
Abstract
This study addresses possible interactions of the vasodilators nitric oxide (NO), calcitonin gene-related peptide (CGRP) and prostaglandins, which may be implicated in the generation of vascular headaches. Local application of the NO donator diethylamine-NONOate (NONOate) to the exposed dura mater encephali of the rat caused dose-dependent increases in meningeal blood flow recorded by laser Doppler flowmetry. Pre-application of the CGRP receptor antagonist CGRP8-37 significantly attenuated the evoked blood flow increases, while the cyclooxygenase inhibitors acetylsalicylic acid and metamizol were only marginally effective. Stimulation of rat dura mater with NONOate in vitro caused increases in CGRP release. NADPH- diaphorase activity indicating NO production was restricted to the endothelium of dural arterial vessels. We conclude that increases in meningeal blood flow caused by NO depend partly on the release and vasodilatory action of CGRP from dural afferents, while prostaglandins are not significantly involved.
Introduction
Vascular headaches such as migraine and cluster headache are typically accompanied by changes of the intracranial blood flow (1, 2). Increases in regional cerebral blood flow, which depend on an increased perfusion of pial blood vessels, have been described in migraine attacks (3). Similar changes in meningeal flow, i.e. an increased perfusion of the dura mater encephali, may occur as well but can be measured only in animal experiments (4). Assuming that the blood flow is directly or indirectly linked to nociceptive processes, the meningeal blood flow may be more relevant than the cerebral flow, since crucial nociceptive processes during vascular headaches seem to take place in the dura mater (5, 6). Experimentally, painful sensations could be evoked by electrical and mechanical stimulation of dural but not pial blood vessels (7).
With regard to this possible link between afferent activation, increased blood flow and vascular headaches, three classes of chemical mediators appear to be most important in the meninges. First, elevated levels of the neuropeptide calcitonin gene-related peptide (CGRP) have been found in the venous outflow from the head, both during attacks of migraine and cluster headache, and during experimental stimulation of the meninges (2, 8, 9). Release of CGRP from rat dura mater can also be provoked by inflammatory mediators and by electrical stimulation of the trigeminal ganglion in vitro (10). It is evident that the CGRP is released from thin afferents which can be visualized forming dense networks of CGRP immunoreactive fibres around meningeal arterial blood vessels (11, 12). Second, the universal vasodilatator nitric oxide (NO) seems to be involved in the generation of severe vascular headaches, since i.v. injection of nitrovasodilators such as nitroglycerin that liberate NO provoke migraine-like pains in migraineurs and different types of headaches in other sensitive persons (13–17). In animal experiments increases in blood flow caused by electrical activation of meningeal afferents are attenuated by inhibition of the NO synthesis (18). Third, prostaglandins can be assumed to be involved in the generation of headaches because inhibition of the prostaglandin synthesis by nonsteroidal anti-inflammatory drugs (NSAIDs) such as acetylsalicylic acid (ASS) is very effective in the treatment of headaches including migraine pain (19). E- and I-type prostaglandins are known to be potent vasodilatators (20) and ASS reduces the activity of trigeminal brain stem neurones with input from the meninges (21).
Considering these data, the question arises in which way these types of mediators interact to increase the intracerebral and meningeal blood flow during vascular headaches that are linked to trigeminovascular activation. An experimental approach using models of intracranial flow in animals appeared promising to solve this question. Previous experiments had shown that increases in cerebral blood flow caused by NO donators were attenuated after trigeminal deafferentation or blockade of CGRP receptors (22), and similar effects on the meningeal flow were recently found in our laboratory. Possibly, the blood flow increasing effect of NO donators is not only due to a direct vasodilatory action of arterial blood vessels, but may also be indirectly brought about by the release of CGRP from perivascular afferents, as has been shown also in other tissues (23, 24). In the skin there is some evidence that prostaglandins are additionally involved in the hyperemic effect of NO (25). With regard to these data we asked to which extent blockade of CGRP receptors and prostaglandin synthesis can limit increases in meningeal blood flow caused by a NO donator locally applied onto the exposed dura mater in the rat. Secondly, we asked if this NO donator is able to release CGRP and prostglandin E2 from the dura mater using an in vitro preparation and direct measurement of mediator release by an enzyme immunoassay. To define the endogenous NO producing structures and to visualize possible changes in their NO producing activities we used nicotinamide adenine dinucleotide phosphate diaphorase (NADPH-diaphorase) histochemistry in control dura mater preparations and in dura mater from animals stimulated with the NO donator.
Methods
Dural arterial blood flow
Anaesthesia and general preparation
Male Wistar rats (Charles River, Germany) with body weights of 300–450 g were used. Experimental procedures, which were performed according to the ethical issues for animal care and treatment were approved by a comittee of the local district government. The animals were initially anaesthetized with 150 mg/kg thiopentone (Trapanal, Byk Gulden, Konstanz, Germany) i.p., followed by additional doses of 25 mg/kg thiopentone when required. Catheters were inserted into the right femoral vein for infusion of solutions and into the right femoral artery for continuous recording of the systemic blood pressure. Depth of anaesthesia was routinely assessed and held at a level in which noxious stimuli (pinching of earlobes and feet) failed to elicit motor reflexes or changes of the systemic arterial pressure, which was 100–120 mmHg (mean). The animals were tracheotomized but breathed spontaneously while the air was enriched by oxygen delivered to the vicinity of the tracheal tube. In some experiments the animals were artificially ventilated with room air and oxygen. In this case, the end-expiratory CO2 was held at 3.5% to suppress spontaneous breathing. No systemic difference was found between spontaneously breathing and ventilated animals regarding systemic blood pressure and dural blood flow. Body temperature of animals was maintained at 37.0–37.5°C with a thermostatically regulated heating plate. For termination of experiments a lethal dose of i.v. thiopentone was given.
Specific preparation and recording of blood flow
The preparation for the recording of dural blood flow has been reported in detail by Kurosawa et al. (4). The head of the animal was fixed in a stereotaxic frame. An incision was made along the midline of the scalp, and the apical and parietal parts of the skull were exposed. Using a dental drill and liquid cooling with drops of saline, a cranial window of 4 × 6 mm was drilled into the parietal bone to expose the dura mater with the medial meningeal artery (MMA). Blood flow was recorded by a laser Doppler system (Moor Instruments, DRT4, time constant 1.0 s) via needle type probes (tip diameter 0.8 mm) positioned over branches of the MMA. The dura mater around the probes was covered with saline and the borders of the trepanized bone were covered with vaseline to hold the solutions within the cranial window.
Drug administration
The NO-donator diethylamine-NONOate (NONOate; Calbiochem, Bad Soden/Ts., Germany) was locally applied at concentrations of 10−5–10−3 M (in 50 µl saline) to the exposed dural surface for 5–10 min Then the dura was washed with saline to remove the NO donator. When the baseline blood flow was restored, the CGRP receptor antagonist CGRP8-37 (Calbiochem, Bad Soden/Ts., Germany) at 10−4 M (in 50 µl saline) was locally applied for 2 min and then removed prior to a second application of the same concentration of the NO donator. When the baseline flow was restored again after wash-out with saline, the cyclooxygenase inhibitors DL-Lysinmono-acetylsalicylate (ASS; Bayer, Leverkusen, Germany) at concentrations of 1 and 10 mg/ml, or metamizol-Na (NovalginR; Hoechst Marion Roussel, Germany) at a concentration of 10 mg/ml (in 50 µl saline each) were locally applied. After 10 min the cyclooxygenase inhibitor was removed and the same concentration of the NO donator was again administered.
Measuring and evaluation of data
Blood flow was measured as the mean flow within periods of 60 s using the averaging program of the DRT4 software (output reading in mV). Flow values after administration of drugs were taken from periods in which the flow reached a maximum and remained at a high level. These flow values were diminished by the basal flow measured before administration of the NO donator to calculate the flow increase. After wash-out of substances, basal flow values were restored in most experiments within 20 min. If the basal flow slightly differed from the previous level, the new baseline was taken to evaluate effects caused by the following substance. Differences between flow increases caused by the NO donator alone and by the NO donator after an antagonist or inhibitor were compared using the analysis of variance (
Release of calcitonin gene-related peptide and prostaglandin E2 in vitro
Preparation
Following the method of Ebersberger et al. (10), immediately after decapitation, the head of male Wistar rats (150–350 g) was halved by a clear cut along the midline using a fine saw. The two cerebral hemispheres were removed without lesioning the dura mater encephali that covered the medial and the posterior cranial fossa. The skull cavities were washed for 30 min at room temperature by superfusion with carbogen-gased synthetic interstitial fluid (SIF) containing: 108 m
Sampling and drug application
Five consecutive samples of the superfusate were collected at periods of 5 min by evacuating the fluid from the skull halves with a pipette without touching the dura. The cavities were then repeatedly refilled with the same volumes of SIF. The collected fluid was cooled on ice in micro test tubes. During the third incubation period 500 µl of NONOate was applied at concentrations of 10−5–10−3 M (in SIF) for 5 min. After drug application, three further samples of eluate were collected from each preparation. All samples were frozen overnight at −20°C for later analysis.
Enzyme immunoassays
For measurements of CGRP and PGE2 concentrations, enzyme immunoassays (EIAs) were used (CGRP: SPIbio Company, France; PGE2: assay adapted in our laboratories). The samples of the two half skulls of each animal were pooled for analysis and diluted with an EIA buffer (five-fold concentrated, ratio 5 : 1) immediately after collection. The minimum detection limit of the assay was 2 pg/ml for CGRP and 30 pg/ml for PGE2. The intra- and inter-assay coefficients of variation were 15–20% for basal release data and 10–15% for values of stimulated release. Standard curves were run with graded CGRP and PGE2 concentrations in SIF.
Data analysis
The concentrations of CGRP and PGE2 were calculated as pg/ml eluate. CGRP and PGE2 concentrations of the second incubation period were taken as baseline values to normalize the data. Means±SEM from groups of identical types of experiments were calculated. An analysis of variance (
Nicotinamide adenine dinucleotide phosphate diaphorase (NADPH-diaphorase) histochemistry
Animals, the dura mater of which was stimulated with NO donator, and control animals were deeply anaesthetized and perfused transcardially with 300 ml saline. The skull was cut in the midline as described above. After removing the brain, the dura mater was fixed locally by superfusion with a fixative containing 4% paraformaldehyde. After fixation for 15 min the parietal dura mater was carefully pulled off from the skull. The whole mounts were preincubated in 0.1
Results
Dural arterial blood flow
Effect of CGRP8-37 on the flow increases caused by NONOate
The NO donator NONOate at concentrations of 10−5 M to 10−3 M was locally applied onto the exposed dura mater while the blood flow in branches of the MMA was measured. NONOate caused concentration-dependent increases in blood flow (Fig. 1a; Fig. 2). The onset latency of these increases was between 20 and 90 s and the maximum of flow increase was reached after 50–200 s (10−3 M). After preapplication of the CGRP receptor antagonist CGRP8-37 at 10−4 M onto the dura the flow increases induced by NONOate at all concentrations(10−5 M to 10−3 M) were significantly smaller (Fig. 1b; Fig. 2). CGRP8-37 was used at a concentration of 10−4 M because in a former study (4) this dose had been shown to reduce the electrically evoked increases in meningeal flow most effectively without changing the basal flow. As a control, a third dose of NONOate was given 30 min after CGRP8-37 causing nearly the same amplitude in flow increase as the first application before CGRP8-37, thus indicating complete recovery. The systemic arterial pressure was not changed by application of any of the substances.

Records of dural blood flow showing three parts of one experiment. Increases in flow caused by local application of (a) the NO donator NONOate, (b) by NONOate after preapplication of CGRP8-37 and (c) by NONOate after preapplication of ASS.

Mean increases (±SEM) in blood flow following local application of NONOate at concentrations of 10−5 M (n=6), 10−4 M (n=8), 10−3 M (n=10) before (▪) and after (□) preapplication of CGRP8-37 (∗P <0.05,
Effects of ASS and metamizol on the flow increases caused by NONOate
NONOate (10−5 M to 10−3 M) was locally applied onto the dura mater causing concentration-dependent increases in meningeal blood flow (Fig. 3). After wash-out with saline, the cyclooxygenase inhibitors ASS (1 mg/ml or 10 mg/ml) or metamizol (10 mg/ml) were pre-applied onto the dura. Both ASS and metamizol caused slight lowering of the basal blood flow that was not significant on average. After ASS at both doses or after metamizol the blood flow increases induced by NONOate at three concentrations (10−5 M to 10−3 M) were smaller (Fig. 1c; Fig. 3). The difference, however, was only significant for NONOate at 10−4 M following ASS at 1 mg/ml (Fig. 3a).
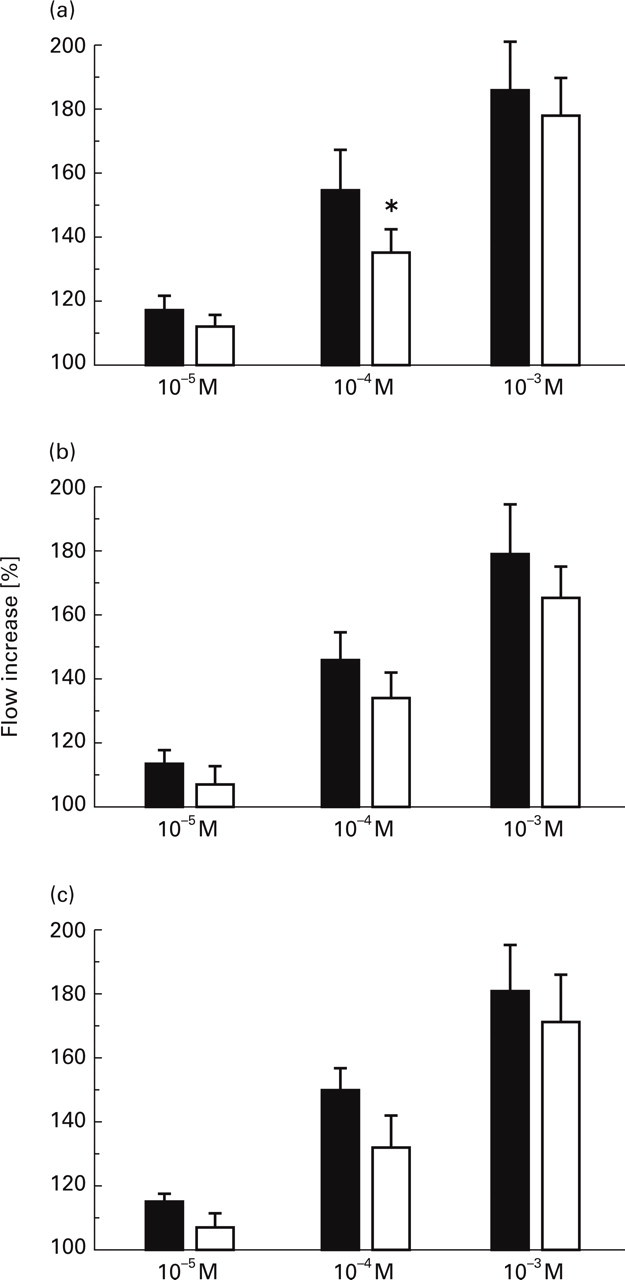
Mean increases (±SEM) in blood flow caused by the NO donor NONOate at concentrations of 10−5 M (n=6), 10−4 M (n=8), 10−3 M (n=10) before (▪) and after (□) pre-application of ASS 1 mg/ml, ASS 10 mg/ml and Metamizol (NovalginR) 10 mg/ml (∗P<0.05,
Release of calcitonin gene-related peptide and prostaglandin E2 in vitro
In these experiments hemisected skulls with adhering dura mater encephali were used as an in vitro preparation to measure CGRP and PGE2 release by an enzyme immuno assay. Skulls were filled with SIF and the release was determined at periods of 5 min. During the third period the dura mater was stimulated with NONOate (solutions of 10−5 M to 10−3 M in SIF). NONOate at all concentrations caused concentration-dependent increases in CGRP release, which were significantly higher than the release before and after the stimulation (Table 1; Fig. 4).
CGRP and PGE2 release (mean±SEM) within 5 min following NONOate at concentrations of 10−5–10−3 M; ∗ significant increase (


Increases in CGRP release (normalized data, mean±SEM) caused by stimulation of the rat dura mater of hemisected skulls with NONOate at concentrations of • 10−5 M, Δ 10−4 M, ▪ 10−3 M, n=6 at all concentrations (∗P<0.05,
The PGE2 release was slightly increased after stimulation with NONOate at all concentrations without concentration-dependency. The stimulated release was rather variable, therefore the mean data were not significantly different to the control values before and after NONOate (Table 1; Fig. 5).

Increases in PGE2 release (normalized data, mean±SEM) caused by stimulation of the rat dura mater of hemisected skulls with NONOate at concentrations of • 10−5 M, Δ 10−4 M, ▪ 10−3 M, n=6 at all concentrations (∗P<0.05,
Localization of NADPH-diaphorase activity in the dura mater
To visualize the localization of NO producing structures in the dura mater, NADPH-diaphorase histochemistry was used. Enzyme activity of NADPH-diaphorase was localized in the wall of arteries and arterioles up to the transition into capillaries, but not in capillary or postcapillary sections of the vasculature (Fig. 6). Nerve fibres or other cells did not indicate any significant NADPH-diaphorase activity. There was no obvious difference in the distribution or intensity of NADPH-diaphorase activities of control animals and animals the dura of which was stimulated with NONOate during the experiments described above.

Light microscopic photomicrograph showing NADPH-diaphorase activity in the branches of the middle meningeal artery in the dura mater of an NO donator stimulated rat. Enzyme activity is localized in the wall of arteries and arterioles up to the transition into capillaries (arrows). Bar: 250 µm.
Discussion
Direct and indirect actions of NO
The results of the present paper suggest that increases in meningeal blood flow caused by local application of NO donators depend partly on the release and vasodilatory action of CGRP from dural afferent fibres. Additionally, a synergistic mechanism of NO and CGRP in smooth muscle cells of dural arterial vessels may be involved, e.g. a permissive effect of intracellular cAMP increase caused by CGRP on the cGMP mechanism induced by NO. Secondly, our results suggest that the release and the vasodilatory action of prostaglandins by NO, as it was suggested to occur in the skin (25) is not significantly involved in this effect. Prostaglandin E2 has been shown experimentally in several tissues to sensitize nociceptive afferents to different stimuli (27, 28), and it is very likely that this occurs also in the meninges (29, 30). Therefore it can be concluded that the efficacy of NSAIDs in inhibiting trigeminal activation (21) and plasma extravasation evoked by trigeminal stimulation in animal experiments (31) is more likely to be a direct inhibitory effect on trigeminal afferents and higher order neurones than an effect on the prostaglandin-dependent vasodilatation. Accordingly, the benefit of prostaglandin synthesis inhibition in the therapy of headaches may be due to this direct inhibitory action (32, 33). Data showing direct effects of NO and CGRP on the activity of trigeminal neurones do not exist so far, but there is evidence that in other tissues NO acts pro-nociceptive after prolonged noxious stimulation or inflammation (34, 35). In the meninges NO synthesis is increased under inflammatory conditions (36, 37), where it causes pathological changes such as the breakdown of the blood–brain barrier and the induction of pro-nociceptive cytokines (38). If NO production is also increased in attacks of primary headaches such as migraine pain and cluster headache, similar mechanisms may contribute to the rapid activation of the trigeminal system. Furthermore, the linkage of NO production and CGRP release may be very relevant under these pathological conditions with regard to the question if the powerful vasodilatation of meningeal blood vessels itself can be a nociceptive stimulus (39).
Origin of endogenous NO in the meninges
Regarding NO as an important mediator in meningeal vasodilatation and possibly as a nociceptive agent, the question arises where NO in meningeal tissues is produced. In a recent study we have presented evidence that NO which contributes both to the maintenance of the basal meningeal blood flow and to increases in flow caused by local electrical stimulation is mainly endothelium-derived, because specific inhibitors of the neuronal and the inducible NO synthetase were not or only marginally effective in inhibiting basal and evoked flow (18). To collect additional evidence for this assumption we used NADPH-diaphorase staining in normal rat dura mater as well as in dura mater from animals stimulated with NO donator. NADPH-diaphorase activity is colocalized with NO synthetase and therefore can be used as a marker for NO producing cells (40, 41). The stainings showed NADPH-diaphorase activity in all arterial segments of the dural vascularization but not in capillaries and venous segments. This suggests that the only cells which considerably contribute to NO production are endothelial cells located in precapillary blood vessels. Other sources of NO production, however, cannot strictly be excluded, since immunohistochemistry using antibodies against NO synthetase have shown this enzyme also in dural nerve fibres and mast cells (42). Finally, the distribution of NADPH-diaphorase activity in dura treated with NO donators was not different to untreated dura, suggesting that there is no up-regulation of NO production by NO induced vasodilatation in the meninges.
Significance of NO mediated vasodilatation in the pathophysiology of vascular headaches
It appears very likely that an increase in NO production may contribute to form a vicious circle in the meninges which involves the release of CGRP and possibly prostaglandins and other mediators (such as histamine) leading to a rapid vasodilatation. An involvement of other nociceptive cascades such as the release of inflammatory mediators following venous leakage and activation of mast cells, or the production of cytokines following attraction of leukocites, could additionally contribute to the sensitization of meningeal nociceptors (30, 43) and trigeminal brainstem neurones (29, 44). It is unclear, however, if these changes occur in vascular headaches, because the data are derived from animal experiments. Meningeal afferents, which have been shown to be very sensitive to mechanical stimuli (30, 44), may finally respond to the vasodilatory and pulsating vasomotor changes of meningeal arterial vessels provoked by the vasodilators, thus causing the typical pressure-like and pulsating pain frequently experienced during vascular headaches.
Footnotes
Acknowledgements
The authors thank Mrs J. Schramm and Mrs B. Vogler for their excellent technical assistance. The study was granted by the Wilhelm-Sander-Stiftung. M. Dux is a fellow of the Alexander von Humboldt-Stiftung.