Figure 3(c)). There was no change in the RF in any of the animals studied after the combination of FeTPPS-CGRP (Figure 3(d)). Similarly, there was no change to neuronal responses to cutaneous facial stimulation after CGRP, for both innocuous brush (F2.4,26.3 = 3.2, p = 0.051; Figure 3(e)) and noxious pinch (F4,44 = 1.0, p = 0.4; Figure 3(f)). Data with statistics are summarized in Table 2.
Behavioral studies
Effects of FeTPPS on CGRP-mediated changes to periorbital mechanical sensitivity
We also conducted behavioral studies, measuring periorbital mechanical thresholds in response to CGRP, and looked for the effects of FeTPPS (30 mg/kg, IP) on the threshold changes. Overall, CGRP (6 µg/kg, IP) caused a significant reduction in periorbital mechanical thresholds over the 5 h on both right (F5,35 = 25.8, p < 0.001, n = 6; Figure 4(a)) and left (F5,35 = 18.9, p < 0.001, n = 6; Figure 4(b)) regions probed. Specifically, this was significant after 1 h (right; t5 = 5.66, p < 0.001, left; t5 = 3.8, p = 0.012) and was maintained for at least 4 h on both sides. This periorbital hypersensitivity was prevented by prior treatment with FeTPPS (right; F5,35 = 2.24, p = 0.082, n = 6, left; F5,35 = 1.3, p = 0.32, n = 6). There was no effect of vehicle control on mechanical sensitivity.

Targeting peroxynitrite prevents CGRP-evoked cutaneous periorbital mechanical hypersensitivity. Time courses changes in cutaneous somatosensory thresholds to von Frey probing on the (a) right and (b) left periorbital regions. CGRP (6 µg/kg, IP, n = 6) mediated a bilateral mechanical hypersensitivity to von Frey probing that was significantly prevented by pre-treatment with FeTPPS (30 mg/kg, IP, n = 6). Vehicle control (normal saline) had no effects.
Discussion
Here we reveal that targeting PN, by accelerating its breakdown, inhibits ongoing and dural-evoked activation of trigeminocervical neurons, a well-validated preclinical migraine model known to predict the efficacy of both abortive and preventive drugs (18,19,27). Further, blocking NO production, with the non-selective NOS inhibitor (L-NAME) similarly inhibits these noxious trigeminocervical responses. This is not unexpected: L-NAME attenuates other dural-evoked trigeminovascular outcomes (40,43), and a non-selective NOS inhibitor was found to more likely provide headache relief compared to placebo in a small-scale clinical trial (8). Neither therapeutic approach affected normal “non-pathological” facial cutaneous nociceptive responses. These last data overlap with studies on spinal nociceptive processing, which conclude that NO, SO or PN have little role in acute, normal physiological nociceptive processing (44,45). Together these data highlight the importance of the RNOS pathway in dural-trigeminovascular migraine mechanisms, and suggest that targeting RNOS beyond NO may also be beneficial in the treatment of migraine.
NO production has been strongly linked to pathophysiological mechanisms of migraine. NO donors like nitroglycerin trigger delayed migraine-like headache only in migraineurs (6). In preclinical studies, NO donors activate and sensitize primary afferent (46) and central dural-trigeminocervical neurons (7,47); the pathway thought to be responsible for migraine-like head pain. Our data confirm previous studies that targeting NO may be beneficial in the treatment of migraine headache, supported by a small double-blind clinical study (8). However, as reviewed recently (48) targeting NO is not without its problems. Non-selective NOS inhibitors cause profound cardiovascular effects, most likely mediated by actions on endothelial NOS present on the vascular endothelium, fundamental for healthy cardiovascular function (49). Therefore, more selective targeting of NOS isoforms is necessary. Selectively targeting inducible NOS, using GW274150, was shown to be no better than placebo as either an abortive or preventive treatment in clinical trials (50,51), a response that is supported by our preclinical studies (40). Targeting neuronal NOS (nNOS) may therefore represent the safest and best opportunity to target NO directly, given its important role in nociceptive sensitization (4). Targeting nNOS is effective in both peripheral and central assays of trigeminovascular nociceptive activation (40,52–54). Despite this, no clinical trials have been completed to support these findings. One molecule, NXN-462 (NeurAxon, Inc), is listed as in phase 2 development for chronic migraine (48), but with no reported data. A combination nNOS inhibitor with a triptan (NXN-188) did not achieve its primary endpoint of pain relief at 2 h in clinical trial, despite a positive response at both 4 and 24 h for secondary endpoints (55). Further studies are likely needed to confirm the viability of nNOS as a target in migraine.
While NO is known independently to be involved in nociceptive mechanisms, it is also involved in the production of PN via a diffusion-controlled reaction with SO (4,5,15). PN is a powerful pro-nociceptive RNOS produced during the development of pain in the spinal dorsal horn and supraspinal neurons. It is thought to be critical for the induction and maintenance of sensitization of spinal neurons in diverse chronic pain states (11,13). Here, we show that accelerating PN breakdown also inhibits noxious dural-trigeminocervical responses, similar to blocking NOS. This suggests that production of PN along the neuronal migraine pain pathway (trigeminocervical and/or supraspinal) may be one of the major mechanisms through which NO is triggering noxious trigeminovascular activation preclinically, and therefore migraine-like headache in patients. The relevance of PN to migraine mechanisms is supported by a clinical study of peripheral blood platelet samples. It demonstrates that in the presence of the NO precursor, L-arginine, PN production was significantly higher in the blood platelet samples of headache-free migraine patients compared to healthy controls (10). This suggests that migraine patients are more responsive to NO precursor molecules that mediate PN production, even outside an attack.
Having established that PN may be involved in mediating neuronal activation along the migraine pain pathway, we also wanted to determine if PN is involved in migraine mechanisms related to sensitization of dural-trigeminovascular neurons, and whether this effect is independent of being the reaction product of NO and SO. CGRP is strongly implicated in migraine mechanisms, and it is thought to have a bidirectional relationship with NO. NO donors, such as nitroglycerin, trigger migraine headache in migraineurs that is accompanied, in the same patients, with increased levels of CGRP in blood plasma samples (56), and NO donor-evoked changes are inhibited by CGRP receptor antagonists (57,58). Some studies support that NO donors mediate CGRP release via activation of TRPA1 in the dural vasculature (59,60). Similarly, CGRP-evoked trigeminovascular responses are attenuated by blocking NO production (40). We therefore used CGRP to promote trigeminovascular changes and compared the effects of targeting either NO or PN. We demonstrate that CGRP mediates activation, and signatures of sensitization, of Aδ-fiber trigeminocervical neurons that is delayed by 60 min, which translated to a reduction of nociceptive periorbital withdrawal thresholds. Similar to our previous study (20), we demonstrate that CGRP has little effect on C-fiber neurons, which likely reflects the lack of CGRP receptor components on C-fiber neurons (61). These data are similar to results found with other known migraine triggers, nitroglycerin (7,62) and pituitary adenylate cyclase-activating peptide (PACAP) (24). Together, the responses are indicative of intracranial headaches that are exacerbated by physical activity (63), and cutaneous cranial allodynia/hyperalgesia; characteristic of the migraine phenotype (64–66). Despite the fact L-NAME and FeTPPS equally inhibited dural-evoked trigeminocervical neuronal responses, L-NAME only partially blocked CGRP-evoked activation. Spontaneous activity of trigeminocervical neurons did not change significantly over time, suggesting that CGRP-mediated activation was blocked. However, the overall response was not significantly different from CGRP alone. In fact, in 6/10 animals CGRP still appeared to promote activation and across all animals CGRP still mediated indicators of central sensitization of dural-trigeminovascular neurons; hypersensitive neuronal responses to intracranial and extracranial somatosensory stimulation and expansion of the cutaneous RF. We therefore conclude that L-NAME had a minimal effect on this subgroup.
While the effect of L-NAME was only partial, FeTPPS completely prevented CGRP-mediated activation of spontaneous trigeminal responses and the resultant hypersensitivity to cranial stimulation and expansion of the cutaneous facial RF. Thus, the development of sensitization was prevented. In a small subset of animals (3/12) there was a 30% increase or higher in spontaneous firing, but this did not ultimately affect the overall response across the group. These data were confirmed by FeTPPS also blocking CGRP-mediated reduction of periorbital withdrawal thresholds, indicative of allodynia/hyperalgesia. PN is known to be a reaction product of NO and SO (4,5), yet our data suggest that CGRP may be mediating the production of PN along the migraine pain pathway independent of NO and that blocking NO production is not sufficient to completely block the effects of CGRP, unlike by catalyzing the breakdown of PN. This might support that the bidirectional relationship between NO and CGRP is more in favor of NO mediating CGRP production than CGRP mediating NO production. Taken together, these data support the idea that production of PN along the migraine pain pathway is perhaps more important in mediating nociceptive trigeminovascular changes related to migraine headache, than NO on its own. As a consequence, accelerating the breakdown of PN may represent a better therapeutic approach.
In summary, we believe these data validate PN as a novel therapeutic target for the treatment of migraine and support the idea that PN production along the migraine pain pathway is involved in the underlying mechanisms of dural-trigeminovascular sensitization, related to migraine pathophysiology. We do accept that a limitation to our overall conclusion is that the study was conducted in male rats only, and so we are cautious to not over-interpret findings to responses in females. That said, previous studies conducted in male rodents, including our own work (7), have proven to be highly translational, predicting therapeutic responses of approved migraine treatments for both men and women. Changes to menstrual hormones are thought to have an important role in a significant proportion of migraine in women, but they are not well understood (33). These data therefore also provide a platform to study the role of peroxynitrite in migraine mechanisms and as a therapeutic target, but specifically in female rodents, and importantly, during different stages of their estrous cycle, to provide us with an understanding of the role of peroxynitrite, as it relates to the complex role of the menstrual hormones, in triggering migraine attacks in women. Furthermore, PN is believed to have no role in normal physiological processes, being produced and involved specifically in pain mechanisms, whereas both SO (learning and memory) and NO (cardiovascular, immune and inflammatory responses) are known to have important roles in various normal physiological functions (4,15,16). The fact FeTPPS did not alter cutaneous responses in naïve (not sensitized by CGRP) animals reflects these data and suggests accelerating the breakdown of PN will not alter normal physiological function. Importantly, drug development programs have already begun focusing on mediating PN breakdown in a diverse range of pain disorders with molecules more suitable as therapeutics, due to their better oral bioavailability. These programs could be readily extended and accelerated to migraine. Overall, the data suggests that accelerating PN breakdown is a relevant mechanism for the treatment of migraine but, importantly, also a potentially viable drug discovery opportunity.
Article highlights
Peroxynitrite production may be involved in mediating nociceptive responses to dural-trigeminovascular neurons. Peroxynitrite may represent a novel therapeutic target for the treatment of migraine and migraine-like headache disorders.
Footnotes
Data availability
The authors confirm that the data supporting the findings of this study are available within the article. Inquiries for additional data are available from the corresponding author, upon reasonable request.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: SA reports personal fees from Amgen, Allergan, and personal fees from Patent/Legal work in headache and orofacial pain, unrelated to this work.
DS reports no competing interest. MRR reports personal fees from Amgen and Allergan, and personal fees from Patent/Legal work in headache and orofacial pain, unrelated to this work.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been supported by start-up funds from University of Maryland Baltimore (SA) and NYU (MRR)